
Multiblock Copolymers of Norbornene and Cyclododecene: Chain Structure and Properties


 , ,
, , 

Abstract
1. Introduction
2. Materials and Methods
3. Results and Discussion
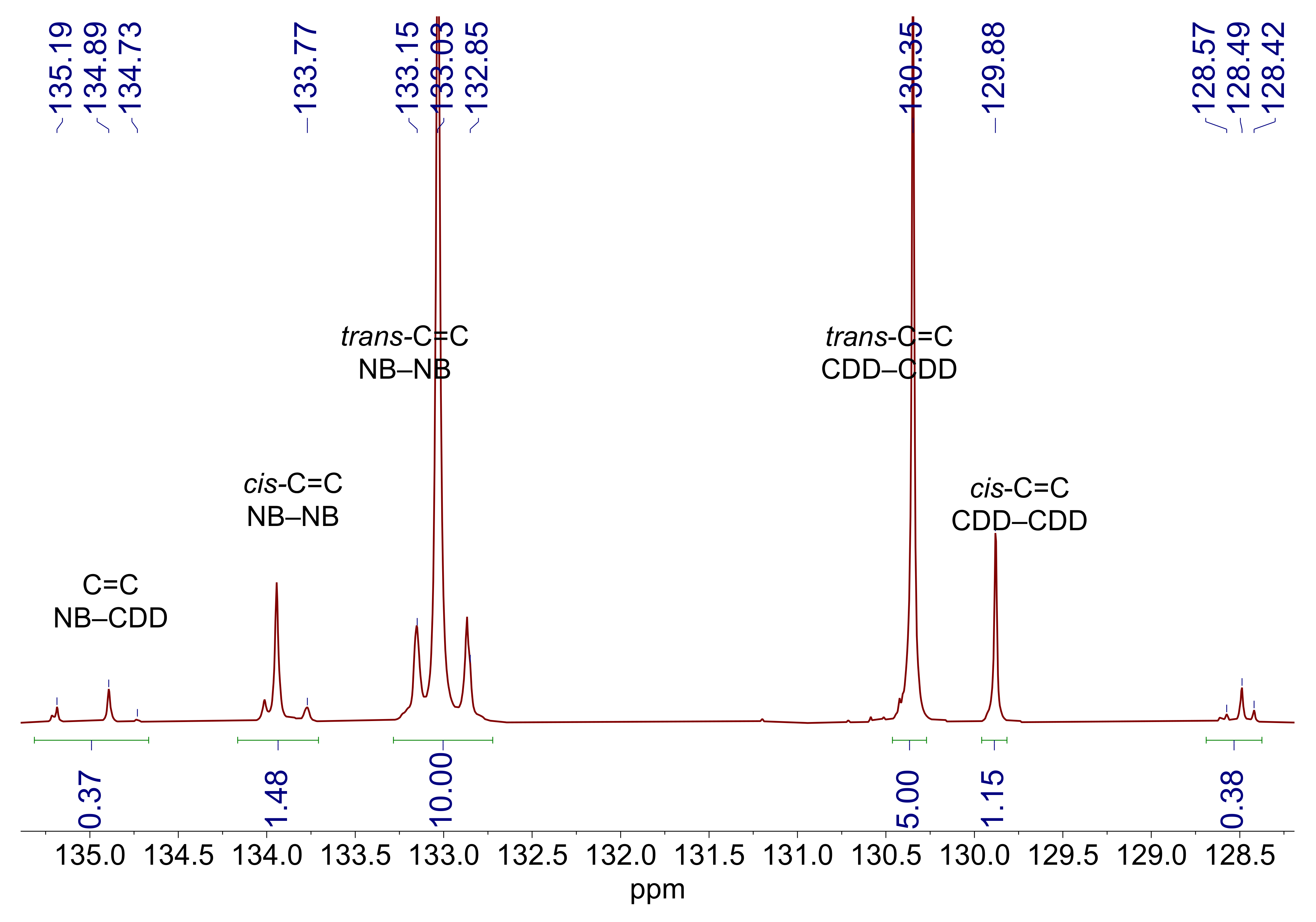
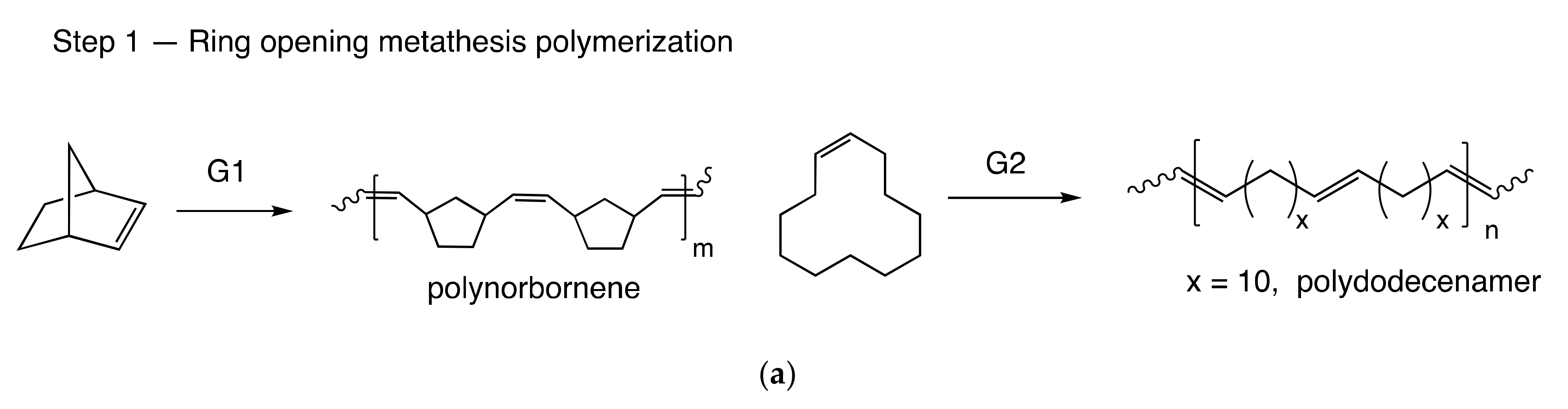
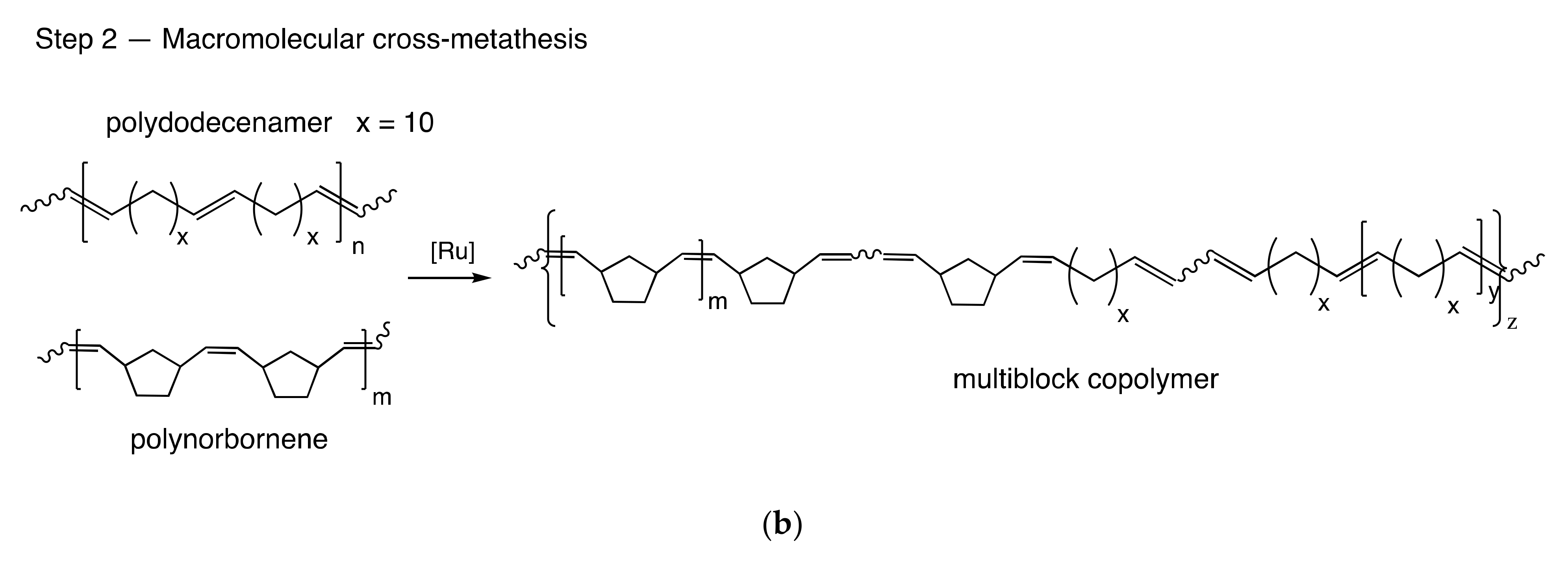
3.1. Copolymer Synthesis and Chain Structure
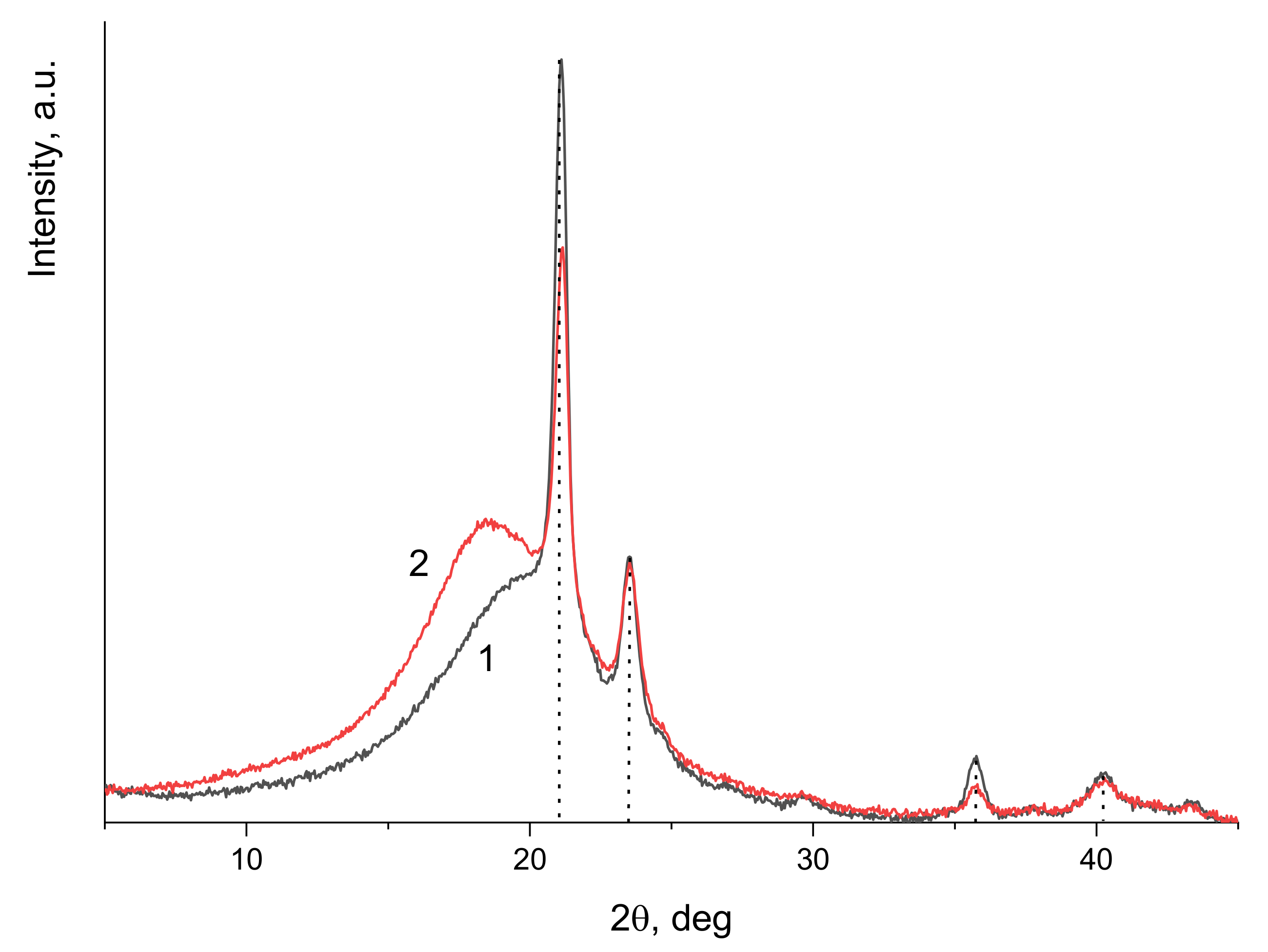
3.2. Copolymer Structure and Crystallinity
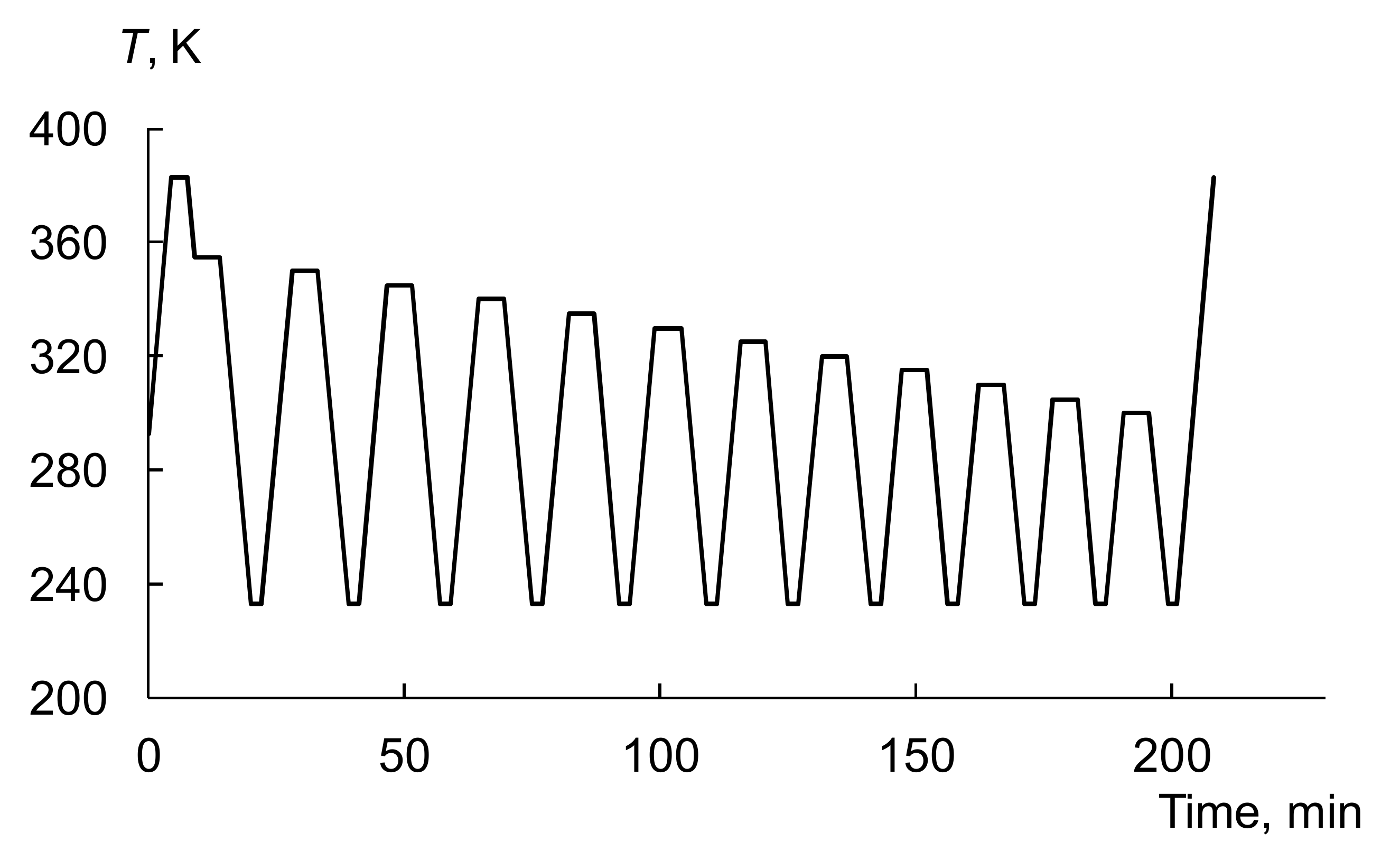
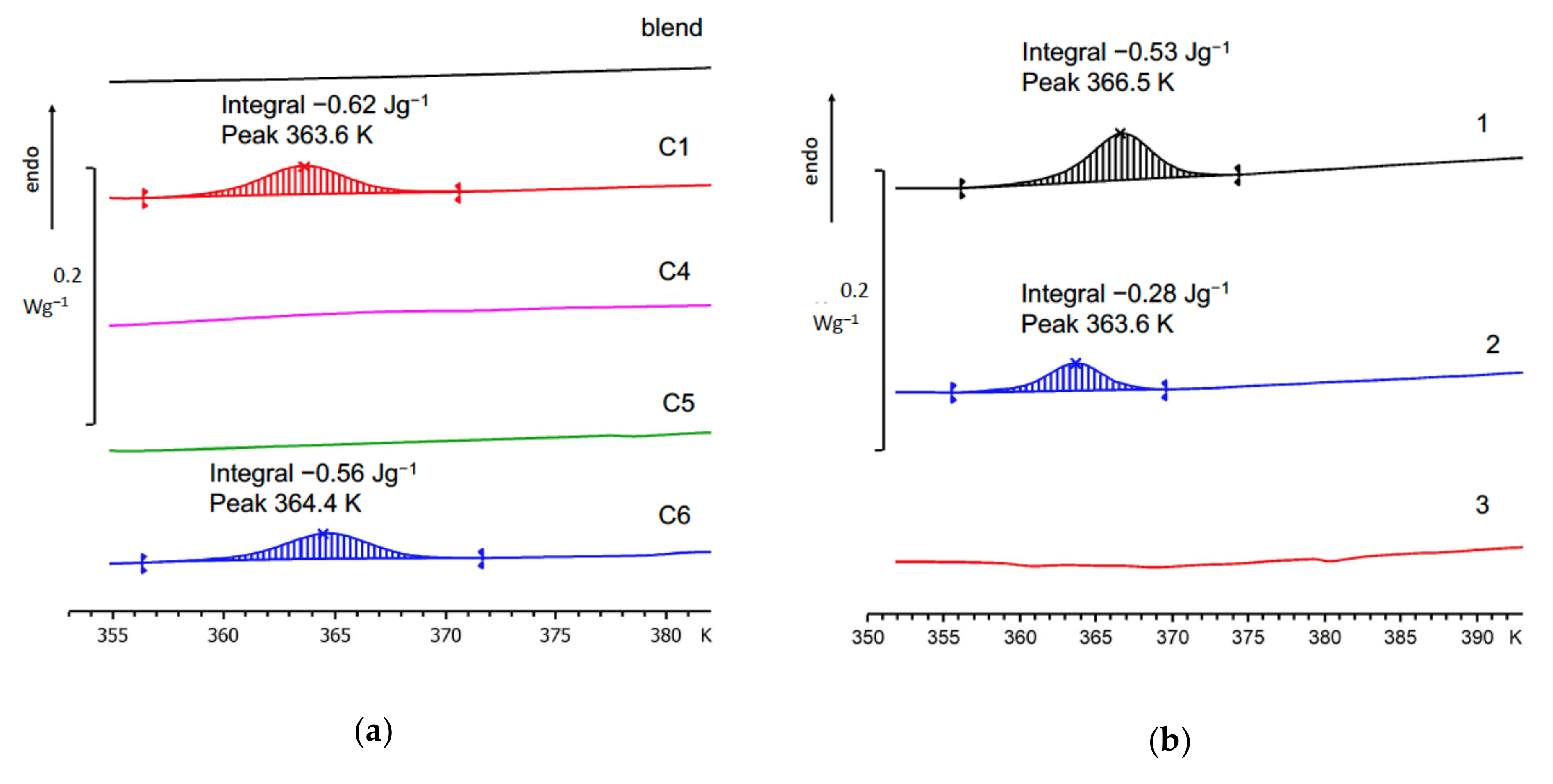
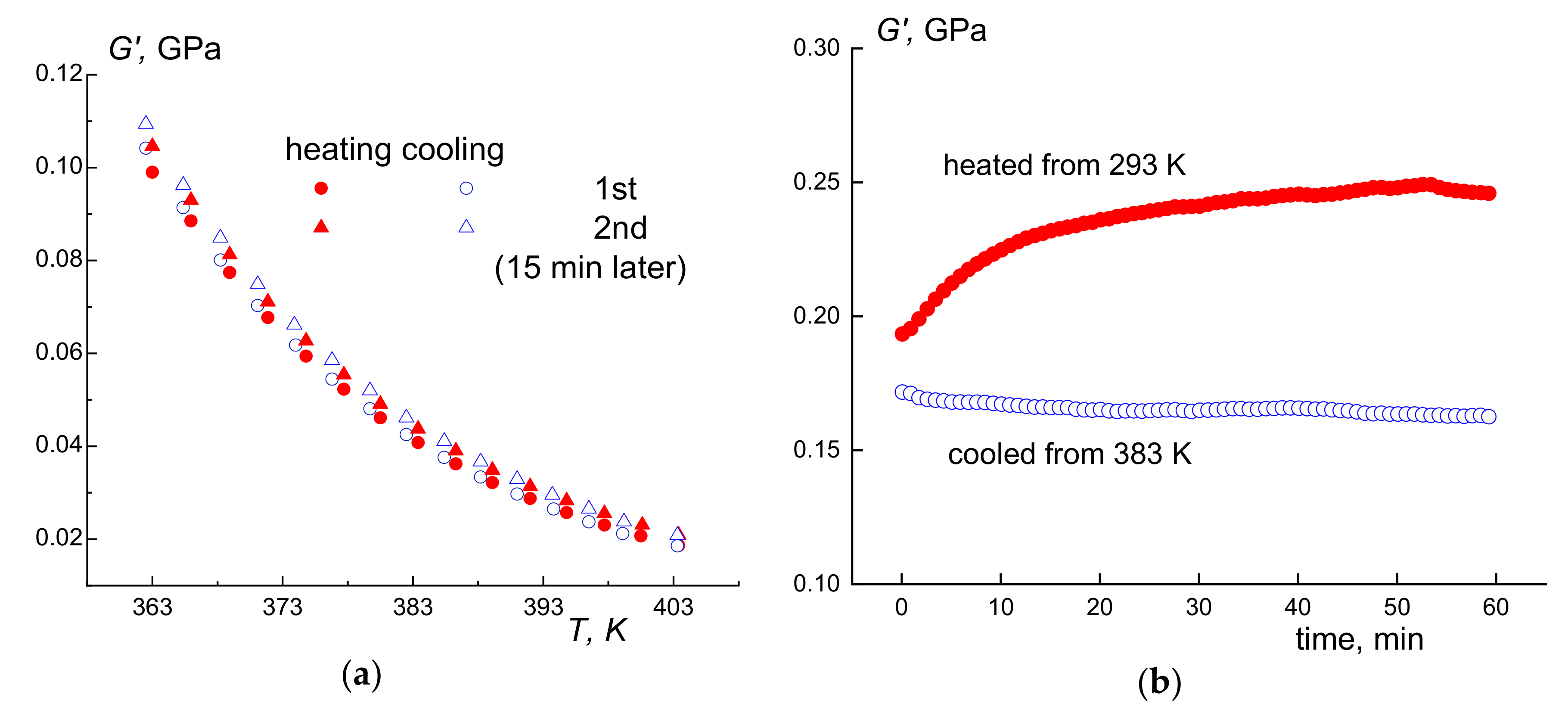
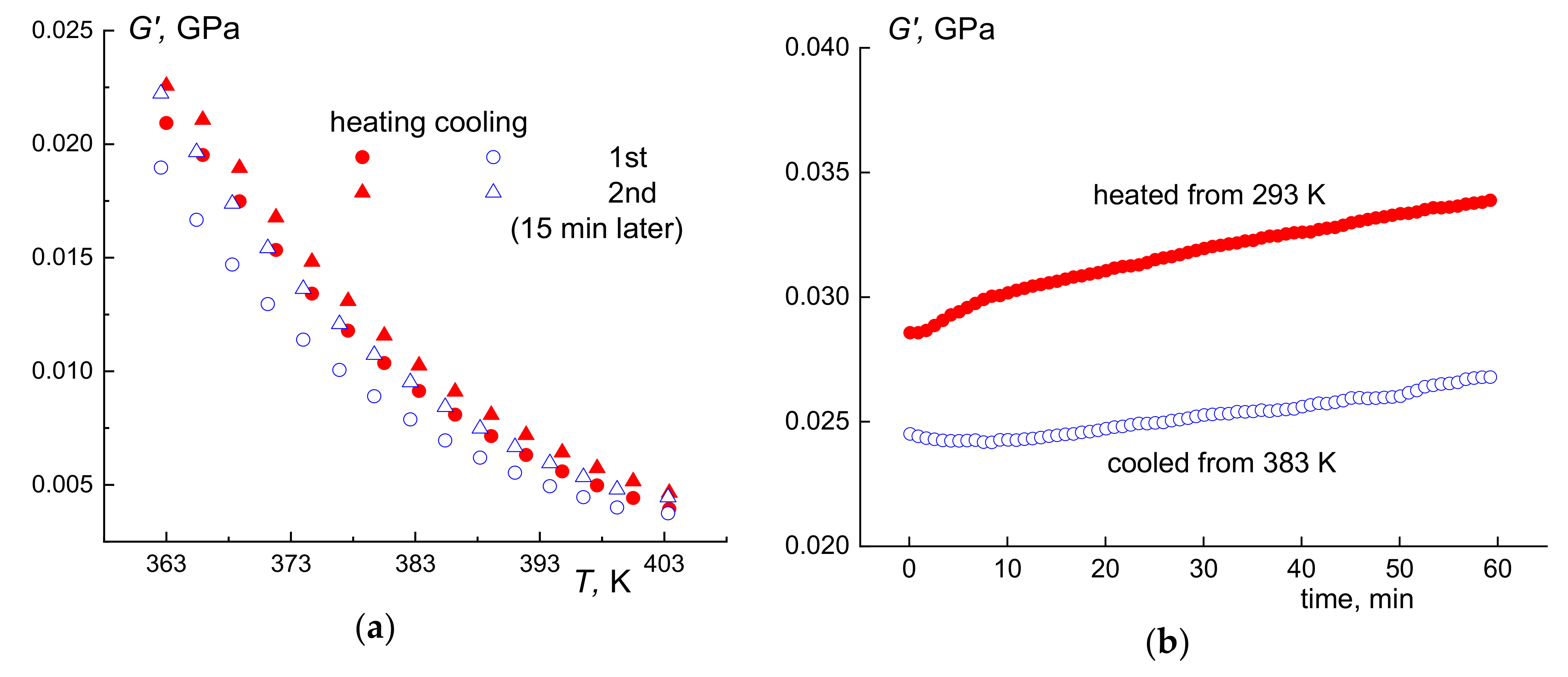
3.3. Thermal Fractionation by DSC
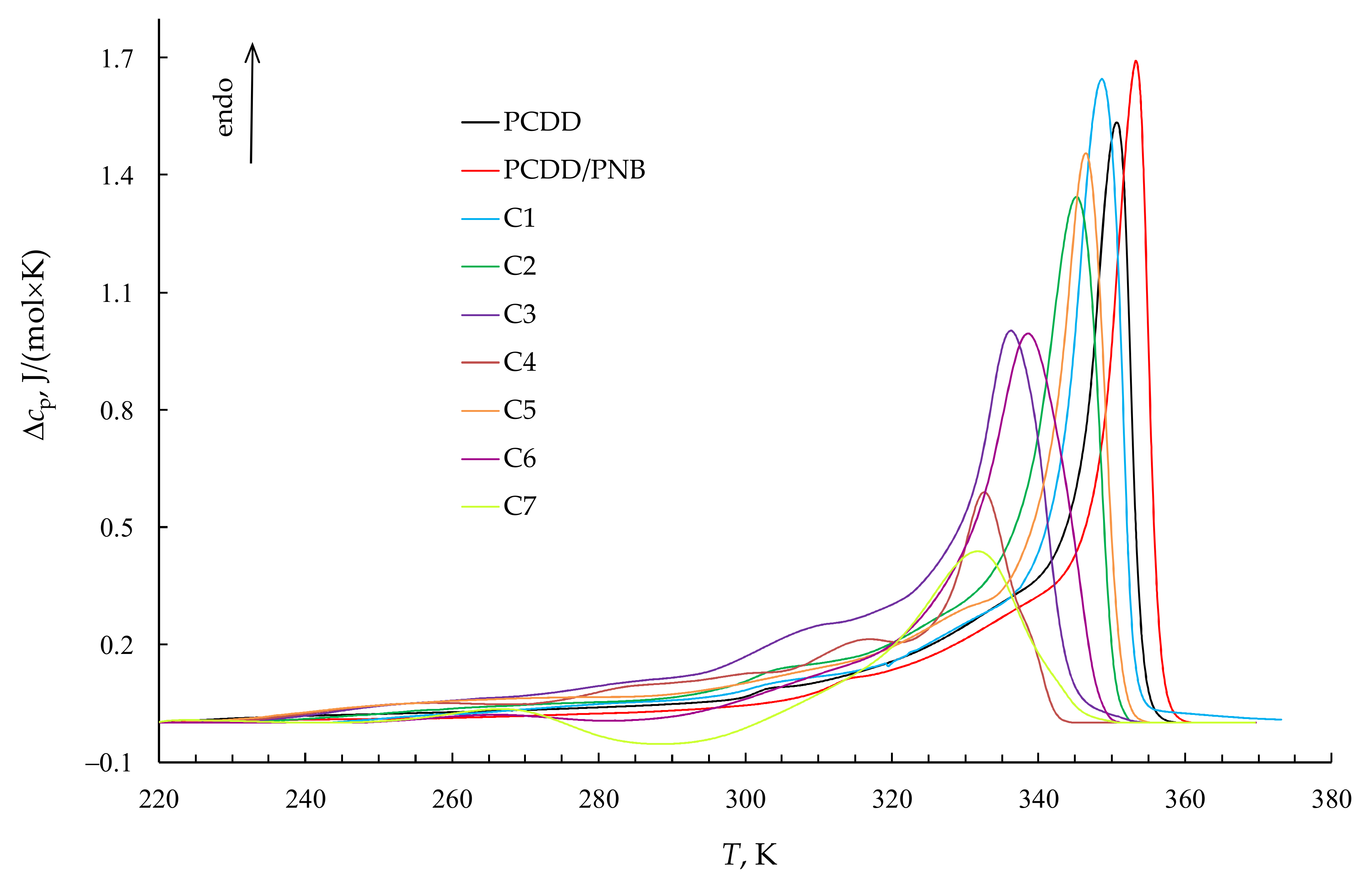
3.4. High-Temperature Endotherm
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jiang, X.; Yu, Y.; Guan, Y.; Liu, T.; Pang, C.; Ma, J.; Gao, H. Random and multiblock PBS copolyesters based on a rigid diol derived from naturally occurring camphor: Influence of chemical microstructure on thermal and mechanical properties. ACS Sustain. Chem. Eng. 2020, 8, 3626–3636. [Google Scholar] [CrossRef]
- De Ballesteros, O.R.; De Rosa, C.; Auriemma, F.; Malafronte, A.; Di Girolamo, R.; Scoti., M. Polymorphism and form II–form I transformation in Ziegler-Natta isotactic 1-butene-ethylene copolymers having a multiblock molecular structure. Polymer 2020, 198, 122460. [Google Scholar] [CrossRef]
- Kelsey, J.; Pickering, N.; Clough, A.; Zhou, J.; White, J.L. Multiblock inverse-tapered copolymers: Glass transition temperatures and dynamic heterogeneity as a function of chain architecture. Macromolecules 2017, 50, 7233–7240. [Google Scholar] [CrossRef]
- Zhang, Q.; Fan, J.; Feng, J.; Lu, X. Regulation of crystalline morphologies and mechanical properties of olefin multiblock copolymers by blending polymer with similar architecture of constituent blocks. Polymer 2015, 73, 139–148. [Google Scholar] [CrossRef]
- Ilyin, S.O.; Malkin, A.Y.; Kulichikhin, V.G.; Denisova, Y.I.; Krentsel, L.B.; Shandryuk, G.A.; Litmanovich, A.D.; Litmanovich, E.A.; Bondarenko, G.N.; Kudryavtsev, Y.V. Effect of chain structure on the rheological properties of vinyl acetate–vinyl alcohol copolymers in solution and bulk. Macromolecules 2014, 47, 4790–4804. [Google Scholar] [CrossRef]
- Beyer, V.P.; Kim, J.; Becer, C.R. Synthetic approaches for multiblock copolymers. Polym. Chem. 2020, 11, 1271–1291. [Google Scholar] [CrossRef]
- Hirao, A.; Matsuo, Y.; Goseki, R. Synthesis of novel block polymers with unusual block sequences by methodology combining living anionic polymerization and designed linking chemistry. J. Polym. Res. 2019, 26, 263. [Google Scholar] [CrossRef]
- Diaz, C.; Mehrkhodavandi, P. Strategies for the synthesis of block copolymers with biodegradable polyester segments. Polym. Chem. 2021, 12, 783–806. [Google Scholar] [CrossRef]
- Gringolts, M.L.; Denisova, Y.I.; Finkelshtein, E.S.; Kudryavtsev, Y.V. Olefin metathesis in multiblock copolymer synthesis. Beilstein J. Org. Chem. 2019, 15, 218–235. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Zhang, Z.; You, Y.-Z. Synthesis of sequence-controlled polymers via sequential multicomponent reactions and interconvertible hybrid copolymerizations. Polym. J. 2020, 52, 33–43. [Google Scholar] [CrossRef]
- Lutz, J.-F. Defining the field of sequence-controlled polymers. Macromol. Rapid Commun. 2017, 38, 1700582. [Google Scholar] [CrossRef]
- Litmanovich, A.D.; Platé, N.A.; Kudryavtsev, Y.V. Reactions in polymer blends: Interchain effects and theoretical problems. Prog. Polym. Sci. 2002, 27, 915–970. [Google Scholar] [CrossRef]
- Zanchin, G.; Leone, G. Polyolefin thermoplastic elastomers from polymerization catalysis: Advantages, pitfalls and future challenges. Prog. Polym. Sci. 2021, 113, 101342. [Google Scholar] [CrossRef]
- Ramaraju, H.; Akman, R.E.; Safranski, D.L.; Hollister, S.J. Designing biodegradable shape memory polymers for tissue repair. Adv. Funct. Mater. 2020, 30, 2002014. [Google Scholar] [CrossRef]
- Terzopoulou, Z.; Papadopoulos, L.; Zamboulis, A.; Papageorgiou, D.G.; Papageorgiou, G.Z.; Bikiaris, D.N. Tuning the properties of furandicarboxylic acid-based polyesters with copolymerization: A review. Polymers 2020, 12, 1209. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Wu, K.B.; Deng, Y.; Yuan, J.; Niu, J. Geared toward applications: A perspective on functional sequence-controlled polymers. ACS Macro Lett. 2021, 10, 243–257. [Google Scholar] [CrossRef]
- Boffito, M.; Sirianni, P.; Di Rienzo, A.M.; Chiono, V. Thermosensitive block copolymer hydrogels based on poly(epsilon-caprolactone) and polyethylene glycol for biomedical applications: State of the art and future perspectives. J. Biomed. Mater. Res. A 2015, 103, 1276–1290. [Google Scholar] [CrossRef] [PubMed]
- Qiang, X.L.; Chakroun, R.; Janoszka, N.; Gröschel, A.H. Self-assembly of multiblock copolymers. Isr. J. Chem. 2019, 59, 945–958. [Google Scholar] [CrossRef]
- Bates, C.M.; Bates, F.S. 50th anniversary perspective: Block polymers—Pure potential. Macromolecules 2017, 50, 3–22. [Google Scholar] [CrossRef]
- Li, S.; Register, R.; Weinhold, J.D.; Landes, B.G. Melt and solid-state structures of polydisperse polyolefin multiblock copolymers. Macromolecules 2012, 45, 5773–5781. [Google Scholar] [CrossRef]
- Lee, I.; Panthani, T.R.; Bates, F.S. Sustainable poly(lactide-b-butadiene) multiblock copolymers with enhanced mechanical properties. Macromolecules 2013, 46, 7387–7398. [Google Scholar] [CrossRef]
- Steube, M.; Plank, M.; Gallei, M.; Frey, H.; Floudas, G. Building bridges by blending: Morphology and mechanical properties of binary tapered diblock/multiblock copolymer blends. Macromol. Chem. Phys. 2021, 222, 2000373. [Google Scholar] [CrossRef]
- Zheng, Y.; Weng, C.; Cheng, C.; Zhao, J.L.; Yang, R.; Zhang, Q.; Ding, M.M.; Tan, H.; Fu, Q. Multiblock copolymers toward segmentation-driven morphological transition. Macromolecules 2020, 53, 5992–6001. [Google Scholar] [CrossRef]
- Lin, Y.; Yakovleva, V.; Chen, H.; Hiltner, A.; Baer, E. Comparison of olefin copolymers as compatibilizers for polypropylene and high-density polyethylene. J. Appl. Polym. Sci. 2009, 113, 1945–1952. [Google Scholar] [CrossRef]
- Zeng, D.; Gupta, R.; Coughlin, E.B.; Hayward, R.C. Assembly of disordered cocontinuous morphologies by multiblock copolymers with random block sequence and length dispersity. ACS Appl. Polym. Mater. 2020, 2, 3282–3290. [Google Scholar] [CrossRef]
- Wagner, N.L.; Timmers, F.J.; Arriola, D.J.; Jueptner, G.; Landes, B.G. Random block copolymers via segment interchange olefin metathesis. Macromol. Rapid Commun. 2008, 29, 1438–1443. [Google Scholar] [CrossRef]
- Otsuka, H.; Muta, T.; Sakada, M.; Maeda, T.; Takahara, A. Scrambling reaction between polymers prepared by step-growth and chain-growth polymerizations: Macromolecular cross-metathesis between 1,4-polybutadiene and olefin-containing polyester. Chem. Commun. 2009, 9, 1073–1075. [Google Scholar] [CrossRef] [PubMed]
- Maeda, T.; Kamimura, S.; Ohishi, T.; Takahara, A.; Otsuka, H. Synthesis of polyethylene/polyester copolymers through main chain exchange reactions via olefin metathesis. Polymer 2014, 55, 6245–6251. [Google Scholar] [CrossRef]
- Ohishi, T.; Suyama, K.; Kamimura, S.; Sakada, M.; Imato, K.; Kawahara, S.; Takahara, A.; Otsuka, H. Metathesis-driven scrambling reactions between polybutadiene or naturally occurring polyisoprene and olefin-containing polyurethane. Polymer 2015, 78, 145–153. [Google Scholar] [CrossRef]
- Daniele, S.; Mariconda, A.; Guerra, G.; Longo, P.; Giannino, L. Single-phase block copolymers by cross-metathesis of 1,4-cis-polybutadiene and 1,4-cis-polyisoprene. Polymer 2017, 130, 143–149. [Google Scholar] [CrossRef]
- Morontsev, А.А.; Gringolts, M.L.; Filatova, M.P.; Peregudov, A.S.; Akmalov, T.R.; Masoud, S.M.; Osipov, S.N.; Denisova, Y.I.; Kudryavtsev, Y.V. Ruthenium-carbene complexes in the synthesis of polybutadiene and its cross-metathesis with polynorbornene. Polym. Sci. Ser. C 2019, 61, 67–77. [Google Scholar] [CrossRef]
- Gringolts, M.L.; Denisova, Y.I.; Shandryuk, G.A.; Krentsel, L.B.; Litmanovich, A.D.; Finkelshtein, E.S.; Kudryavtsev, Y.V. Synthesis of norbornene–cyclooctene copolymers by the cross-metathesis of polynorbornene with polyoctenamer. RSC Adv. 2015, 5, 316–319. [Google Scholar] [CrossRef]
- Denisova, Y.I.; Gringolts, M.L.; Peregudov, A.S.; Krentsel, L.B.; Litmanovich, E.A.; Litmanovich, A.D.; Finkelshtein, E.S.; Kudryavtsev, Y.V. Cross-metathesis of polynorbornene with polyoctenamer: A kinetic study. Beilstein J. Org. Chem. 2015, 11, 1796–1808. [Google Scholar] [CrossRef] [PubMed]
- Denisova, Y.I.; Roenko, A.V.; Adzhieva, O.A.; Gringolts, M.L.; Shandryuk, G.A.; Peregudov, A.S.; Finkelshtein, E.S.; Kudryavtsev, Y.V. Facile synthesis of norbornene-ethylene-vinyl acetate/vinyl alcohol multiblock copolymers by the olefin cross-metathesis of polynorbornene with poly(5-acetoxy-1-octenylene). Polym. Chem. 2020, 11, 7063–7077. [Google Scholar] [CrossRef]
- Roenko, A.V.; Denisova, Y.I.; Gringolts, M.L.; Peregudov, A.S.; Shandryuk, G.A.; Finkelshtein, E.S.; Kudryavtsev, Y.V. Cross-metathesis between polynorbornene and poly-(5,6-epoxy-1-octenamer). Polym. Sci. Ser. C 2019, 61, 110–121. [Google Scholar] [CrossRef]
- Denisova, Y.I.; Gringolts, M.L.; Roenko, A.V.; Shandryuk, G.A.; Finkelshtein, E.S.; Kudryavtsev, Y.V. New multiblock copolymers of norbornene and 5-hydroxycyclooctene. Mendeleev Commun. 2017, 27, 416–418. [Google Scholar] [CrossRef]
- Denisova, Y.I.; Gringolts, M.L.; Krentsel, L.B.; Shandryuk, G.A.; Peregudov, A.S.; Finkelshtein, E.S.; Kudryavtsev, Y.V. Synthesis of new multiblock copolymers via cross-metathesis reaction of polytrimethylsilylnorbornene and polycyclooctene. Polym. Sci. Ser. B 2017, 59, 412–420. [Google Scholar] [CrossRef]
- Denisova, Y.I.; Zhigarev, V.A.; Gringolts, M.L.; Shandryuk, G.A.; Peregudov, A.S.; Finkelshtein, E.S.; Kudryavtsev, Y.V. Cyclododecene in the olefin metathesis: Polymerization and macromolecular cross-metathesis with polynorbornene. Polym. Sci. Ser. C 2019, 61, 122–135. [Google Scholar] [CrossRef]
- Keller, A.; Martuscelli, E. Studies on polyalkenamers. Part I. Single crystal growth and chain folding in polydodecenamer and polydecenamer. Makromol. Chem. 1971, 141, 189–198. [Google Scholar] [CrossRef]
- Keller, A.; Martuscelli, E. Studies on polyalkenamers. II. Degradation of polydodecenamer and polydecenamer single crystals. Makromol. Chem. 1972, 151, 169–187. [Google Scholar] [CrossRef]
- Martuscelli, E.; Vittoria, V. Melting and annealing of oriented single crystal aggregates of trans-polydodecenamer. Polymer 1972, 13, 360–364. [Google Scholar] [CrossRef]
- Van Werden, K.; Holland-Moritz, K. Vibrational spectroscopic studies on different modifications of poly(trans-alkenylene)s. II: Poly(trans-octenylene) and poly(trans-dodecenylene). Colloid Polym. Sci. 1981, 259, 731–740. [Google Scholar] [CrossRef]
- Haag, H.; Nordsiek, K.-H.; Streck, R.; Zerpner, D. Polydodecenamers as reinforcing resins for elastomers. US Patent 4,048,262, 13 September 1977. [Google Scholar]
- Lerum, M.F.Z.; Chen, W. Surface-initiated ring-opening metathesis polymerization in the vapor phase: An efficient method for grafting cyclic olefins with low strain energies. Langmuir 2011, 27, 5403–5409. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Qin, H.; Mather, P.T. Review of progress in shape-memory polymers. J. Mater. Chem. 2007, 17, 1543–1558. [Google Scholar] [CrossRef]
- Onbulak, S.; Hillmyer, M.A. Precision ethylene-styrene copolymers through the ring opening metathesis polymerization of 3-phenyl cyclododecenes. Polym. Chem. 2021, 12, 1681–1691. [Google Scholar] [CrossRef]
- Finkelshtein, E.S.; Bermeshev, M.V.; Gringolts, M.L.; Starannikova, L.E.; Yampolskii, Y.P. Substituted polynorbornenes as promising materials for gas separation membranes. Russ. Chem. Rev. 2011, 80, 341–362. [Google Scholar] [CrossRef]
- Müller, A.J.; Arnal, M.L. Thermal fractionation of polymers. Prog. Polym. Sci. 2005, 30, 559–603. [Google Scholar] [CrossRef]
- Müller, A.J.; Michell, R.M.; Perez, R.A.; Lorenzo, A.T. Successive Self-nucleation and Annealing (SSA): Correct design of thermal protocol and applications. Eur. Polym. J. 2015, 65, 132–154. [Google Scholar] [CrossRef]
- Fernández-d’Arlas, B.; Maiz, J.; Pérez-Camargo, R.A.; Baumann, R.-P.; Pöselt, E.; Dabbous, R.; Stribeck, A.; Müller, A.J. SSA fractionation of thermoplastic polyurethanes. Polym. Cryst. 2021, 4, e10148. [Google Scholar]
- Shandryuk, G.A.; Denisova, Y.I.; Gringolts, M.L.; Krentsel, L.B.; Litmanovich, A.D.; Finkelshtein, E.S.; Kudryavtsev, Y.V. Peculiarities of crystallization in the multiblock copolymers of norbornene and cyclooctene. Eur. Polym. J. 2017, 86, 143–153. [Google Scholar] [CrossRef]
- Natta, G.; Bassi, I.W. The crystal structure of trans polydodecenamer. Eur. Polym. J. 1967, 3, 43–55. [Google Scholar] [CrossRef]
- Natta, G.; Bassi, I.W.; Fagherazzi, G. The monoclinic structure of even trans-polyalkenamers. Eur. Polym. J. 1967, 3, 339–352. [Google Scholar] [CrossRef]
- Gianotti, G.; Capizzi, A. Thermodynamic data for some even trans polyalkenamers. Eur. Polym. J. 1970, 6, 743–749. [Google Scholar] [CrossRef]
- Kudryavtsev, Y.V.; Chertovich, A.V.; Guseva, D.V.; Litmanovich, A.D. Early stages of interchange reactions in polymer blends. Macromol. Symp. 2007, 254, 188–195. [Google Scholar] [CrossRef]
- Mandelkern, L. Crystallization of Polymers. Equilibrium Concepts; Cambridge University Press: Cambridge, UK, 2002; Volume 1. [Google Scholar]
- Askadskii, A.A. Computational Materials Science of Polymers; Cambridge International: Cambridge, UK, 2003. [Google Scholar]
- Flory, P.J. Theory of crystallization in copolymers. Trans. Faraday Soc. 1955, 51, 848–857. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | NB/CDD, mol | G1, % mol a | Cross-Reaction Time, h | Mw × 10−3, g/mol | Ð | CDD cis/trans | φ*NB-CDD, ×100% | φCDD, ×100% | LCDD/Ltr-CDDb |
|---|---|---|---|---|---|---|---|---|---|
| PNB | 1:0 | - | - | 307 | 2.0 | - | 0 | 0 | -/- |
| PCDD | 0:1 | - | - | 406 | 2.5 | 15/85 | 0 | 100 | 967/6.7 |
| PNB/PCDD | 1:1 | - | - | 344/325 | 1.9/2.2 | 14/86 | 0 | 55 | 890/7.1 |
| NB–CDD C1 | 1:1 | 0.33 | 5 | 142 | 2.4 | 16/84 | 3.0 | 51 | 45/5.4 |
| NB–CDD C2 | 1:1 | 0.33 | 24 | 169 | 2.1 | 19/81 | 3.3 | 50 | 34/4.6 |
| NB–CDD C3 | 1:1 | 1 | 5 | 83 | 2.4 | 21/79 | 10 | 51 | 9.9/3.5 |
| NB–CDD C4 | 1:1 | 5 | 0.67 | 26 | 1.5 | 21/79 | 11 | 50 | 8.5/3.4 |
| NB–CDD C5 | 1:2 | 0.33 | 5 | 162 | 1.9 | 19/81 | 3.0 | 63 | 48/4.8 |
| NB–CDD C6 | 2:1 | 0.33 | 5 | 114 | 2.1 | 18/82 | 3.5 | 36 | 17/4.5 |
| NB–CDD C7 | 3:1 | 0.17 | 5 | 126 | 1.7 | 23/77 | 2.0 | 24 | 12/3.8 |
| Sample | 2θ, deg | Reflection Indices, hkl | Lattice Spacing d, Å | FWHM, deg | Crystallite Size L, Å | DCstr, % a |
|---|---|---|---|---|---|---|
| PCDD | 21.1 | 110 | 4.20 | 0.49 | 178 | 25 |
| 23.5 | 200 | 3.78 | 0.62 | 138 | ||
| 35.7 | 020 | 2.51 | 0.68 | 126 | ||
| 40.2 | 310 | 2.24 | 1.33 | 64 | ||
| NB–CDD C1 | 21.1 | 110 | 4.20 | 0.54 | 162 | 16.5 |
| 23.6 | 200 | 3.81 | 0.77 | 108 | ||
| 35.7 | 020 | 2.51 | 0.66 | 130 | ||
| 40.2 | 310 | 2.24 | 1.08 | 79 |
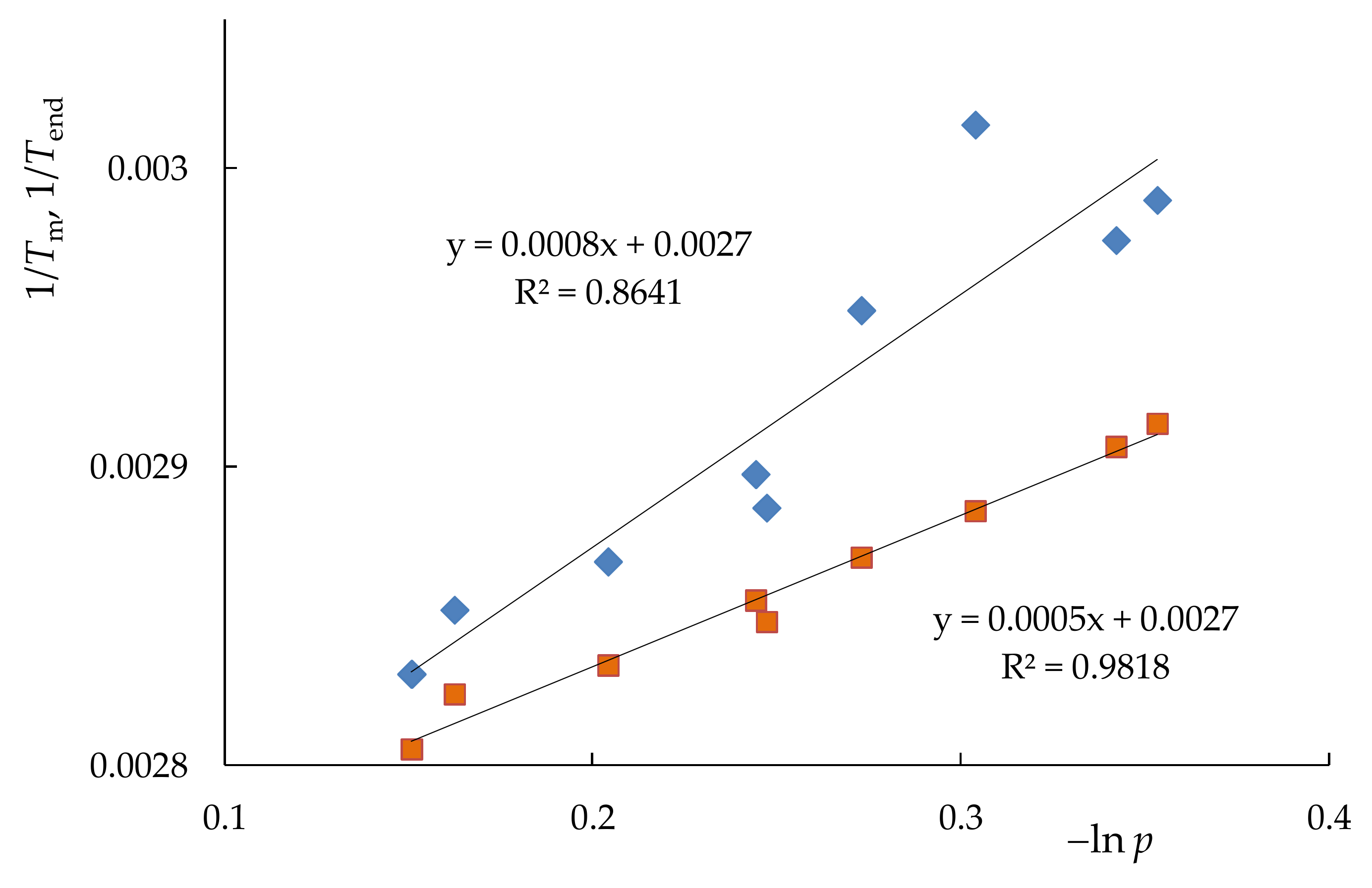
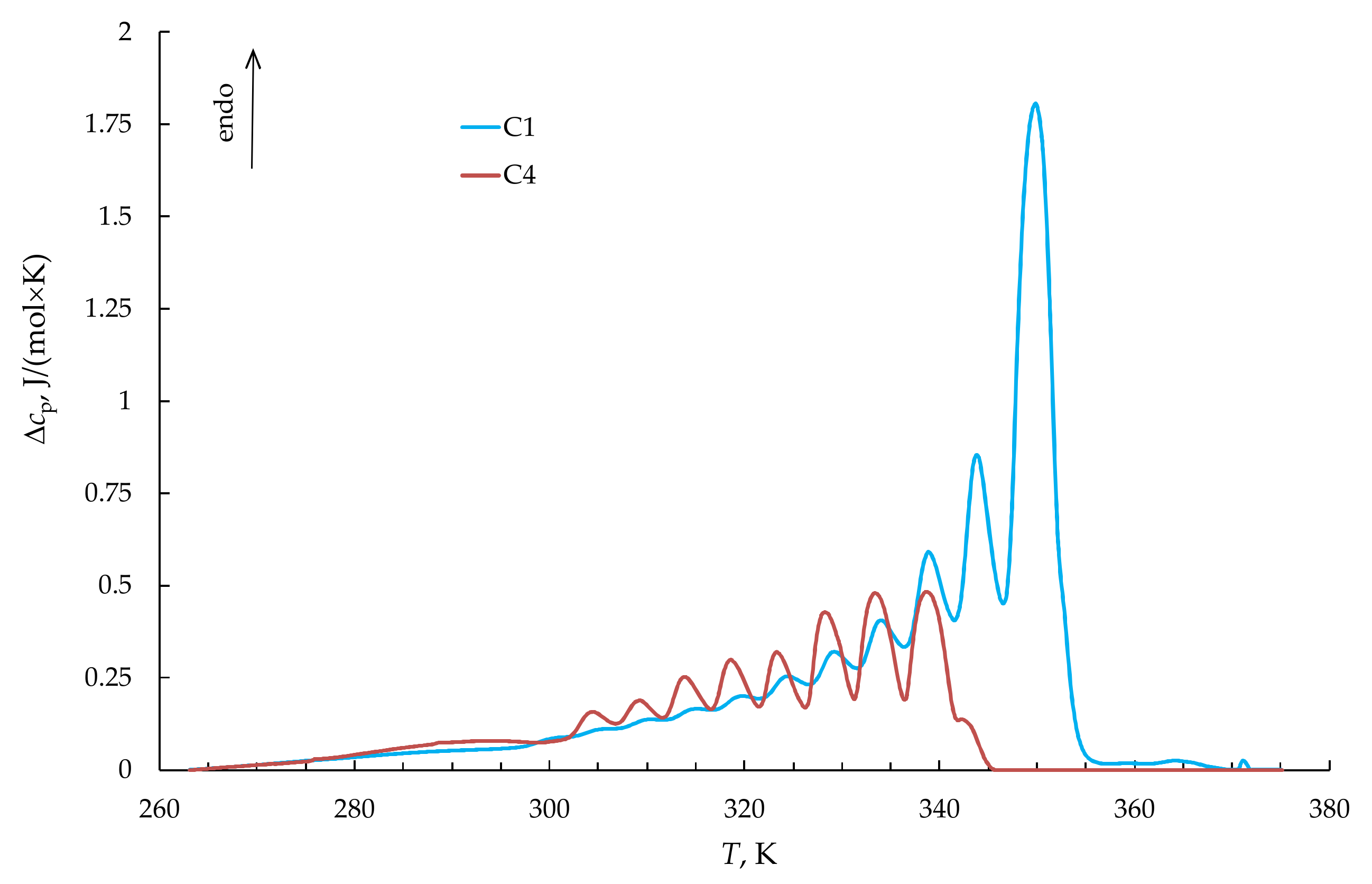
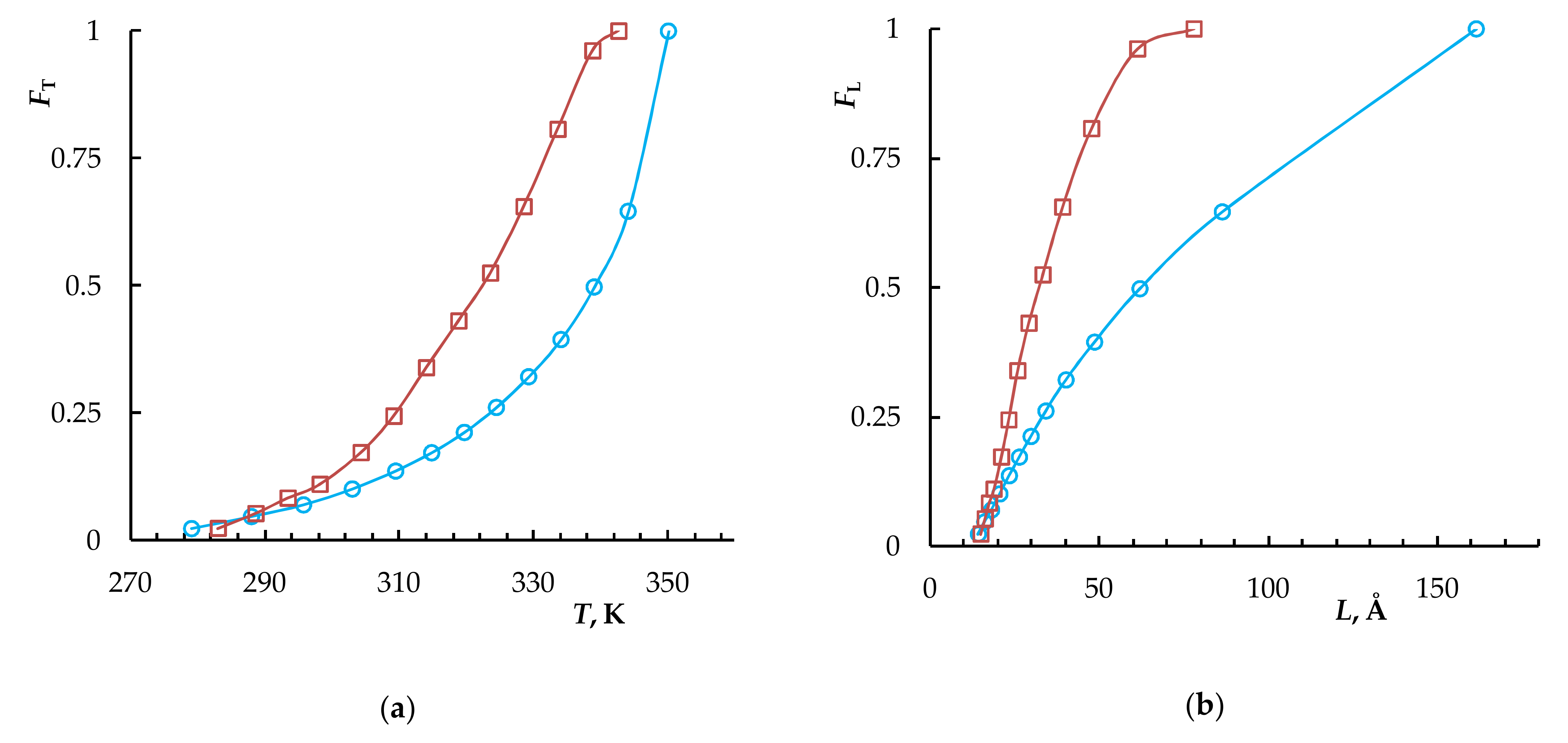
| Sample | Tg, K | Tm, K | Tend, K | ΔH, J/g | DCcal | Lm, Å | Lend, Å | p | T(c)m0, K |
|---|---|---|---|---|---|---|---|---|---|
| PNB | 310 | - | - | - | - | - | - | - | - |
| PCDD | 223 | 351 | 354 | 91.2 | 46 | 172 | 382 | 0.850 | 352.6 |
| PNB/PCDD | 303 | 353 | 356.5 | 62.0 | 47 | 302 | 2102 | 0.860 | 352.8 |
| NB–CDD C1 | 243 | 349 | 353 | 67.3 | 53 | 131 | 268 | 0.815 | 351.5 |
| NB–CDD C2 | 252 | 345 | 350 | 62.8 | 53 | 94 | 165 | 0.783 | 350.3 |
| NB–CDD C3 | 243 | 336 | 344 | 56.3 | 48.5 | 53 | 86 | 0.710 | 347.6 |
| NB–CDD C4 | 241 | 334.5 | 343 | 39.8 | 35 | 50 | 80 | 0.702 | 347.3 |
| NB–CDD C5 | 243 | 346.5 | 351 | 68.3 | 50 | 106 | 190 | 0.791 | 350.5 |
| NB–CDD C6 | 259 | 339 | 348.5 | 49.2 | 53 | 61 | 131 | 0.780 | 350.5 |
| NB–CDD C7 | 261 | 332 | 347 | 21.5 | 34 | 44 | 107 | 0.738 | 348.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Denisova, Y.I.; Shandryuk, G.A.; Arinina, M.P.; Levin, I.S.; Zhigarev, V.A.; Gringolts, M.L.; Finkelshtein, E.S.; Malkin, A.Y.; Kudryavtsev, Y.V. Multiblock Copolymers of Norbornene and Cyclododecene: Chain Structure and Properties. Polymers 2021, 13, 1756. https://doi.org/10.3390/polym13111756
Denisova YI, Shandryuk GA, Arinina MP, Levin IS, Zhigarev VA, Gringolts ML, Finkelshtein ES, Malkin AY, Kudryavtsev YV. Multiblock Copolymers of Norbornene and Cyclododecene: Chain Structure and Properties. Polymers. 2021; 13(11):1756. https://doi.org/10.3390/polym13111756
Chicago/Turabian StyleDenisova, Yulia I., Georgiy A. Shandryuk, Marianna P. Arinina, Ivan S. Levin, Vsevolod A. Zhigarev, Maria L. Gringolts, Eugene Sh. Finkelshtein, Alexander Ya. Malkin, and Yaroslav V. Kudryavtsev. 2021. "Multiblock Copolymers of Norbornene and Cyclododecene: Chain Structure and Properties" Polymers 13, no. 11: 1756. https://doi.org/10.3390/polym13111756
APA StyleDenisova, Y. I., Shandryuk, G. A., Arinina, M. P., Levin, I. S., Zhigarev, V. A., Gringolts, M. L., Finkelshtein, E. S., Malkin, A. Y., & Kudryavtsev, Y. V. (2021). Multiblock Copolymers of Norbornene and Cyclododecene: Chain Structure and Properties. Polymers, 13(11), 1756. https://doi.org/10.3390/polym13111756