Heat Shock Protein 90 (Hsp90)-Inhibitor-Luminespib-Loaded-Protein-Based Nanoformulation for Cancer Therapy



Abstract

1. Introduction
2. Materials and Methods
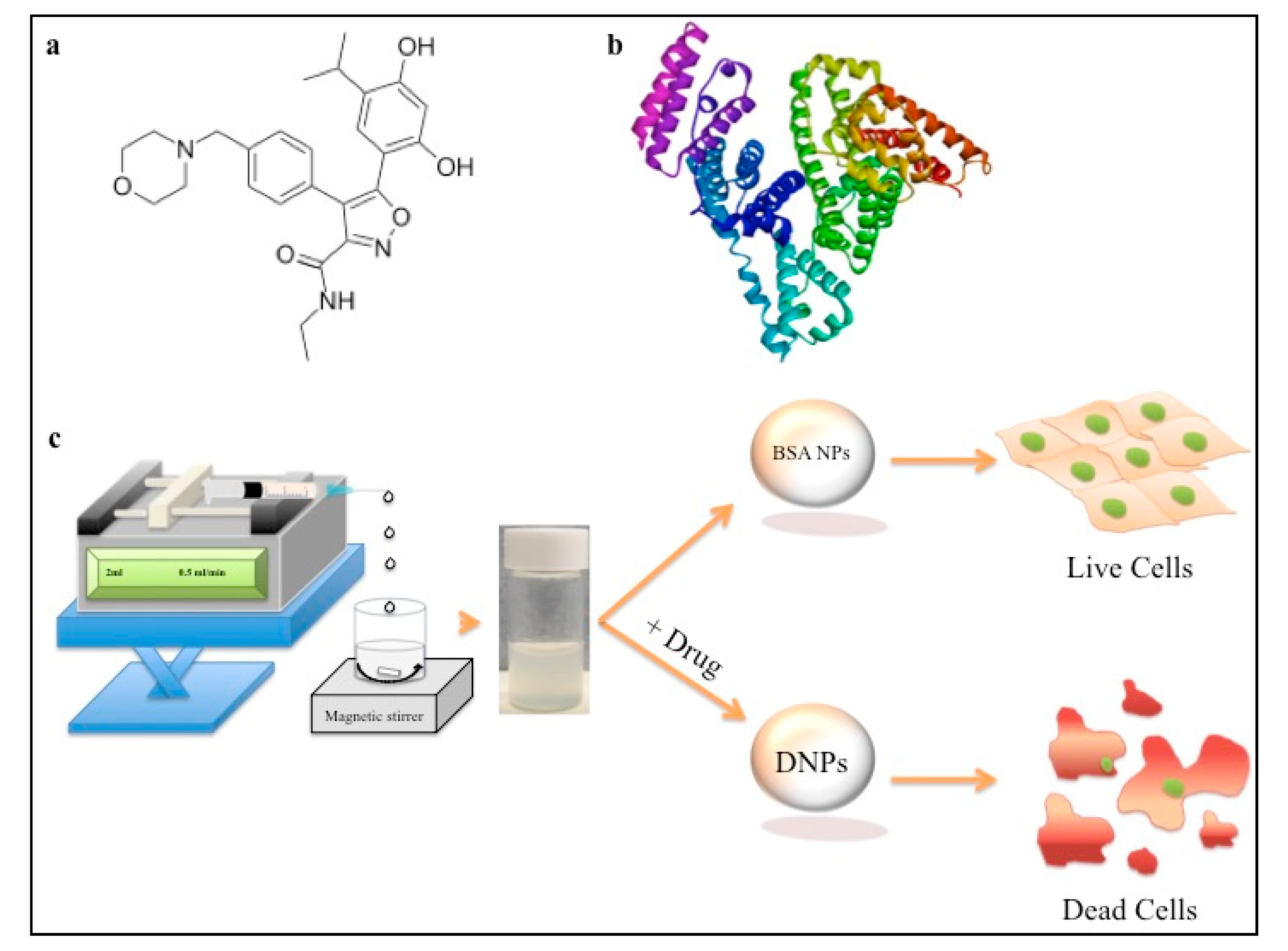
2.1. Synthesis of BSA-Luminespib Nanoconjugates
2.2. Luminespib BSA Particle Characterization Studies
2.3. In Silico Molecular Docking
2.4. Luminespib-BSA Interaction Studies
2.4.1. Fluorescence Quenching Studies for Drug BSA Complex
2.4.2. UV-Vis Absorption Studies
2.4.3. Thin-Layer Chromatography (TLC)
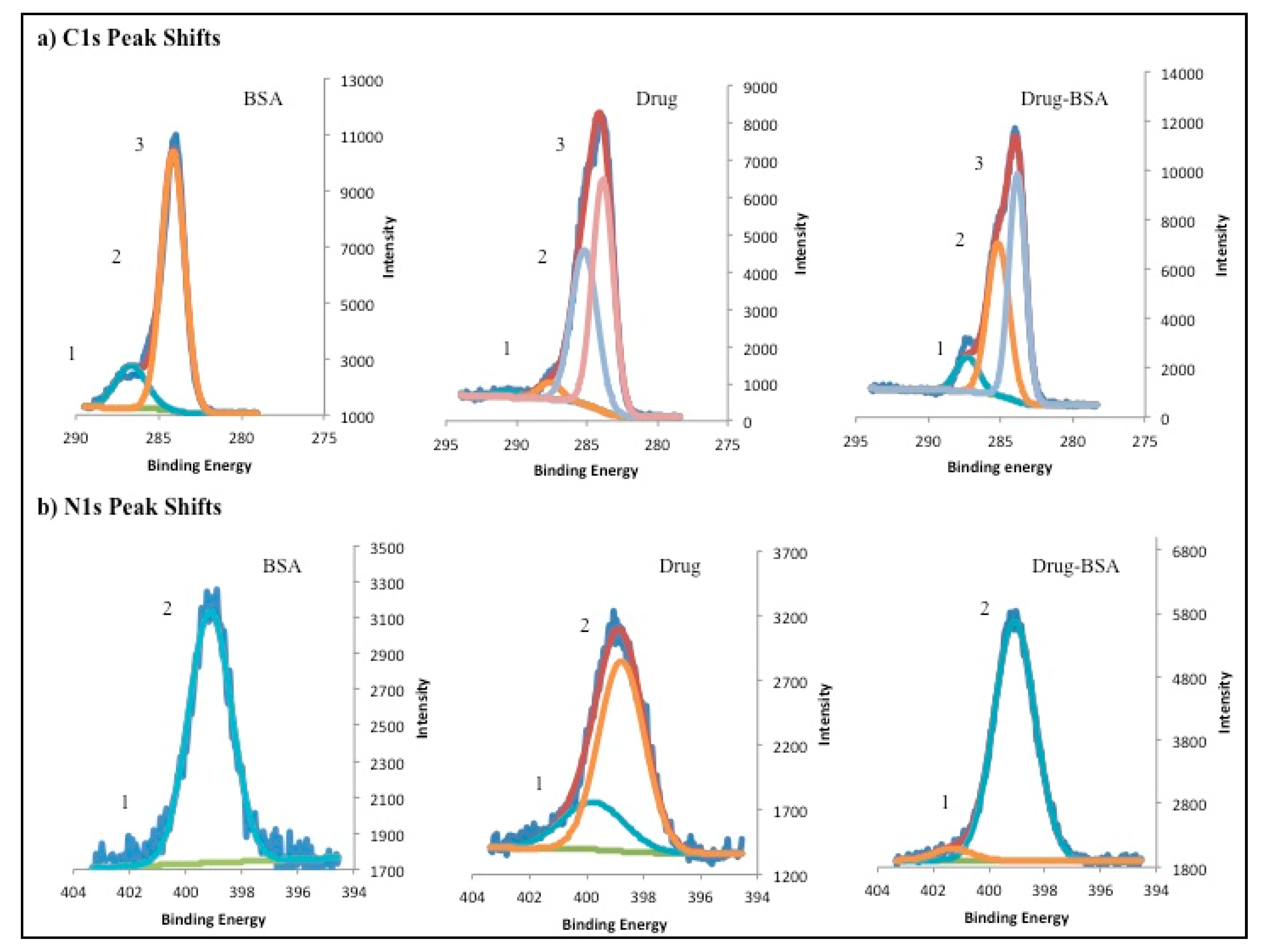
2.4.4. X-ray Photoelectron Spectroscopy Studies (XPS)
2.5. Encapsulation and Drug Loading Evaluation
2.6. Stability of DNPs
2.7. In Vitro Drug Release for DNPs
2.8. Cell Culture and In Vitro Cytotoxicity Studies
3. Results and Discussion
3.1. Synthesis, Particle Size Characterization and Encapsulation of BSA Luminespib NPs
3.2. BSA Luminespib Interaction Studies
3.3. X-ray Photo Electron Spectroscopy, TLC and Stability Studies
3.4. In Vitro Drug Release Studies
3.5. In Vitro Cytotoxicity Studies
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Malvezzi, M.; Carioli, G.; Bertuccio, P.; Rosso, T.; Boffetta, P.; Levi, F.; La Vecchia, C.; Negri, E. European cancer mortality predictions for the year 2016 with focus on leukaemias. Ann. Oncol. 2016, 27, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Xiao, L.; Wang, L.; Ruden, D.M. Hsp90 inhibitors and drug resistance in cancer: The potential benefits of combination therapies of Hsp90 inhibitors and other anti-cancer drugs. Biochem. Pharmacol. 2012, 83, 995–1004. [Google Scholar] [CrossRef] [PubMed]
- Reshkin, S.J.; Stock, C.; Taylor, S.; Walsh, M.; Muddathir, A.K.; Verduzco, D.; Bashir, A.; Mohammed, O.Y.; Elhassan, G.O.; Harguindey, S.; et al. Resistance to cancer chemotherapy: Failure in drug response from ADME to P-gp. Cancer Cell Int. 2015, 15, 71. [Google Scholar] [CrossRef]
- Jego, G.; Hazoume, A.; Seigneuric, R.; Garrido, C. Targeting heat shock proteins in cancer. Cancer Lett. 2013, 332, 275–285. [Google Scholar] [CrossRef]
- Sőti, C.; Nagy, E.; Giricz, Z.; Vigh, L.; Csermely, P.; Ferdinandy, P. Heat shock proteins as emerging therapeutic targets. Br. J. Pharmacol. 2005, 146, 769–780. [Google Scholar] [CrossRef]
- Neckers, L.M.; Workman, P. Hsp90 molecular chaperone inhibitors: Are we there yet? Clin. Cancer Res. 2012, 18, 64–76. [Google Scholar] [CrossRef]
- Rochani, A.K.; Girija, A.R.; Borah, A.; Maekawa, T.; Kumar, D.S. Heat-shock protein 90–targeted nano anticancer therapy. J. Pharm. Sci. 2016, 105, 1454–1466. [Google Scholar] [CrossRef]
- Rochani, A.K.; Singh, M.; Tatu, U. Heat shock protein 90 inhibitors as broad spectrum anti-infectives. Curr. Pharm. Des. 2013, 19, 377–386. [Google Scholar] [CrossRef]
- Modi, S.; Stopeck, A.; Linden, H.; Solit, D.; Chandarlapaty, S.; Rosen, N.; D’Andrea-Carlino, G.; Dickler, M.; Moynahan, M.E.; Sugarman, S.; et al. HSP90 inhibition is effective in breast cancer: A phase II trial of tanespimycin (17-AAG) plus trastuzumab in patients with HER2-positive metastatic breast cancer progressing on trastuzumab. Clin. Cancer Res. 2011, 17, 5132–5139. [Google Scholar] [CrossRef]
- Barril, X.; Brough, P.; Drysdale, M.; Hubbard, R.E.; Massey, A.; Surgenor, A.; Wright, L. Structure-based discovery of a new class of Hsp90 inhibitors. Bioorg. Med. Chem. Lett. 2005, 15, 5187–5191. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, T.; Schwartz, S.J.; Sun, D. New developments in Hsp90 inhibitors as anti-cancer therapeutics: Mechanisms, clinical perspective and more potential. Drug Resist. Updat. 2009, 12, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Brough, P.A.; Aherne, W.; Barril, X.; Borgognoni, J.; Boxall, K.; Cansfield, J.E.; Cheung, K.-M.J.; Collins, I.; Davies, N.G.M.; Drysdale, M.J.; et al. 4,5-Diarylisoxazole Hsp90 Chaperone Inhibitors: Potential Therapeutic Agents for the Treatment of Cancer. J. Med. Chem. 2008, 51, 196–218. [Google Scholar] [CrossRef] [PubMed]
- Pharmaceuticals, N. Phase I-II Study to Determine the Maximum Tolerated Dose (MTD) of AUY922 in Advanced Solid Malignancies, and Efficacy in HER2+ or ER+ Locally Advanced or Metastatic Breast Cancer Patients. Available online: https://www.clinicaltrials.gov/ct2/show/NCT00526045?term=AUY922+breast+cancer&rank=1 (accessed on 10 August 2020).
- Minami, C.A.; Chung, D.U.; Chang, H.R. Management options in triple-negative breast cancer. Breast Cancer Basic Clin. Res. 2011, 5, 175–199. [Google Scholar] [CrossRef] [PubMed]
- Moser, C.; Lang, S.A.; Hackl, C.; Wagner, C.; Scheiffert, E.; Schlitt, H.J.; Geissler, E.K.; Stoeltzing, O. Targeting HSP90 by the novel inhibitor NVP-AUY922 reduces growth and angiogenesis of pancreatic cancer. Anticancer Res. 2012, 32, 2551–2561. [Google Scholar] [PubMed]
- Jensen, M.R.; Schoepfer, J.; Radimerski, T.; Massey, A.J.; Guy, C.T.; Brueggen, J.; Quadt, C.; Buckler, A.; Cozens, R.; Drysdale, M.J.; et al. NVP-AUY922: A small molecule HSP90 inhibitor with potent antitumor activity in preclinical breast cancer models. Breast Cancer Res. 2008, 10, R33. [Google Scholar] [CrossRef] [PubMed]
- Sessa, C.; Shapiro, G.I.; Bhalla, K.N.; Britten, C.; Jacks, K.S.; Mita, M.; Papadimitrakopoulou, V.; Pluard, T.; Samuel, T.A.; Akimov, M.; et al. First-in-human phase I dose-escalation study of the HSP90 inhibitor AUY922 in patients with advanced solid tumors. Clin. Cancer Res. 2013, 19, 3671–3680. [Google Scholar] [CrossRef]
- Johnson, M.L.; Yu, H.A.; Hart, E.M.; Weitner, B.B.; Rademaker, A.W.; Patel, J.D.; Kris, M.G.; Riely, G.J. Phase I/II study of HSP90 inhibitor AUY922 and erlotinib for EGFR-mutant lung cancer with acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors. J. Clin. Oncol. 2015, 33, 1666–1673. [Google Scholar] [CrossRef]
- Kong, A.; Rea, D.; Ahmed, S.; Beck, J.T.; López, R.L.; Biganzoli, L.; Armstrong, A.C.; Aglietta, M.; Alba, E.; Campone, M.; et al. Phase 1B/2 study of the HSP90 inhibitor AUY922 plus trastuzumab in metastatic HER2-positive breast cancer patients who have progressed on trastuzumab-based regimen. Oncotarget 2016, 7, 37680–37692. [Google Scholar] [CrossRef]
- Rosenholm, J.M.; Mamaeva, V.; Sahlgren, C.M.; Lindén, M. Nanoparticles in targeted cancer therapy: Mesoporous silica nanoparticles entering preclinical development stage. Nanomedicine 2012, 7, 111–120. [Google Scholar] [CrossRef]
- Parveen, S.; Sahoo, S.K. Polymeric nanoparticles for cancer therapy. J. Drug Target. 2008, 16, 108–123. [Google Scholar] [CrossRef]
- Rochani, A.K.; Balasubramanian, S.; Girija, A.R.; Raveendran, S.; Borah, A.; Nagaoka, Y.; Nakajima, Y.; Maekawa, T.; Kumar, D.S. Dual mode of cancer cell destruction for pancreatic cancer therapy using Hsp90 inhibitor loaded polymeric nano magnetic formulation. Int. J. Pharm. 2016, 511, 648–658. [Google Scholar] [CrossRef] [PubMed]
- Misak, H.E.; Asmatulu, R.; Gopu, J.S.; Man, K.-P.; Zacharias, N.M.; Wooley, P.H.; Yang, S.-Y. Albumin-based nanocomposite spheres for advanced drug delivery systems. Biotechnol. J. 2013, 9, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Elzoghby, A.O.; Samy, W.M.; Elgindy, N.A. Albumin-based nanoparticles as potential controlled release drug delivery systems. J. Control. Release 2012, 157, 168–182. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.X.; Kim, H.-Y.; Dass, C. Probing three-dimensional structure of bovine serum albumin by chemical cross-linking and mass spectrometry. J. Am. Soc. Mass Spectrom. 2004, 15, 1237–1247. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Wang, D.; Li, X.; Lu, Y. Exploring the binding mechanism of dihydropyrimidinones to human serum albumin: Spectroscopic and molecular modeling techniques. Colloids Surf. B Biointerfaces 2011, 84, 272–279. [Google Scholar] [CrossRef]
- Desai, N. Nanoparticle albumin-bound paclitaxel (Abraxane®). In Albumin in Medicine; Springer: Singapore, 2016; pp. 101–119. [Google Scholar]
- Hao, H.; Ma, Q.; Huang, C.; He, F.; Yao, P. Preparation, characterization, and in vivo evaluation of doxorubicin loaded BSA nanoparticles with folic acid modified dextran surface. Int. J. Pharm. 2013, 444, 77–84. [Google Scholar] [CrossRef]
- Li, J.-M.; Chen, W.; Wang, H.; Jin, C.; Yu, X.-J.; Lu, W.-Y.; Cui, L.; Fu, D.-L.; Ni, Q.-X.; Hou, H.-M. Preparation of albumin nanospheres loaded with gemcitabine and their cytotoxicity against BXPC-3 cells in vitro. Acta Pharmacol. Sin. 2009, 30, 1337–1343. [Google Scholar] [CrossRef]
- Tao, C.; YU, C.; De, T.K.; Everett, N.; Frankel, T.; Ci, S.; Trieu, V.; Soon-Shiong, P.; Desai, N. Preparation of Nanoparticle Albumin Bound 17AAG (nab-17AAG) Suitable for Intravenous Administration. Available online: http://cancerres.aacrjournals.org/content/65/9_Supplement/336.3 (accessed on 10 August 2020).
- Merodio, M.; Arnedo, A.; Renedo, M.; Irache, J.M. Ganciclovir-loaded albumin nanoparticles: Characterization and in vitro release properties. Eur. J. Pharm. Sci. 2001, 12, 251–259. [Google Scholar] [CrossRef]
- Sripriyalakshmi, S.; Anjali, C.H.; George, P.D.; Rajith, B.; Ravindran, A. BSA nanoparticle loaded atorvastatin calcium—A new facet for an old drug. PLoS ONE 2014, 9, e86317. [Google Scholar] [CrossRef]
- Green, J.L.; Moon, R.W.; Whalley, D.; Bowyer, P.W.; Wallace, C.; Rochani, A.; Nageshan, R.K.; Howell, S.A.; Grainger, M.; Jones, H.M.; et al. Imidazopyridazine inhibitors of plasmodium falciparum calcium-dependent protein kinase 1 also target cyclic GMP-dependent protein kinase and heat shock protein 90 to kill the parasite at different stages of intracellular development. Antimicrob. Agents Chemother. 2015, 60, 1464–1475. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Solis, F.J.; Wets, R.J.-B. Minimization by random search techniques. Math. Oper. Res. 1981, 6, 19–30. [Google Scholar] [CrossRef]
- Galisteo-González, F.; Molina-Bolívar, J.A. Systematic study on the preparation of BSA nanoparticles. Colloids Surf. B Biointerfaces 2014, 123, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Langer, K.; Balthasar, S.; Vogel, V.; Dinauer, N.; Von Briesen, H.; Schubert, D. Optimization of the preparation process for human serum albumin (HSA) nanoparticles. Int. J. Pharm. 2003, 257, 169–180. [Google Scholar] [CrossRef]
- Sadeghi, R.; Moosavi-Movahedi, A.A.; Emam-Jomeh, Z.; Kalbasi, A.; Razavi, S.H.; Karimi, M.; Kokini, J. The effect of different desolvating agents on BSA nanoparticle properties and encapsulation of curcumin. J. Nanoparticle Res. 2014, 16. [Google Scholar] [CrossRef]
- Yu, Z.; Yu, M.; Zhang, Z.; Hong, G.; Xiong, Q. Bovine serum albumin nanoparticles as controlled release carrier for local drug delivery to the inner ear. Nanoscale Res. Lett. 2014, 9, 343. [Google Scholar] [CrossRef]
- Shahabadi, N.; Hadidi, S. Molecular modeling and spectroscopic studies on the interaction of the chiral drug venlafaxine hydrochloride with bovine serum albumin. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2014, 122, 100–106. [Google Scholar] [CrossRef]
- Paul, B.K.; Ray, D.; Guchhait, N. Unraveling the binding interaction and kinetics of a prospective anti-HIV drug with a model transport protein: Results and challenges. Phys. Chem. Chem. Phys. 2013, 15, 1275–1287. [Google Scholar] [CrossRef]
- Paul, B.K.; Ghosh, N.; Mukherjee, S. Interplay of multiple interaction forces: Binding of norfloxacin to human serum albumin. J. Phys. Chem. B 2015, 119, 13093–13102. [Google Scholar] [CrossRef]
- Rajendiran, N.; Thulasidhasan, J. Interaction of sulfanilamide and sulfamethoxazole with bovine serum albumin and adenine: Spectroscopic and molecular docking investigations. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2015, 144, 183–191. [Google Scholar] [CrossRef]
- Tatoulian, M.; Cavalli, F.; Lorang, G.; Amouroux, J.; Arefi-khonsari, F. Polymer Surface Modification: Relevance to Adhesion; VSP BV: Zeist, The Netherlands, 2000; Volume 2. [Google Scholar]
- Dementjev, A.; De Graaf, A.; Van De Sanden, M.; Maslakov, K.; Naumkin, A.; Serov, A. X-Ray photoelectron spectroscopy reference data for identification of the C3N4 phase in carbon–nitrogen films. Diam. Relat. Mater. 2000, 9, 1904–1907. [Google Scholar] [CrossRef]
- Singh, P.; Singh, H.; Castro-Aceituno, V.; Ahn, S.; Kim, Y.-J.; Yang, D.C. Bovine serum albumin as a nanocarrier for the efficient delivery of ginsenoside compound K: Preparation, physicochemical characterizations and in vitro biological studies. RSC Adv. 2017, 7, 15397–15407. [Google Scholar] [CrossRef]
- Cheng, K.; Sun, S.; Gong, X. Preparation, characterization, and antiproliferative activities of biotin-decorated docetaxel-loaded bovine serum albumin nanoparticles. Braz. J. Pharm. Sci. 2018, 54. [Google Scholar] [CrossRef]
- Gonzalez-Angulo, A.M.; Meric-Bernstam, F.; Chawla, S.; Falchook, G.; Hong, D.; Akcakanat, A.; Chen, H.; Naing, A.; Fu, S.; Wheler, J.; et al. Weekly nab-Rapamycin in patients with advanced nonhematologic malignancies: Final results of a phase I trial. Clin. Cancer Res. 2013, 19, 5474–5484. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Values |
|---|---|
| Z-average particle size (nm) | 222.433 ± 1.150 |
| Zeta potential average (mV) | −30.63 ± 1.365 |
| Poly dispersity index (PDI) | 0.133 ± 0.014 |
| Encapsulation efficiency (%) | 48.22 ± 1.948 |
| Drug loading | 4.28 ± 1.94 |
| Hydrogen Bond | Polar | Cation-pi | Hydrophobic | Other |
|---|---|---|---|---|
| TYR147 (−0.6974) | ARG458 (−1.526) | TYR451 (−0.6994) | ALA193 (−0.5145) | LEU189 (−1.6106) |
| SER428 (−0.04736) | HIS145 (−1.1971) | PRO146 (−0.4804) | THR190 (−0.6233) | |
| SER192 (−0.4233) | ARG196 (−1.0906) | ILE455 (−0.4311) | ||
| GLU424 (−0.5991) | ||||
| ASP108 (−0.479) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
K. Rochani, A.; Balasubramanian, S.; Ravindran Girija, A.; Maekawa, T.; Kaushal, G.; Kumar, D.S. Heat Shock Protein 90 (Hsp90)-Inhibitor-Luminespib-Loaded-Protein-Based Nanoformulation for Cancer Therapy. Polymers 2020, 12, 1798. https://doi.org/10.3390/polym12081798
K. Rochani A, Balasubramanian S, Ravindran Girija A, Maekawa T, Kaushal G, Kumar DS. Heat Shock Protein 90 (Hsp90)-Inhibitor-Luminespib-Loaded-Protein-Based Nanoformulation for Cancer Therapy. Polymers. 2020; 12(8):1798. https://doi.org/10.3390/polym12081798
Chicago/Turabian StyleK. Rochani, Ankit, Sivakumar Balasubramanian, Aswathy Ravindran Girija, Toru Maekawa, Gagan Kaushal, and D. Sakthi Kumar. 2020. "Heat Shock Protein 90 (Hsp90)-Inhibitor-Luminespib-Loaded-Protein-Based Nanoformulation for Cancer Therapy" Polymers 12, no. 8: 1798. https://doi.org/10.3390/polym12081798
APA StyleK. Rochani, A., Balasubramanian, S., Ravindran Girija, A., Maekawa, T., Kaushal, G., & Kumar, D. S. (2020). Heat Shock Protein 90 (Hsp90)-Inhibitor-Luminespib-Loaded-Protein-Based Nanoformulation for Cancer Therapy. Polymers, 12(8), 1798. https://doi.org/10.3390/polym12081798