
High-Strength GO/PA66 Nanocomposite Fibers via In Situ Precipitation and Polymerization


Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of GO Aqueous Dispersion
2.3. Preparation of GO Ethanol Dispersion by Solvent Replacement
2.4. Preparation of GO/PA66 Salt Nanocomposites by In Situ Precipitation
2.5. Fabrication of GO/PA66 Nanocomposite Fibers by In Situ Polymerization
2.6. Characterizations
3. Results and Discussion
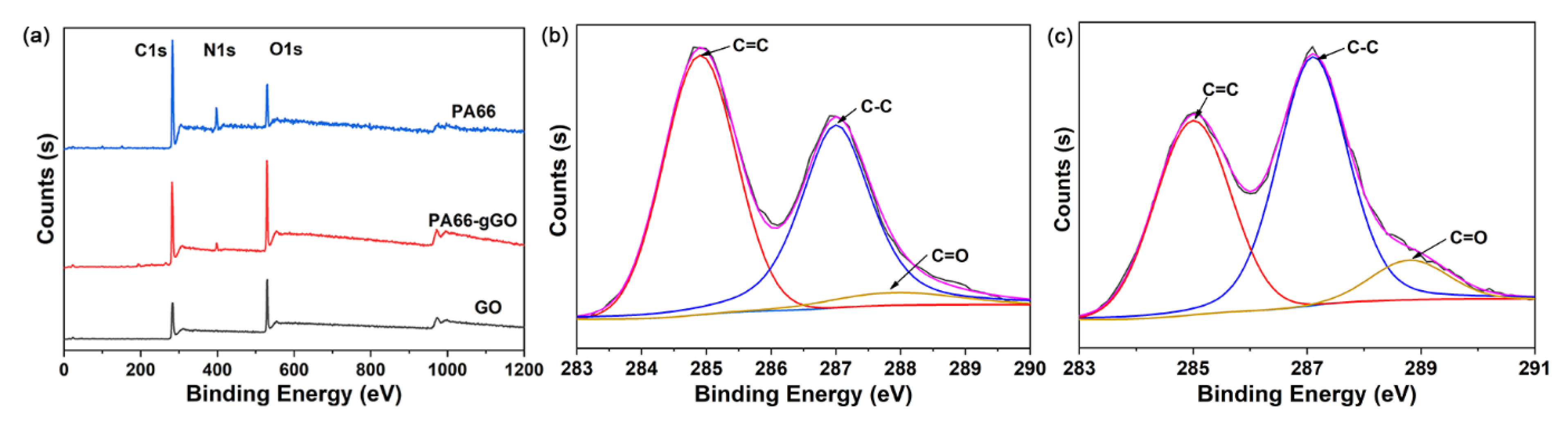
3.1. Formation of GO/PA66 Salt Nanocomposites
3.2. In Situ Polymerization of GO/PA66 Salt Nanocomposites
3.3. Properties of GO/PA66 Nanocomposite Fibers
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Spina, R.; Cavalcante, B. Evaluation of grinding of unfilled and glass fiber reinforced polyamide 6,6. Polymers 2020, 12, 2288. [Google Scholar] [CrossRef]
- Cakal Sarac, E.; Haghighi Poudeh, L.; Berktas, I.; Saner Okan, B. Scalable fabrication of high-performance graphene/polyamide 66 nanocomposites with controllable surface chemistry by melt compounding. J. Appl. Polym. Sci. 2021, 138, 1–12. [Google Scholar] [CrossRef]
- Zhang, J.; Gao, X.; Zhang, X.; Liu, H.; Zhang, H.; Zhang, X. Polyamide 66 and amino-functionalized multi-walled carbon nanotube composites and their melt-spun fibers. J. Mater. Sci. 2019, 54, 11056–11068. [Google Scholar] [CrossRef]
- Zhang, H.; Lu, J.; Yang, H.; Lang, J.; Yang, H. Comparative study on the flame-retardant properties and mechanical properties of PA66 with different dicyclohexyl hypophosphite acid metal salts. Polymers 2019, 11, 1956. [Google Scholar] [CrossRef]
- Lu, M.; Liao, J.; Dong, J.; Wu, J.; Qiu, H.; Zhou, X.; Li, J.; Jiang, D.; He, T.C.; Quan, Z. An effective treatment of experimental osteomyelitis using the antimicrobial titanium/silver-containing nHP66 (nano-hydroxyapatite/polyamide-66) nanoscaffold biomaterials. Sci. Rep. 2016, 6, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Mirvakili, S.M.; Hunter, I.W. Multidirectional Artificial Muscles from Nylon. Adv. Mater. 2017, 29, 1–7. [Google Scholar] [CrossRef]
- Haines, C.S.; Lima, M.D.; Li, N.; Spinks, G.M.; Foroughi, J.; Madden, J.D.W.; Kim, S.H.; Fang, S.; De Andrade, M.J.; Göktepe, F.; et al. Artificial muscles from fishing line and sewing thread. Science 2014, 343, 868–872. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.S.; Li, S.M.; Hsiao, S.T.; Liao, W.H.; Chen, P.H.; Yang, S.Y.; Tien, H.W.; Ma, C.C.M.; Hu, C.C. Integration of tailored reduced graphene oxide nanosheets and electrospun polyamide-66 nanofabrics for a flexible supercapacitor with high-volume- and high-area-specific capacitance. Carbon 2014, 73, 87–98. [Google Scholar] [CrossRef]
- Oh, J.Y.; Choi, Y.S.; Yang, S.J.; Kim, J.; Choi, H.S.; Choi, G.D.; Yun, C.H.; Lee, B.K.; Park, C.R. Effect of microstructure and morphological properties of carbon nanotubes on the length reduction during melt processing. Compos. Sci. Technol. 2015, 112, 42–49. [Google Scholar] [CrossRef]
- Dericiler, K.; Sadeghi, H.M.; Yagci, Y.E.; Sas, H.S.; Okan, B.S. Experimental and numerical investigation of flow and alignment behavior of waste tire-derived graphene nanoplatelets in PA66 matrix during melt-mixing and injection. Polymers 2021, 13, 949. [Google Scholar] [CrossRef] [PubMed]
- Cho, B.G.; Lee, J.E.; Hwang, S.H.; Han, J.H.; Chae, H.G.; Park, Y. Bin Enhancement in mechanical properties of polyamide 66-carbon fiber composites containing graphene oxide-carbon nanotube hybrid nanofillers synthesized through in situ interfacial polymerization. Compos. Part A Appl. Sci. Manuf. 2020, 135, 105938. [Google Scholar] [CrossRef]
- Geim, A.K.; Novoselov, K.S. The rise of graphene. Nat. Mater. 2007, 6, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Wei, X.; Kysar, J.W.; Hone, J. Measurement of the Elastic Properties and Intrinsic Strength of Monolayer Graphene. Science 2008, 321, 385–388. [Google Scholar] [CrossRef]
- Orlita, M.; Faugeras, C.; Plochocka, P.; Neugebauer, P.; Martinez, G.; Maude, D.K.; Barra, A.L.; Sprinkle, M.; Berger, C.; De Heer, W.A.; et al. Approaching the dirac point in high-mobility multilayer epitaxial graphene. Phys. Rev. Lett. 2008, 101, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Balandin, A.A.; Ghosh, S.; Bao, W.; Calizo, I.; Teweldebrhan, D.; Miao, F.; Lau, C.N. Superior thermal conductivity of single-layer graphene. Nano Lett. 2008, 8, 902–907. [Google Scholar] [CrossRef]
- Young, R.J.; Liu, M.; Kinloch, I.A.; Li, S.; Zhao, X.; Vallés, C.; Papageorgiou, D.G. The mechanics of reinforcement of polymers by graphene nanoplatelets. Compos. Sci. Technol. 2018, 154, 110–116. [Google Scholar] [CrossRef]
- Xu, X.; Chen, J.; Zhou, J.; Li, B. Thermal Conductivity of Polymers and Their Nanocomposites. Adv. Mater. 2018, 30, 1–10. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Petkovich, N.D.; Liu, K.; Qian, Y.; Macosko, C.W.; Stein, A. Unsaturated polyester resin toughening with very low loadings of GO derivatives. Polymer 2017, 110, 149–157. [Google Scholar] [CrossRef]
- Park, Y.T.; Qian, Y.; Chan, C.; Suh, T.; Nejhad, M.G.; Macosko, C.W.; Stein, A. Epoxy toughening with low graphene loading. Adv. Funct. Mater. 2015, 25, 575–585. [Google Scholar] [CrossRef]
- Wang, Y.Z.; Gao, X.F.; Liu, H.H.; Zhang, J.; Zhang, X.X. Green fabrication of functionalized graphene via one-step method and its reinforcement for polyamide 66 fibers. Mater. Chem. Phys. 2020, 240, 122288. [Google Scholar] [CrossRef]
- Papadopoulou, E.L.; Pignatelli, F.; Marras, S.; Marini, L.; Davis, A.; Athanassiou, A.; Bayer, I.S. Nylon 6,6/graphene nanoplatelet composite films obtained from a new solvent. RSC Adv. 2016, 6, 6823–6831. [Google Scholar] [CrossRef]
- Cho, B.G.; Lee, S.; Hwang, S.H.; Han, J.H.; Chae, H.G.; Park, Y. Bin Influence of hybrid graphene oxide-carbon nanotube as a nano-filler on the interfacial interaction in nylon composites prepared by in situ interfacial polymerization. Carbon 2018, 140, 324–337. [Google Scholar] [CrossRef]
- Ji, X.; Xu, Y.; Zhang, W.; Cui, L.; Liu, J. Review of functionalization, structure and properties of graphene/polymer composite fibers. Compos. Part A Appl. Sci. Manuf. 2016, 87, 29–45. [Google Scholar] [CrossRef]
- Dallinger, A.; Kindlhofer, P.; Greco, F.; Coclite, A.M. Multiresponsive Soft Actuators Based on a Thermoresponsive Hydrogel and Embedded Laser-Induced Graphene. ACS Appl. Polym. Mater. 2021, 3, 1809–1818. [Google Scholar] [CrossRef]
- Cataldi, P.; Steiner, P.; Raine, T.; Lin, K.; Kocabas, C.; Young, R.J.; Bissett, M.; Kinloch, I.A.; Papageorgiou, D.G. Multifunctional Biocomposites Based on Polyhydroxyalkanoate and Graphene/Carbon Nanofiber Hybrids for Electrical and Thermal Applications. ACS Appl. Polym. Mater. 2020, 2, 3525–3534. [Google Scholar] [CrossRef]
- Cseri, L.; Baugh, J.; Alabi, A.; AlHajaj, A.; Zou, L.; Dryfe, R.A.W.; Budd, P.M.; Szekely, G. Graphene oxide-polybenzimidazolium nanocomposite anion exchange membranes for electrodialysis. J. Mater. Chem. A 2018, 6, 24728–24739. [Google Scholar] [CrossRef]
- Duan, X.; Yu, B.; Yang, T.; Wu, Y.; Yu, H.; Huang, T. In situ polymerization of nylon 66/reduced graphene oxide nanocomposites. J. Nanomater. 2018, 2018. [Google Scholar] [CrossRef]
- Li, D.; Müller, M.B.; Gilje, S.; Kaner, R.B.; Wallace, G.G. Processable aqueous dispersions of graphene nanosheets. Nat. Nanotechnol. 2008, 3, 101–105. [Google Scholar] [CrossRef]
- Hong, B.J.; Compton, O.C.; An, Z.; Eryazici, I.; Nguyen, S.T. Successful stabilization of graphene oxide in electrolyte solutions: Enhancement of biofunctionalization and cellular uptake. ACS Nano 2012, 6, 63–73. [Google Scholar] [CrossRef]
- Dhayal, V.; Hashmi, S.Z.; Kumar, U.; Choudhary, B.L.; Kuznetsov, A.E.; Dalela, S.; Kumar, S.; Kaya, S.; Dolia, S.N.; Alvi, P.A. Spectroscopic studies, molecular structure optimization and investigation of structural and electrical properties of novel and biodegradable Chitosan-GO polymer nanocomposites. J. Mater. Sci. 2020, 55, 14829–14847. [Google Scholar] [CrossRef]
- Alammar, A.; Park, S.H.; Ibrahim, I.; Deepak, A.; Holtzl, T.; Dumée, L.F.; Lim, H.N.; Szekely, G. Architecting neonicotinoid-scavenging nanocomposite hydrogels for environmental remediation. Appl. Mater. Today 2020, 21, 100878. [Google Scholar] [CrossRef]
- Kinloch, I.A.; Suhr, J.; Lou, J.; Young, R.J.; Ajayan, P.M. Composites with carbon nanotubes and graphene: An outlook. Science 2018, 362, 547–553. [Google Scholar] [CrossRef]
- He, D.; Shen, L.; Zhang, X.; Wang, Y.; Bao, N.; Kung, H.H. An efficient and eco-friendly solution-chemical route for preparation of ultrastable reduced graphene oxide suspensions. AIChE J. 2014, 60, 2757–2764. [Google Scholar] [CrossRef]
- Li, C.; Guo, Y.; Shen, L.; Ji, C.; Bao, N. Scalable concentration process of graphene oxide dispersions via cross-flow membrane filtration. Chem. Eng. Sci. 2019, 200, 127–137. [Google Scholar] [CrossRef]
- Yoon, K.Y.; An, S.J.; Chen, Y.; Lee, J.H.; Bryant, S.L.; Ruoff, R.S.; Huh, C.; Johnston, K.P. Graphene oxide nanoplatelet dispersions in concentrated NaCl and stabilization of oil/water emulsions. J. Colloid Interface Sci. 2013, 403, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.Y.; Lu, H.; Zhuang, Z.; Wang, X.P.; Fang, Q.F. Nano-hydroxyapatite/poly(L-lactic acid) composite synthesized by a modified in situ precipitation: Preparation and properties. J. Mater. Sci. Mater. Med. 2010, 21, 3077–3083. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, A.C.; Meyer, J.C.; Scardaci, V.; Casiraghi, C.; Lazzeri, M.; Mauri, F.; Piscanec, S.; Jiang, D.; Novoselov, K.S.; Roth, S.; et al. Raman spectrum of graphene and graphene layers. Phys. Rev. Lett. 2006, 97, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Zhang, X.; Gao, X.; Liu, H.; Zhang, X. Fabrication of high-strength PET fibers modified with graphene oxide of varying lateral size. J. Mater. Sci. 2020, 55, 8940–8953. [Google Scholar] [CrossRef]
- Min, C.; Nie, P.; Song, H.J.; Zhang, Z.; Zhao, K. Study of tribological properties of polyimide/graphene oxide nanocomposite films under seawater-lubricated condition. Tribol. Int. 2014, 80, 131–140. [Google Scholar] [CrossRef]
- Wang, C.-Y.; Li, G.; Zhao, X.-Y.; Jiang, J.-M. High solubility, low-dielectric constant, and optical transparency of novel polyimides derived from 3,3′,5,5′-tetramethyl-4,4′-diaminodiphenyl-4″-isopropyltoluene. J. Polym. Sci. Part A Polym. Chem. 2009, 47, 3309–3317. [Google Scholar] [CrossRef]
- Zangmeister, R.A.; Morris, T.A.; Tarlov, M.J. Characterization of polydopamine thin films deposited at short times by autoxidation of dopamine. Langmuir 2013, 29, 8619–8628. [Google Scholar] [CrossRef]
- Sheng, X.; Mo, R.; Ma, Y.; Zhang, X.; Zhang, L.; Wu, H. Waterborne Epoxy Resin/Polydopamine Modified Zirconium Phosphate Nanocomposite for Anticorrosive Coating. Ind. Eng. Chem. Res. 2019, 58, 16571–16580. [Google Scholar] [CrossRef]
- Wan, Y.J.; Tang, L.C.; Gong, L.X.; Yan, D.; Li, Y.B.; Wu, L.B.; Jiang, J.X.; Lai, G.Q. Grafting of epoxy chains onto graphene oxide for epoxy composites with improved mechanical and thermal properties. Carbon 2014, 69, 467–480. [Google Scholar] [CrossRef]
- Ramesh, C.; Keller, A.; Eltink, S.J.E.A. Studies on the crystallization and melting of nylon-6,6: 1. The dependence of the Brill transition on the crystallization temperature. Polymer 1994, 35, 2483–2487. [Google Scholar] [CrossRef]
- Zhang, G.; Yan, D. Crystallization kinetics and melting behavior of nylon 10,10 in nylon 10,10-montmorillonite nanocomposites. J. Appl. Polym. Sci. 2003, 88, 2181–2188. [Google Scholar] [CrossRef]
- Garcia, D.; Starkweather, H.W. Hydrogen bonding in nylon 66 and model compounds. J. Polym. Sci. Polym. Phys. Ed. 1985, 23, 537–555. [Google Scholar] [CrossRef]
- Morimune-Moriya, S.; Yada, S.; Kuroki, N.; Ito, S.; Hashimoto, T.; Nishino, T. Strong reinforcement effects of nanodiamond on mechanical and thermal properties of polyamide 66. Compos. Sci. Technol. 2020, 199, 108356. [Google Scholar] [CrossRef]
- Basavaraj, E.; Ramaraj, B. A study on mechanical, thermal, and wear characteristics of nylon 66/molybdenum disulfide composites reinforced with glass fibers. Polym. Compos. 2012, 33, 1570–1577. [Google Scholar] [CrossRef]
- Abdallah, W.; Yilmazer, U. Polyamide 66 nanocomposites based on organoclays treated with thermally stable phosphonium salts. J. Appl. Polym. Sci. 2013, 127, 772–783. [Google Scholar] [CrossRef]
- Zhou, K.; Jiang, S.; Shi, Y.; Liu, J.; Wang, B.; Hu, Y.; Gui, Z. Multigram-scale fabrication of organic modified MoS2nanosheets dispersed in polystyrene with improved thermal stability, fire resistance, and smoke suppression properties. RSC Adv. 2014, 4, 40170–40180. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Tm (°C) | Tc (°C) | ΔHc (J/g) | Xc (%) |
|---|---|---|---|---|
| PA66-GO-0 | 260.5 | 228.4 | 53.86 | 28.5 |
| PA66-GO-0.1 | 261.1 | 228.8 | 48.15 | 25.6 |
| PA66-GO-0.2 | 261.3 | 231.1 | 50.59 | 26.9 |
| PA66-GO-0.3 | 261.8 | 234.0 | 49.62 | 26.3 |
| PA66-GO-0.4 | 262.1 | 234.3 | 52.63 | 27.9 |
| PA66-GO-0.5 | 262.5 | 234.5 | 55.09 | 29.2 |
| Samples | E’ (MPa) | Tan δ | Tg (°C) |
|---|---|---|---|
| PA66-GO-0 | 1345 | 0.120 | 43.2 |
| PA66-GO-0.1 | 1475 | 0.110 | 51.6 |
| PA66-GO-0.2 | 1691 | 0.081 | 56.1 |
| PA66-GO-0.3 | 1900 | 0.076 | 57.5 |
| PA66-GO-0.4 | 1317 | 0.073 | 58.4 |
| PA66-GO-0.5 | 970 | 0.072 | 59.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gu, A.; Wu, J.; Shen, L.; Zhang, X.; Bao, N. High-Strength GO/PA66 Nanocomposite Fibers via In Situ Precipitation and Polymerization. Polymers 2021, 13, 1688. https://doi.org/10.3390/polym13111688
Gu A, Wu J, Shen L, Zhang X, Bao N. High-Strength GO/PA66 Nanocomposite Fibers via In Situ Precipitation and Polymerization. Polymers. 2021; 13(11):1688. https://doi.org/10.3390/polym13111688
Chicago/Turabian StyleGu, Ao, Jian Wu, Liming Shen, Xiaoyan Zhang, and Ningzhong Bao. 2021. "High-Strength GO/PA66 Nanocomposite Fibers via In Situ Precipitation and Polymerization" Polymers 13, no. 11: 1688. https://doi.org/10.3390/polym13111688
APA StyleGu, A., Wu, J., Shen, L., Zhang, X., & Bao, N. (2021). High-Strength GO/PA66 Nanocomposite Fibers via In Situ Precipitation and Polymerization. Polymers, 13(11), 1688. https://doi.org/10.3390/polym13111688