
Determination of Cellulose Degree of Polymerization in Historical Papers with High Lignin Content



Abstract
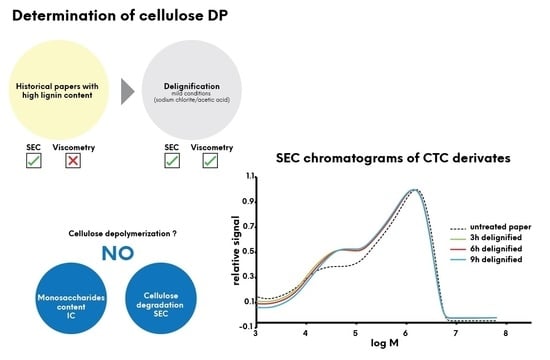
1. Introduction
2. Materials and Methods
2.1. Paper Samples
2.2. Fiber Furnish Analyses
2.3. Paper Delignification
2.4. pH Measurements
2.5. Kappa Number
2.6. Viscometry
2.7. Determination of Weight Average Molecular Mass
2.8. Determination of Monosaccharides
3. Results and Discussion
3.1. Determination of Kappa Number and Solubility in CED
3.2. Stability of Cellulose during Delignification
3.3. Viscometric Determination of Cellulose Average DPv of Delignified Paper Samples
3.4. Reliability of the Results
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Łojewski, T.; Zięba, K.; Knapik, A.; Bagniuk, J.; Lubańska, A.; Łojewska, J. Evaluating paper degradation progress. Cross-linking between chromatographic, spectroscopic and chemical results. Appl. Phys. A 2010, 100, 809–821. [Google Scholar] [CrossRef]
- Zaccaron, S.; Henniges, U.; Potthast, A.; Rosenau, T. How alkaline solvents in viscosity measurements affect data for oxidatively damaged celluloses. Cuoxam and Cadoxen. Carbohydr. Polym. 2020, 240, 116251. [Google Scholar] [CrossRef]
- Strlič, M.; Liu, Y.; Lichtblau, D.A.; De Bruin, G.; Knight, B.; Winther, T.; Kralj Cigić, I.; Brereton, R.G. Development and mining of a database of historic European paper properties. Cellulose 2020, 27, 8287–8299. [Google Scholar] [CrossRef]
- Małachowska, E.; Dubowik, M.; Boruszewski, P.; Łojewska, J.; Przybysz, P. Influence of lignin content in cellulose pulp on paper durability. Sci. Rep. 2020, 10, 19998. [Google Scholar] [CrossRef]
- Vizárová, K.; Kirschnerová, S.; Kačík, F.; Briškárová, A.; Šutý, Š.; Katuščák, S. Relationship between the decrease of degree of polymerisation of cellulose and the loss of groundwood pulp paper mechanical properties during accelerated ageing. Chem. Pap. 2012, 66, 1124–1129. [Google Scholar] [CrossRef]
- Schmidt, J.A.; Rye, C.S.; Gurnagul, N. Lignin inhibits autoxidative degradation of cellulose. Polym. Degrad. Stab. 1995, 49, 291–297. [Google Scholar] [CrossRef]
- Brown, N.; Lichtblau, D.; Fearn, T.; Strlič, M. Characterisation of 19th and 20th century Chinese paper. Herit. Sci. 2017, 5, 47. [Google Scholar] [CrossRef]
- Łojewski, T.; Zięba, K.; Łojewska, J. Size exclusion chromatography and viscometry in paper degradation studies. New Mark-Houwink coefficients for cellulose in cupri-ethylenediamine. J. Chromatogr. A 2010, 42, 6462–6468. [Google Scholar] [CrossRef]
- Dupont, A.L.; Benaud, O.; Mortha, G. Chlorite-based pretreatments of cellulosic samples prior to SEC analysis. In Book of Abstracts, Proceedings of the 27th International Symposium on Polymer Analysis and Characterization, Les Diablerets, Switzerland, 15–18 June 2014; ISPAC: Ormont-Dessus, Suisse, 2014; p. 51. Available online: https://ispac-conferences.org/Data/Sites/1/ISPAC2014/documents/ispac-2014-book-of-abstracts.pdf (accessed on 16 June 2021).
- Conner, A.H. Size Exclusion Chromatography of Cellulose and Cellulose Derivatives. In Handbook of Size Exclusionchromatography; Chromatographic Science Series; Wu, C., Ed.; Marcel Dekker, Inc.: New York, UY, USA, 1995; Volume 69, pp. 331–352. [Google Scholar]
- Potthast, A.; Radosta, S.; Saake, B.; Lebioda, S.; Heinze, T.; Henniges, U.; Isogai, A.; Koschella, A.; Kosma, P.; Rosenau, T.; et al. Comparison testing of methods for gel permeation chromatography of cellulose: Coming closer to a standard protocol. Cellulose 2015, 22, 1591–1613. [Google Scholar] [CrossRef]
- Kaminska, E. Determination of degree of polymerization of cellulose in ligneous papers. In Materials Issues in Art and Archaeology V, Proceeding of the Materials Research Society Symposium, Boston, MA, USA, 2–6 December 1996; Vandiver, P.V., Druzik, J.R., Eds.; Materials Research Society: Warrendale, MI, USA, 1997; Volume 462, pp. 45–51. [Google Scholar]
- Strlič, M.; Kolar, J.; Pihlar, B. Methodology and analytical techniques in paper stability studies. In Ageing and Stabilisation of Paper; Strlič, M., Kolar, J., Eds.; National and University Library: Ljubljana, Slovenia, 2005; pp. 27–47. [Google Scholar]
- Dupont, A.L.; Réau, D.; Bégin, O.; Paris-Lacombe, S.; Tétreault, J.; Mortha, G. Accurate molar masses of cellulose for the determination of degradation rates in complex paper samples. Carbohydr. Polym. 2018, 202, 172–185. [Google Scholar] [CrossRef] [PubMed]
- Kačík, F.; Kačíková, D.; Jablonský, M.; Katuščák., S. Cellulose degradation in newsprint paper ageing. Polym. Degrad. Stab. 2009, 94, 1509–1514. [Google Scholar] [CrossRef]
- Dupont, A.L.; Mortha, G. Comparative evaluation of size-exclusion chromatography and viscometry for the characterisation of cellulose. J. Chromatogr. A 2004, 1026, 129–141. [Google Scholar] [CrossRef] [PubMed]
- SCAN-CM 15:88. Viscosity in Cupri-Ethylenediamine (CED) Solution; SCAN: Stockholm, Sweden, 1998. [Google Scholar]
- Tappi T 230 om-08. Viscosity of Pulp (Capillary Viscometer Method); (Revision of T230 om-08, draft No. 3); TAPPI: Atlanta, GA, USA, 2013; Available online: https://www.tappi.org/content/tag/sarg/t230.pdf (accessed on 6 June 2021).
- Hubbell, C.A.; Ragauskas, A. Effect of acid-chlorite delignification on cellulose degree of polymerization. Bioresour. Technol. 2010, 101, 7410–7415. [Google Scholar] [CrossRef]
- Kumar, R.; Mago, G.; Balan, V.; Wyman, C.E. Physical and chemical characterizations of corn stover and poplar solids resulting from leading pretreatment technologies. Bioresour. Technol. 2009, 100, 3948–3962. [Google Scholar] [CrossRef]
- Singh, O.P. Kinetics and mechanism of hypochlorite oxidation of cellulose. Text. Dyer Print. 1982, 15, 35–38. [Google Scholar]
- Mortha, G.; Marcon, J.; Dallérac, D.; Marlin, N.; Vallée, C.; Charon, N.; Le Masle, A. Depolymerization of cellulose during cold acidic chlorite treatment. Holzforschung 2015, 69, 731–736. [Google Scholar] [CrossRef]
- Kumar, R.; Hu, F.; Hubbell, C.A.; Ragauskas, A.J.; Wyman, C.E. Comparison of laboratory delignification methods, their selectivity, and impacts on physiochemical characteristics of cellulosic biomass. Bioresour. Technol. 2013, 130, 372–381. [Google Scholar] [CrossRef]
- Čabalová, I.; Kačík, F.; Gojný, J.; Češek, B.; Milichovský, M.; Mikala, O.; Tribulová, T.; Ďurkovič, J. Changes in the chemical and physical properties of paper documents due to natural ageing. BioResources 2017, 12, 2618–2634. [Google Scholar] [CrossRef]
- Stol, R.; Pedersoli, J.L., Jr.; Poppe, H.; Kok, W.T. Application of size exclusion electrochromatography to the microanalytical determination of the molecular mass from objects of cultural and historical value. Anal. Chem. 2002, 74, 2314–2320. [Google Scholar] [CrossRef]
- ISO 9184-1Paper, Board and Pulps-Fibre Furnish Analysis; ISO: Geneva, Switzerland, 1990.
- TAPPI T 509 om-02. Hydrogen Ion Concentration (pH) of Paper Extracts (Cold Extraction Method); TAPPI: Atlanta, GA, USA, 2002. [Google Scholar]
- Matija, S.; Kolar, J.; Kočar, D.; Drnovšek, T.; Šelih, V.S.; Susič, R.; Pihlar, B. What is the pH of alkaline paper? e-PS 2004, 1, 35–47. [Google Scholar]
- TAPPI/ANSI T 236 om-13. Kappa Number of Pulp; TAPPI: Atlanta, GA, USA, 2013. [Google Scholar]
- ISO 5351:2004. Pulps-Determination of Limiting Viscosity Number in Cupri-Ethylenediamine (CED) Solution; ISO: Geneva, Switzerland, 2004. [Google Scholar]
- Evans, R.; Wallis, A. Cellulose molecular weights determined by viscometry. J. Appl. Polym. Sci. 1989, 37, 2331–2340. [Google Scholar] [CrossRef]
- Kolar, J.; Malešič, J.; Kočar, D.; Strlič, M.; de Bruin, G.; Koleša, D. Characterisation of paper containing iron gall ink using size exclusion chromatography. Polym. Degrad. Stab. 2012, 97, 2212–2216. [Google Scholar] [CrossRef]
- TAPPI. Kappa Number of Pulp TAPPI/ANSI T 236 om-13. Available online: https://imisrise.tappi.org/TAPPI/Products/01/T/0104T236.aspx (accessed on 9 January 2021).
- ISO 302:2015. Pulps—Determination of Kappa Number; ISO: Geneva, Switzerland, 2015. [Google Scholar]
- Łojewska, J.; Rabin, I.; Pawcenis, D.; Bagniuk, J.; Aksamit-Koperska, M.A.; Sitarz, M.; Missori, M.; Krutzsch, M. Recognizing ancient papyri by a combination of spectroscopic, diffractional and chromatographic analytical tools. Sci. Rep. 2017, 7, 46236. [Google Scholar] [CrossRef] [PubMed]
- Kačík, F.; Podzimek, Š.; Vizárová, K.; Kačíková, D.; Čabalová, L. Characterization of cellulose degradation during accelerated ageing by SEC-MALS, SEC-DAD, and A4F-MALS methods. Cellulose 2016, 23, 357–366. [Google Scholar] [CrossRef]
- Łojewski, T.; Zięba, K.; Kołodziej, A.; Łojewska, J. Following cellulose depolymerization in paper: Comparison of size exclusion chromatography techniques. Cellulose 2011, 18, 1349. [Google Scholar] [CrossRef]
- Balažic, A.; Kolar, J.; Strlič, M.; Žagar, E. Size exclusion chromatography of cellulose. In Proceedings of the Durability of paper and writing 2: Book of abstracts, Ljubljana, Slovenia, 7–9 July 2008; Strlič, M., Kolar, J., Eds.; Faculty of Chemistry and Chemical Technology: Ljubljana, Slovenia, 2008; pp. 33–35. [Google Scholar]
- Strlič, M.; Kolar, J.; Žigon, M.; Pihlar, B. Evaluation of size-exclusion chromatography and viscometry for the determination of molecular masses of oxidised cellulose. J. Chromatogr. A 1998, 805, 93–99. [Google Scholar] [CrossRef]
- Ahn, K.; Zaccaron, S.; Rosenau, T.; Potthast, A. How Alkaline Solvents in Viscosity Measurements Affect Data for Oxidatively Damaged Celluloses: Cupri-Ethylenediamine. Biomacromolecules 2019, 20, 4117–4125. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample No. | Year of Publication | pH Value | Groundwood Fiber Content (%) | Bleached Chemical Pulp (%) | Cotton Fibers (%) |
|---|---|---|---|---|---|
| 1 | 1983 | 5.1 | 85 | 15 | |
| 2 | 1960 | 6.4 | 66 | 34 | |
| 3 | 1958 | 6.2 | 65 | 35 | |
| 4 | 1903 | 4.8 | 64 | 36 | |
| 5 | 1982 | 5.3 | 84 | 14 | 2 |
| 6 | 1987 | 5.6 | 45 | 26.6 | 28.5 |
| 7 | 1953 | 6.9 | 58 | 42 | |
| 8 | 1965 | 5.8 | 64 | 36 | |
| 9 | 1889 | 4.9 | 81 | 19 | |
| 10 | 1899 | 5.1 | 61 | 39 | |
| 11 | 1898 | 5.3 | 58 | 42 | |
| 12 | 1896 | 5.3 | 68 | 32 | |
| 13 | 1909 | 5.5 | 35 | 65 | |
| 14 | 1911 | 4.9 | 66 | 34 | |
| 15 | 1887 | 4.6 | 57 | 20.5 | 22.5 |
| Sample No. | Time of Delignification (h) | Kappa Number | Solubility in CED |
|---|---|---|---|
| 1 | 0 | above 100 | |
| 3 | 44 | ||
| 6 | 38 | ||
| 9 | 19 | + | |
| 2 | 0 | 97 | |
| 6 | 19 | + | |
| 3 | 0 | 97 | |
| 6 | 18 | + | |
| 4 | 0 | 72 | |
| 6 | 8 | + | |
| 5 | 0 | above 100 | |
| 6 | 42 | ||
| 9 | 18 | + | |
| 6 | 0 | 75 | |
| 6 | 16 | + | |
| 7 | 0 | 76 | |
| 6 | 20 | + | |
| 8 | 0 | 92 | |
| 6 | 32 | ||
| 9 | 11 | + | |
| 9 | 0 | 81 | |
| 6 | 25 | ||
| 9 | 7 | + | |
| 10 | 0 | 62 | |
| 6 | 10 | + | |
| 11 | 0 | 57 | |
| 6 | 16 | + | |
| 12 | 0 | 81 | |
| 6 | 20 | + | |
| 13 | 0 | 19 | + |
| 6 | 3 | + | |
| 14 | 0 | 24 | |
| 6 | 3 | + | |
| 15 | 0 | 82 | |
| 6 | 23 | + |
| Monosaccharide | First Filtrate, 3 h | First Filtrate, 6 h | Second Filtrate, 9 h |
|---|---|---|---|
| glucose | 0.11 mg·L−1 | 0.12 mg·L−1 | <LOD |
| mannose | nd | nd | nd |
| galactose | nd | <LOD | nd |
| xylose | nd | <LOD | nd |
| arabinose | <LOD | 1.16 mg·L−1 | nd |
| Sample No. | Mw Before Delignification (mg·L−1) | Mw After Delignification (mg·L−1) | Mw Difference (%) |
|---|---|---|---|
| 1 | 989,500 | 971,000 | 1 |
| 2 | 1,117,000 | 1,026,000 | 6 |
| 3 | 1,127,000 | 1,027,000 | 7 |
| 4 | 486,000 | 527,500 | 6 |
| 5 | 867,000 | 997,000 | 10 |
| 6 | 1,011,500 | 963,000 | 3 |
| 7 | 939,000 | 900,500 | 3 |
| 8 | 914,500 | 803,000 | 9 |
| 9 | 755,000 | 727,500 | 3 |
| 10 | 663,000 | 685,500 | 2 |
| 11 | 695,500 | 735,000 | 4 |
| 12 | 1,040,500 | 970,000 | 5 |
| 13 | 471,000 | 447,000 | 4 |
| 14 | 351,000 | 350,500 | below 1 |
| 15 | 363,000 | 412,000 | 9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malešič, J.; Kraševec, I.; Kralj Cigić, I. Determination of Cellulose Degree of Polymerization in Historical Papers with High Lignin Content. Polymers 2021, 13, 1990. https://doi.org/10.3390/polym13121990
Malešič J, Kraševec I, Kralj Cigić I. Determination of Cellulose Degree of Polymerization in Historical Papers with High Lignin Content. Polymers. 2021; 13(12):1990. https://doi.org/10.3390/polym13121990
Chicago/Turabian StyleMalešič, Jasna, Ida Kraševec, and Irena Kralj Cigić. 2021. "Determination of Cellulose Degree of Polymerization in Historical Papers with High Lignin Content" Polymers 13, no. 12: 1990. https://doi.org/10.3390/polym13121990
APA StyleMalešič, J., Kraševec, I., & Kralj Cigić, I. (2021). Determination of Cellulose Degree of Polymerization in Historical Papers with High Lignin Content. Polymers, 13(12), 1990. https://doi.org/10.3390/polym13121990