
Lattice Vibrations and Time-Dependent Evolution of Local Phonon Modes during Exciton Formation in Conjugated Polymeric Molecules



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methodology
3. Results and Discussion
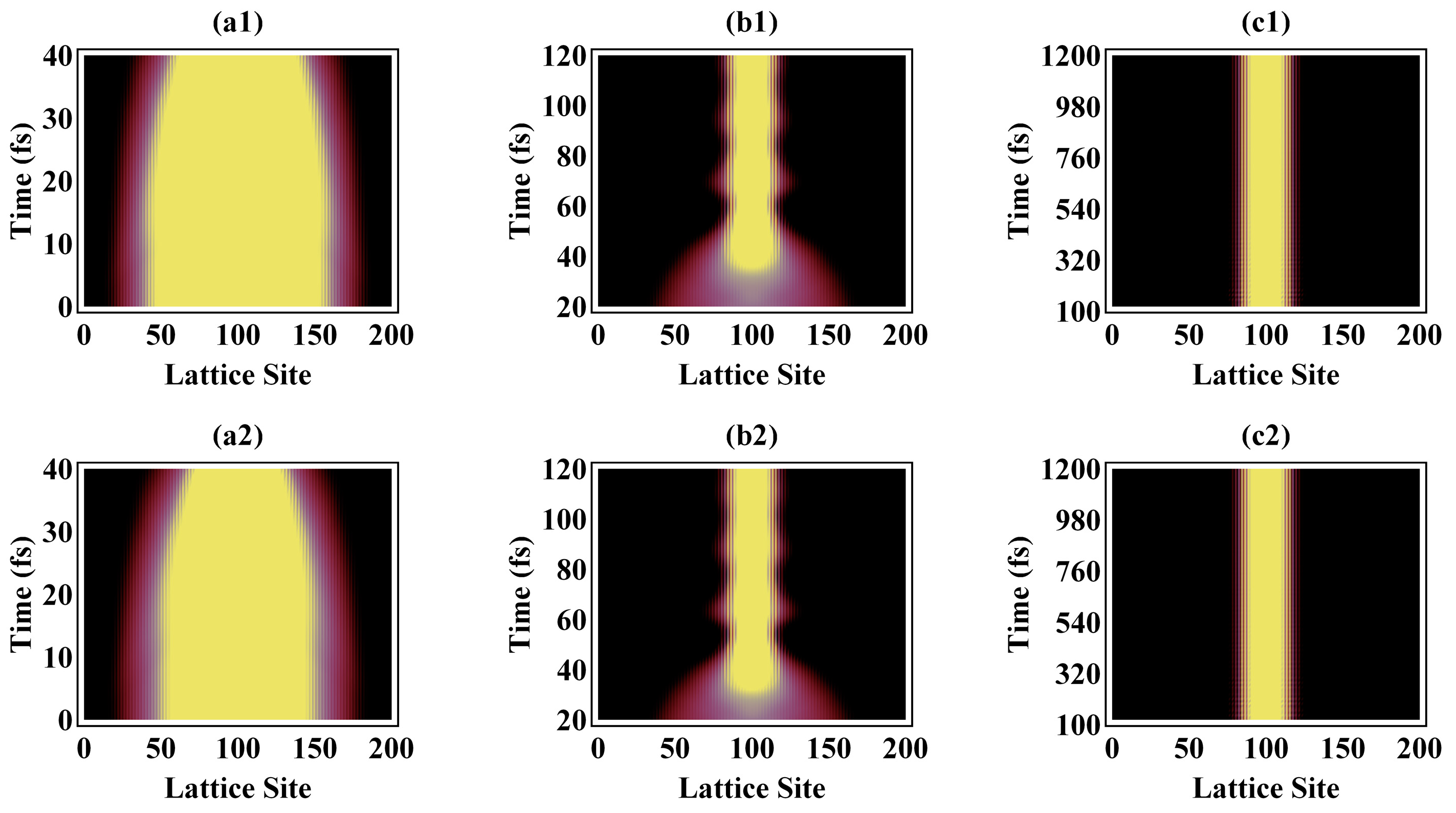
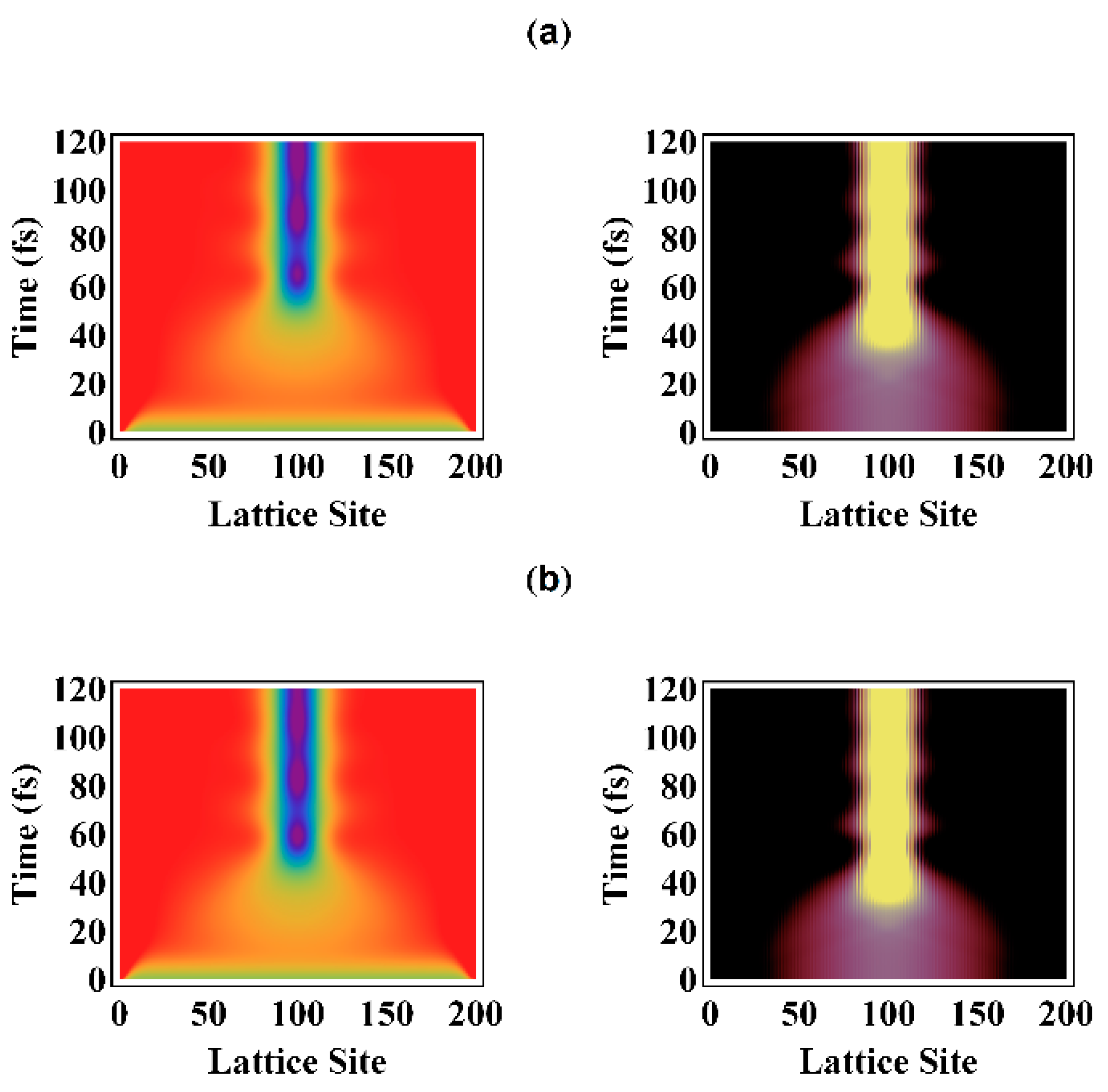
3.1. Electron Localization and Photoinduced Exciton Formation
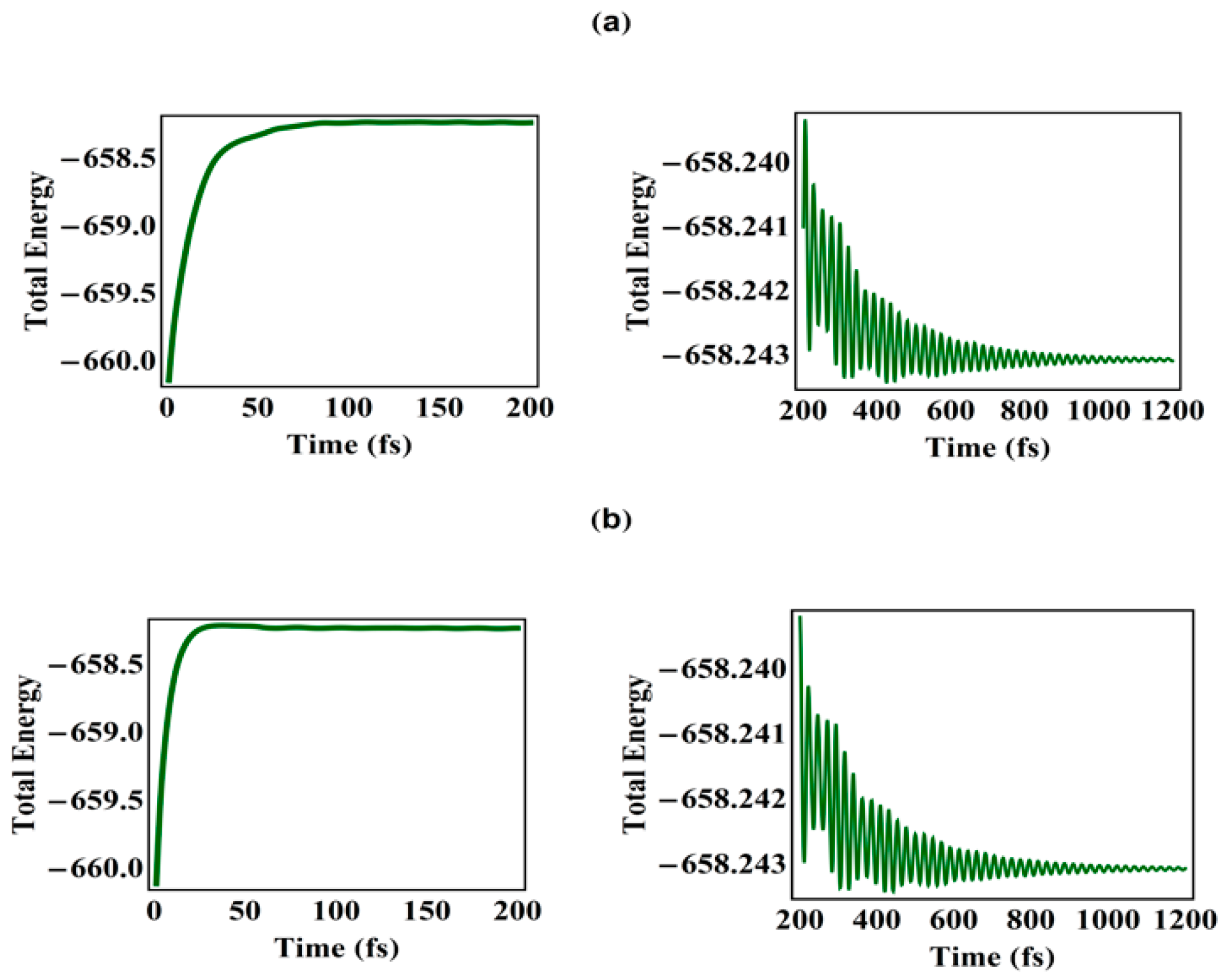
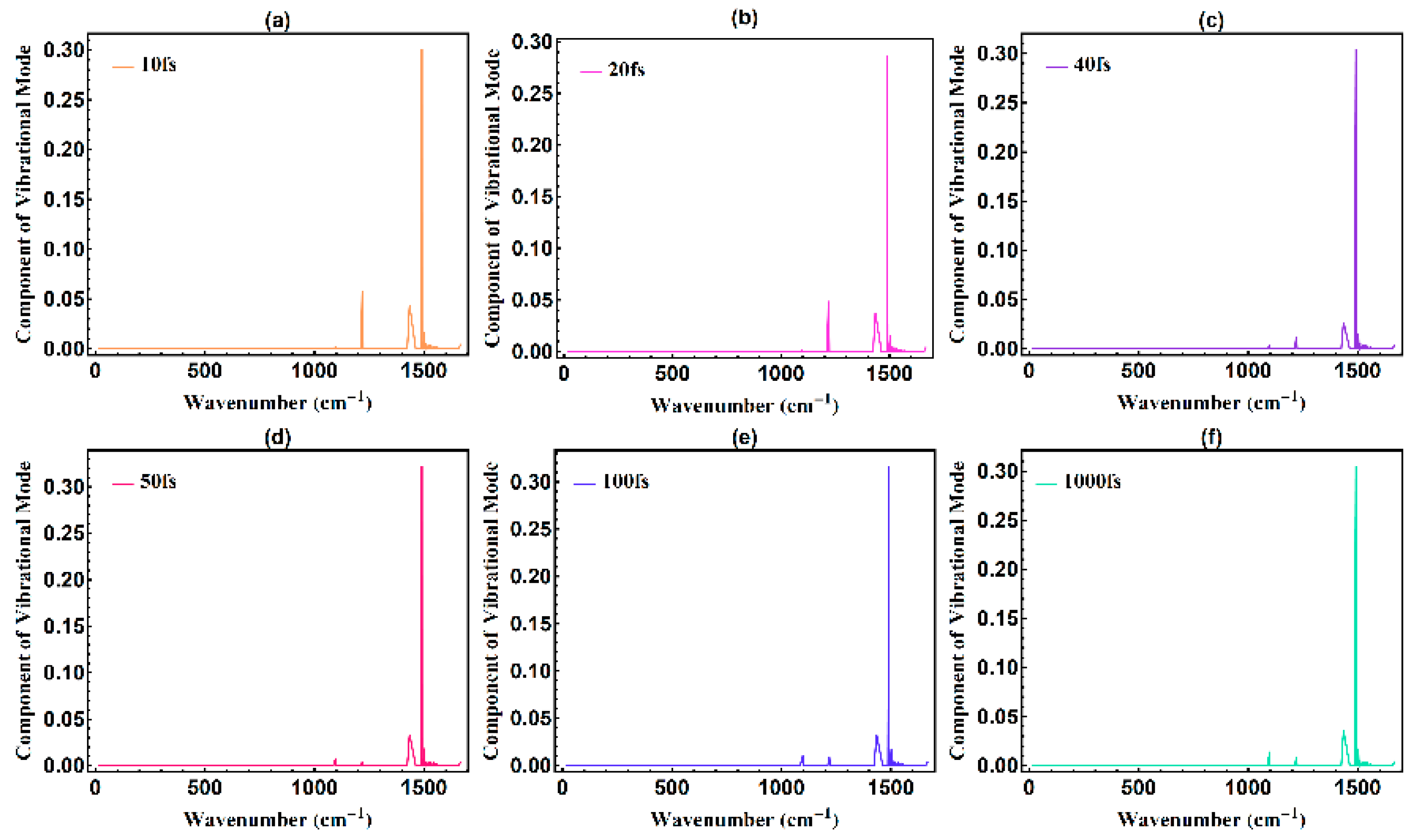
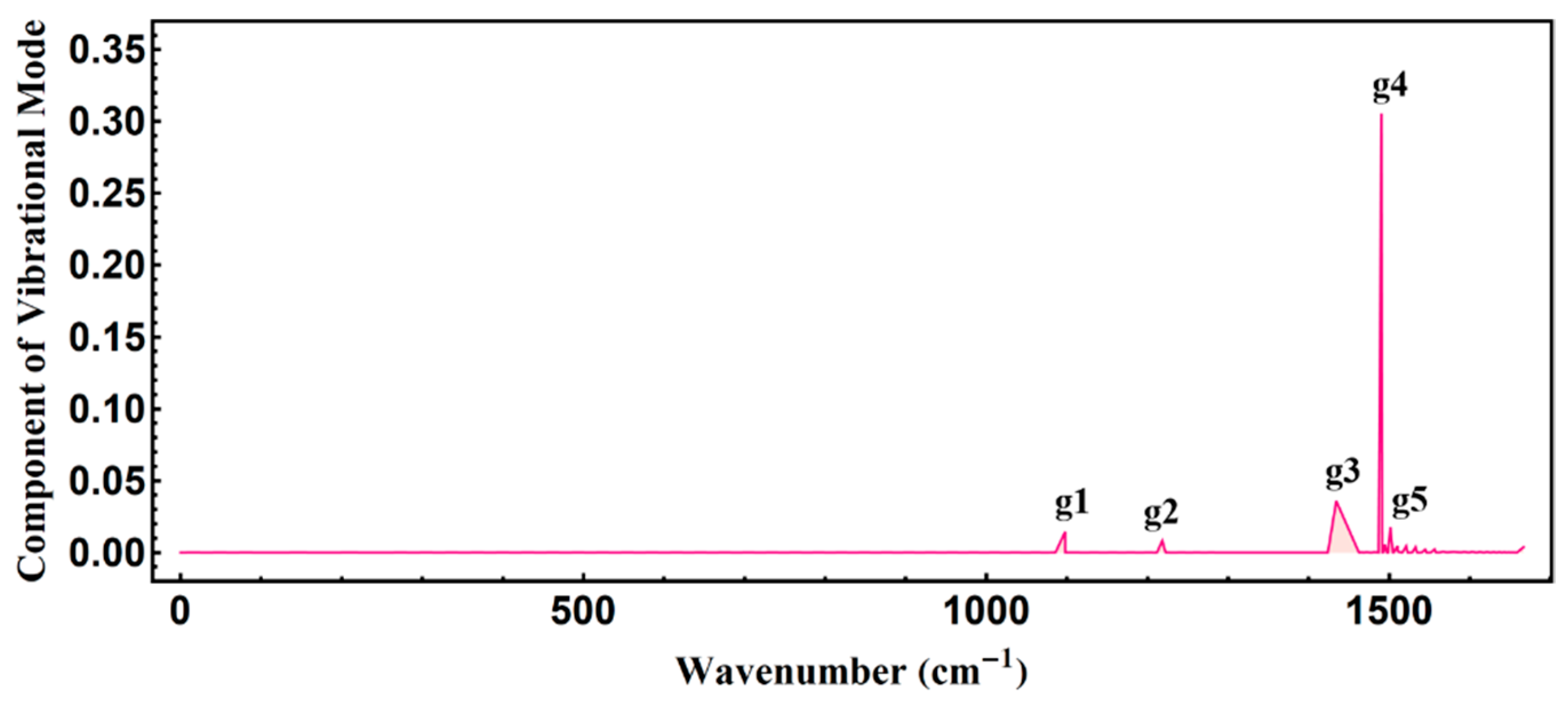
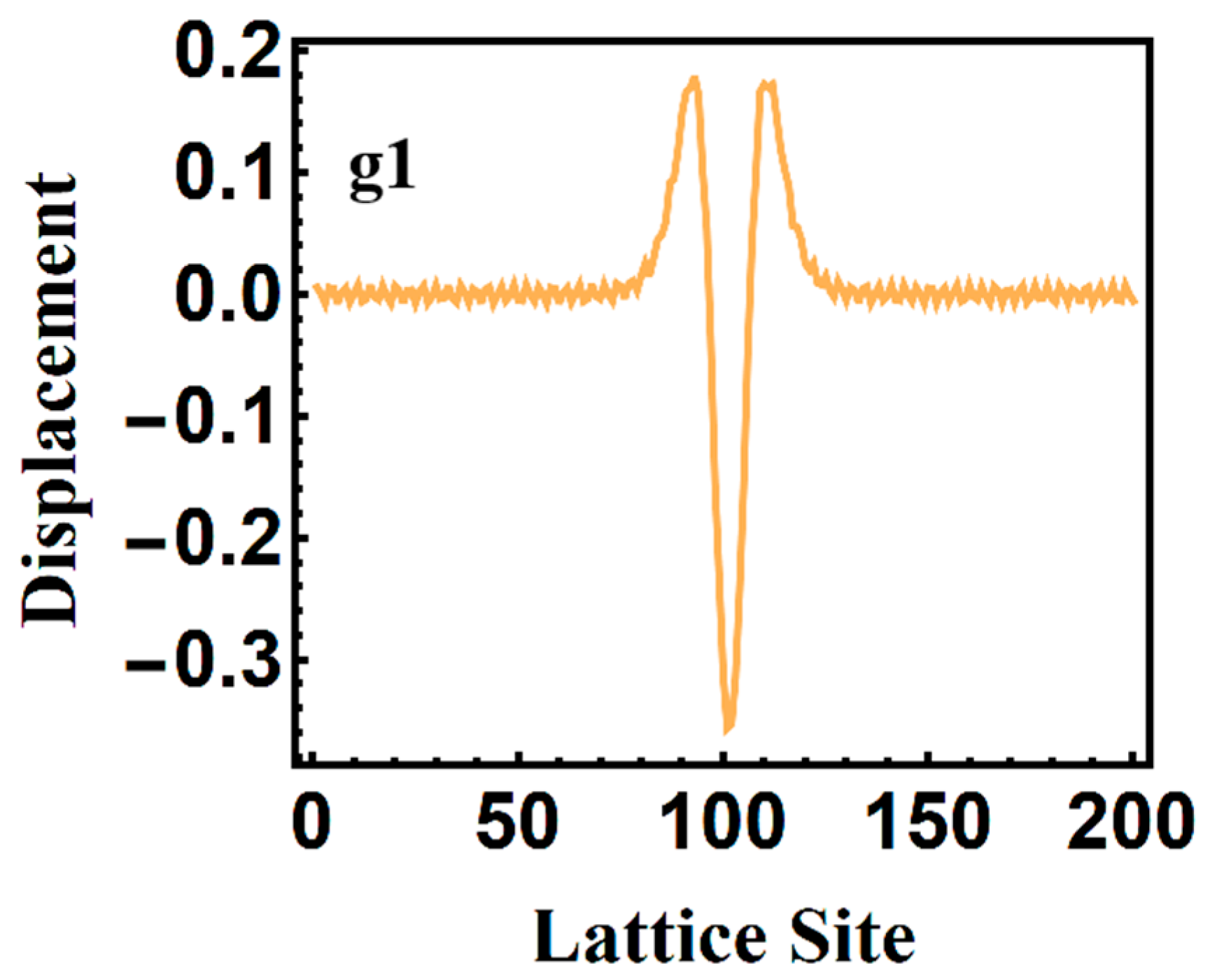
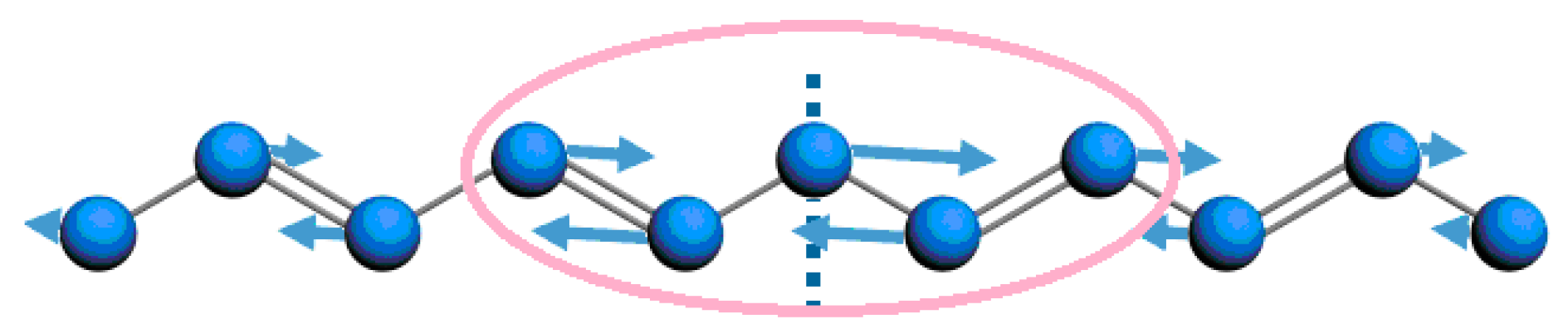
3.2. Temporal Evolution of Alternating Bonds
4. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Wang, C.; Zhuang, L.Q.; Chen, R.A.; Li, S.; George, T.F. Localization and relaxation of singlet exciton formation in conjugated polymers under photoexcitation. J. Phys. Chem. B 2013, 117, 3258–3263. [Google Scholar] [CrossRef]
- Burroughes, J.H.; Bradley, D.D.C.; Brown, A.R.; Marks, R.N.; Mackay, K.; Friend, R.H.; Burns, P.L.; Holmes, A.B. Light-emitting diodes based on conjugated polymers. Nature 1990, 347, 539–541. [Google Scholar] [CrossRef]
- Friend, R.H.; Gymer, R.W.; Holmes, A.B.; Burroughes, J.H.; Marks, N.; Taliani, C.; Bradley, D.D.C.; Dos Santos, D.A.; Bredas, J.L.; Logdlund, M.; et al. Electroluminescence in conjugated polymers. Nature 1999, 397, 121–128. [Google Scholar] [CrossRef]
- Phillips, R.T.; Hayes, G.R.; Samuel, I.D.W.; Phillips, R.T. Ultrafast synamics of photoexcitations in conjugated polymers. Synth. Met. 1997, 84, 889–890. [Google Scholar]
- Hayes, G.R.; Samuel, I.D.W.; Phillips, R.T. Exciton dynamics in electroluminescent polymers studied by femtosecond time-resolved photoluminescence spectroscopy. Phys. Rev. B 1995, 52, 11569–11572. [Google Scholar] [CrossRef]
- Grage, M.M.L.; Pullerits, T.; Ruseckas, A.; Theander, M.; Inganäs, O.; Sundström, V. Conformational disorder of a substituted polythiophene in solution revealed by excitation transfer. Chem. Phys. Lett. 2001, 339, 96–102. [Google Scholar] [CrossRef]
- Grage, M.M.L.; Zaushitsyn, Y.; Yartsev, A.; Chachisvilis, M.; Sundström, V.; Pullerits, T. Ultrafast excitation transfer and trapping in a thin polymer film. Phys. Rev. B 2003, 67, 205207. [Google Scholar] [CrossRef]
- Parkinson, P.; Müller, C.; Stingelin, N.; Johnston, M.B.; Herz, L.M. Role of Ultrafast torsional relaxation in the emission from polythiophene aggregates. J. Phys. Chem. Lett. 2010, 1, 117–118. [Google Scholar] [CrossRef]
- Banerji, N.; Cowan, S.; Leclerc, M.; Vauthey, E.; Heeger, A.J. Exciton formation, relaxation, and decay in PCDTBT. J. Am. Chem. Soc. 2010, 132, 17459–17470. [Google Scholar] [CrossRef]
- Banerji, N.; Cowan, S.; Vauthey, E.; Heeger, A.J. Ultrafast relaxation of the poly (3-hexylthiophene) emission spectrum. J. Phys. Chem. C 2011, 115, 9726–9739. [Google Scholar] [CrossRef]
- Banerji, N.; Seifter, J.; Wang, M.; Vauthey, E.; Wudl, F.; Heeger, A.J. Ultrafast spectroscopic investigation of a fullerene poly (3-hexylthiophene) dyad. Phys. Rev. B 2011, 84, 075206. [Google Scholar] [CrossRef]
- Guo, Z.; Lee, D.; Gao, H.; Huang, L. Exciton structure and dynamics in solution aggregates of a low-bandgap copolymer. J. Phys. Chem. B 2015, 119, 7666–7672. [Google Scholar] [CrossRef]
- Karabunarliev, S.; Bittner, E.R. Electroluminescence yield in donor−acceptor copolymers and diblock polymers: A comparative theoretical study. J. Phys. Chem. B 2004, 108, 10219–10225. [Google Scholar] [CrossRef]
- Wu, G.; Li, Z.; Zhang, X.; Lu, G. Charge separation and exciton dynamics at polymer/ZnO interface from first-principles simulations. J. Phys. Chem. Lett. 2014, 5, 2649–2656. [Google Scholar] [CrossRef][Green Version]
- Müller, J.G.; Atas, E.; Tan, C.; Schanze, K.S.; Kleiman, V.D. The role of exciton hopping and direct energy transfer in the efficient quenching of conjugated polyelectrolytes. J. Am. Chem. Soc. 2006, 128, 4007–4016. [Google Scholar] [CrossRef]
- Oh-e, M.; Yokoyama, H.; Yorozuya, S.; Akagi, K.; Belkin, M.A.; Shen, Y.R. Sum-frequency vibrational spectroscopy of a helically structured conjugated polymer. Phys. Rev. Lett. 2004, 93, 267402. [Google Scholar] [CrossRef]
- Walter, M.J.; Lupton, J.M.; Becker, K.; Feldmann, J.; Gaefke, G.; Höger, S. Simultaneous Raman and fluorescence spectroscopy of single conjugated polymer chains. Phys. Rev. Lett. 2007, 98, 137401. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Wu, Z.; Lei, T.; Yu, Y.; Yuan, F.; Ning, S.; Jiao, B.; Hou, X. Theoretical insight into the deep-blue amplified spontaneous emission of new organic semiconductor molecules. Org. Electron. 2014, 15, 3144–3153. [Google Scholar] [CrossRef]
- Jensen, S.A.; Mics, Z.; Ivanov, I.; Varol, H.S.; Turchinovich, D.; Koppens, F.H.L.; Bonn, M.; Tielrooij, K.J. Competing ultrafast energy relaxation pathways in photoexcited graphene. Nano Lett. 2014, 14, 5839–5845. [Google Scholar] [CrossRef]
- Gao, K.; Xie, S.; Li, Y.; Xia, C. Realization of the population inversion in a conjugated polymer by a single or double stimulating pulse. Org. Electron. 2014, 15, 1965–1971. [Google Scholar] [CrossRef]
- Gao, K.; Liu, X.; Liu, D.; Xie, S. Charge carrier generation through reexcitations of an exciton in poly (p-phenylene vinylene) molecules. Phys. Rev. B 2007, 75, 205412. [Google Scholar] [CrossRef]
- Pandya, R.; Alvertis, A.M.; Gu, Q.; Sung, J.; Legrand, L.; Kéher, D.; Barisien, T.; Chin, A.W.; Schnedermann, C.; Rao, A. Exciton Diffusion in Highly-Ordered One Dimensional Conjugated Polymers: Effects of Back-Bone Torsion, Electronic Symmetry, Phonons and Annihilation. J. Phys. Chem. Lett. 2021, 12, 3669–3678. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Shi, H.; Luo, J.; Shen, J.; Li, S.; George, T.F. Lattice Vibrations and Time-Dependent Evolution of Local Phonon Modes during Exciton Formation in Conjugated Polymeric Molecules. Polymers 2021, 13, 1724. https://doi.org/10.3390/polym13111724
Zhang Y, Shi H, Luo J, Shen J, Li S, George TF. Lattice Vibrations and Time-Dependent Evolution of Local Phonon Modes during Exciton Formation in Conjugated Polymeric Molecules. Polymers. 2021; 13(11):1724. https://doi.org/10.3390/polym13111724
Chicago/Turabian StyleZhang, Yusong, Huayan Shi, Junteng Luo, Jianguo Shen, Sheng Li, and Thomas F. George. 2021. "Lattice Vibrations and Time-Dependent Evolution of Local Phonon Modes during Exciton Formation in Conjugated Polymeric Molecules" Polymers 13, no. 11: 1724. https://doi.org/10.3390/polym13111724
APA StyleZhang, Y., Shi, H., Luo, J., Shen, J., Li, S., & George, T. F. (2021). Lattice Vibrations and Time-Dependent Evolution of Local Phonon Modes during Exciton Formation in Conjugated Polymeric Molecules. Polymers, 13(11), 1724. https://doi.org/10.3390/polym13111724