1. Introduction
Gene therapy, which is based on the delivery of nucleic acids to target cells in the patient’s body, is a promising approach for cancer treatment. Currently, viral delivery systems are most commonly used in gene therapy systems. They provide an efficient means of transport of genetic material into the target cells owing to their ability to transfer their genome into the cells of the host. However, viral carriers have several disadvantages, including cytotoxicity, potential carcinogenicity, difficulties in production and storage, as well as immunogenicity-related problems associated with the re-administration of such drugs [
1,
2]. In this regard, non-viral delivery systems, based on cationic polymers, cationic lipids or peptides, and nuclear DNA-binding proteins, such as histones, are promising alternatives. Nuclear proteins have a number of additional advantages, including biocompatibility, biodegradability, ability to deliver the cargo specifically to the nucleus due to intrinsic nuclear localization signal (NLS), and, finally, ease of desired sequence changes.
Positively charged histones bind nonspecifically to DNA and neutralize its negative charge, thus allowing the DNA to enter cells through a negatively charged cell membrane. Subsequently, their NLS facilitates the entry of the DNA into the nucleus [
3]. We have previously confirmed the ability of the human histone H2A to bind to pDNA [
4] and transfect eukaryotic cells [
5].
The efficiency and specificity of DNA delivery into certain cells may be increased using ligands that selectively bind to receptors exposed on the cell surface and induce internalization of these receptors [
6,
7]. In tumors, the cancer cells are usually considered the main target of gene therapy. The first cancer gene therapy drugs approved for clinical use (Gendicine, Oncorine, and Imlygic) were administered using intratumoral injection and were directed at cancer cells. However, it is also possible to use gene delivery targeted at tumor stromal cells, such as cancer-associated fibroblasts (CAFs) [
8]. Stromal cells modified through gene delivery can serve as a long-term source of the antitumor compounds inside the tumor. In addition, the delivery efficiency of stroma-targeted therapy may be higher than that of delivery into cancer cells owing to the stability of stromal cell markers. Moreover, the stroma-targeted carriers would be more universal than the cancer cell-targeted carriers and, therefore, applicable to various types of tumors. At present, the number of studies on the use of CAF-targeted nucleic acid delivery is limited. In a previous study [
9], lipid-coated protamine complexes loaded with pDNA encoding the secreted form of the tumor necrosis factor-related apoptosis-inducing ligand (sTRAIL) were used. Anisamide was conjugated on the surface of the particles as a ligand for the sigma-receptor, which is overexpressed on both CAFs and cancer cells. The study demonstrated the effectiveness of the system in a murine tumor model. As a result of the triple administration of the complexes, more than half of the CAFs expressed the therapeutic gene and produced cytokine, which, in turn, induced apoptosis of neighboring tumor cells, but not the CAFs. Moreover, residual CAFs were reverted to a non-activated state by sTRAIL. Another example of CAF-targeted nucleic acid delivery is a recent work [
10], where the authors used peptide nanoparticles bearing CXCL12-targeted siRNA. These nanoparticles incorporated monoclonal anti-fibroblast activation protein α (FAPα) antibodies and were targeted specifically toward CAFs because they overexpress the FAPα receptor. The authors demonstrated that the absorption of the complexes was FAP-dependent and that tumor angiogenesis, as well as tumor cell invasion and migration, was significantly inhibited in vitro. In addition, these effects led to the suppression of metastasis in vivo. Thus, CAF-targeted gene delivery represents a promising approach to cancer therapy.
The β-type platelet-derived growth factor receptor, PDGFRβ, is one of the well-studied CAF markers [
11]. A wide variety of ligands are known to bind to PDGFRβ, many of which are linear [
12,
13] and cyclic [
14] peptides or antibody mimetic (affibody) molecules [
11]. These ligands have also been used to deliver cytotoxic proteins and radioisotopes to the tumor stroma. However, they have not been used as a part of a gene delivery system. The linear peptide YG2 (YIPLPPPRRPFFK) binds to PDGFRβ-positive cells and accumulates inside them [
13]. We hypothesized that the fusion of histone H2A with YG2 increases the efficiency of DNA delivery into CAFs. To test this hypothesis, we generated histone H2A fused with YG2 and analyzed the effect of the YG2 peptide on the efficiency of histone H2A-mediated DNA delivery into PDGFRβ-positive eukaryotic cells.
2. Materials and Methods
2.1. Materials
Plasmid pET30a(+) was obtained from Novagen (Madison, WI, USA), plasmid pGL3-BV was obtained from Promega (Madison, WI, USA), Escherichia coli DH5α strain was obtained from Invitrogen (Carlsbad, CA, USA), E. coli BL21(DE3) strain was obtained from Novagen. DMEM/F12, RPMI-1640, Fetal Bovine Serum (FBS), Trypan Blue Stain (0.4%), and Lipofectamine® 2000 were obtained from Invitrogen. Antibiotic-Antimycotic (100×), Trypsin-EDTA, and Opti-MEM were obtained from GibcoTM (Carlsbad, CA, USA); 5× passive lysis buffer (PLB) was obtained from Promega. All the oligonucleotide primers were synthesized by Evrogen (Moscow, Russia). All other chemical reagents were obtained commercially as reagent grade products.
2.2. pDNA Production
Plasmid encoding human histone H2A (H2A-pET30) was previously obtained [
5]. The gene encoding histone H2A fused with YG2 using the (Gly)
3Ser linker and flanked by NdeI and HindIII restriction sites was generated with H2A-pET30 using a 2-step PCR and cloned into the pET30a(+) vector, resulting in the H2A-YG2-pET30 plasmid. PCR was performed using Encyclo polymerase (Evrogen) and H2A for and YG1 rev primers in the first step and H2A for and YG2 rev primers in the second step. The primer sequences are presented in
Table 1. The fidelity of the H2A-YG2-encoding sequence was confirmed using Sanger sequencing.
The bicistronic plasmid pCMV-EGFP-P2A-luc2, encoding reporter proteins EGFP (enhanced green fluorescent protein) and firefly luciferase connected through a P2A peptide, was generated previously [
15] based on the
EGFP gene from pEGFP-N1 (Clontech Laboratories Inc. Mountain View, CA, USA), the
luc2 gene from pGL4.10 [luc2] (Promega), and P2A sequence [
16].
All plasmids were transformed into E. coli DH5α strain for further manipulation. Plasmids H2A-pET30 and H2A-YG2-pET30 were extracted from bacterial cells using the Plasmid Miniprep kit (Evrogen) and then transformed into E. coli BL21(DE3) strain for protein synthesis. Plasmid pCMV_EGFP_P2A_luc2 was extracted from 200 mL of overnight culture using the EndoFree Plasmid Maxi kit (QIAgen, Venlo, The Netherlands). The DNA concentration was determined using a NanoDropTM 2000 (Thermo ScientificTM, Wilmington, DE, USA). The pGL3-BV plasmid was obtained from Promega.
2.3. Recombinant Histone Preparation
Bacterial cells containing H2A-pET30 or H2A-YG2-pET30 plasmid were propagated in the LB medium. Protein expression was induced by the addition of IPTG (isopropyl β-d-1-thiogalactopyranoside) to the final concentration of 1 mM. After 3 h of incubation at 37 °C, cells were harvested and lysed by sonication in lysis buffer (50 mM Tris-HCl, pH 7.5, 6 M urea, 1 mM EDTA, 200 mM NaCl, and 1 mM phenylmethanesulfonyl fluoride). The obtained lysate was centrifuged at 4000 rpm for 40 min to pellet the insoluble debris. To purify the clarified lysate from endotoxin, it was treated with HCl (final concentration of 0.25 M) for 1 h at 25 °C, centrifuged at 4000 rpm for 40 min to discard the insoluble pellet, and the pH of the solution was adjusted to 7.5 using 3 M Tris-base. Target proteins were purified in two steps using ion-exchange chromatography (SP Sepharose High-Performance resin (GE Healthcare, Danderyd, Sweden), 0.2–1 M NaCl gradient elution), followed by reversed-phase HPLC (Supelco BIO Wide Pore C18 column, Sigma-Aldrich, St. Louis, MO, USA, 5–80% acetonitrile gradient). H2A or H2A-YG2-containing fractions from the second chromatography were combined and dehydrated using a SpeedVac Concentrator (Savant Instruments Inc., Farmingdale, NY, USA). Subsequently, the proteins were dissolved in PBS buffer, and the protein concentration was quantified using the Protein Assay Dye Reagent Concentrate (Bio-Rad, Hercules, CA, USA) according to the manufacturer’s protocol. Protein purity was estimated electrophoretically.
2.4. Binding of H2A-YG2 to pDNA
H2A and H2A-YG2 were complexed with 1 μg of pGL3-BV plasmid at N/P (nitrogen to phosphate; charge) ratios of 0.5:1, 1:1, 2:1, 3:1, and 4:1 in PBS. The N/P ratios were calculated based on the number of arginine and lysine residues in the protein sequence and the number of phosphate groups in the DNA. The resulting complexes were incubated at 25 °C for 30 min, and then 1/5 of the resulting mixes were subjected to electrophoresis on a 0.8% agarose gel (1 × TAE). Next, the pDNA mobility was visualized using ethidium bromide staining.
2.5. Measurement of Size and Zeta Potential for Histone:pDNA Complexes
The complexes of histones H2A and H2A-YG2 with 20 μg of pCMV_EGFP_P2A_luc2 plasmid at N/P ratios of 2.5:1, 5:1, 10:1, and 15:1 were prepared in PBS (pH 7.4). Particle sizes of these complexes were measured by dynamic light scattering using a Brookhaven 90plus particle size analyzer (Brookhaven Instruments, Holtsville, NY, USA). Measurements were performed in a plastic cuvette at 25 °C in ten runs of 20 s duration each and analyzed in MSD (multimodal size distribution) analysis using 90Plus Particle sizing software. Zeta potential of the particles was measured by electrophoretic light scattering using a Brookhaven 90plus particle size analyzer with the BI-PALS module (Brookhaven Instruments, Holtsville, NY, USA) in the same conditions and calculated using PALS Zeta Potential Analyzer software (Ver, 5.78, Brookhaven Instruments, Holtsville, NY, USA).
2.6. Cell Culture
293T (human embryonic kidney transformed with SV40 T antigen, ATCC® CRL-3216™), A375 (human melanoma, ATCC® CRL-1619™), CT 26 (mouse colon carcinoma, ATCC® CRL-2638™), and NIH/3T3 (mouse embryonic fibroblasts, ATCC®, CRL-1658™) cell lines were obtained from ATCC® (Manassas, VA, USA).
293T, A375, and NIH/3T3 cell lines were cultured in DMEM/F12 medium (1:1) supplemented with 10% FBS and antibiotic-antimycotic solution in a 5% CO2 incubator at 37 °C under 95% humidity.
The CT 26 cell line was cultured in RPMI-1640 medium supplemented with 12.5% FBS and antibiotic-antimycotic solution in a 5% CO2 incubator at 37 °C under 95% humidity.
All the cell lines were subcultured every 3–4 days using trypsin-EDTA. For mRNA content characterization and transfection experiments, cells were collected with trypsin-EDTA, stained with trypan blue stain (Invitrogen), and counted using the Countess II FL Automated Cell Counter (Invitrogen).
2.7. Analysis of PDGFRB mRNA Expression in Cell Lines Using RT-PCR
A total of 1 × 106 cells were collected using trypsin-EDTA, homogenized using QIAshredder (QIAgen, Venlo, The Netherlands), and the total RNA was isolated using an RNeasy Plus Mini kit (QIAgen). The quality of the isolated RNA was evaluated by electrophoresis on a 1% agarose gel containing ethidium bromide. The RNA was reverse-transcribed with random decamer primers and Mint cDNA Synthesis kit (Evrogen). Real-time PCR was performed using qPCR mix-HS SYBR (Evrogen) according to the manufacturer’s instructions with a final primer concentration of 300 nM and 10 ng of the matrix. Primers Psmb2 for, Psmb2 rev, Psmb7 for, Psmb7 rev, Pdgfrb for, and Pdgfrb rev were used for murine cell lines, while primers HPRT for, HPRT rev, 18S for, 18S rev, PDGFRB for, and PDGFRB rev were used for human cell lines. PCR was performed using the LightCycler® 96 real-time PCR cycler (Roche, Basel, Switzerland). All reactions were performed in triplicate according to the following protocol: heating at 95 °C for 5 min, followed by 45 cycles of denaturation at 95 °C for 10 s, annealing at 60 °C for 10 s, and extension at 72 °C for 10 s. Gene expression was quantified in N0 units using LinRegPCR software (Heart Failure Research Center, Amsterdam, The Netherlands), and the relative expression of PDGFRB (Pdgfrb) was calculated by dividing N0 for PDGFRB (Pdgfrb) by the geometric mean of N0 of housekeeping genes proteasome subunit beta type-2 and 7 (Psmb2 and Psmb7), respectively, for murine cell lines and hypoxanthine-guanine phosphoribosyltransferase (HPRT) and 18S ribosomal RNA (18S rRNA) for human cell lines.
2.8. Transfection and Internalization Studies
To compare the internalization and transfection efficiency of H2A-YG2 and H2A, NIH/3T3 and 293T cell lines were transfected with the different carriers. The pCMV-EGFP-P2A-luc2 plasmid encoding EGFP and firefly luciferase was used for these experiments.
The transfection efficiency was analyzed using luciferase activity assay and flow cytometry. To evaluate luciferase activity, NIH/3T3 (105 cells/well) and 293T (1.5 × 105 cells/well) cells were plated in 24-well plates in growth medium and incubated in a 5% CO2 incubator at 37 °C for 24 h. The next day, when the cells reached 40–60% confluence, the transfection complexes containing H2A or H2A-YG2 in various protein to pDNA N/P charge ratios (2.5, 5, 10, and 15) were prepared in PBS (1 μg of pDNA/well; final volume of 125 μL for two wells) and incubated at 25 °C for 30 min. Transfection with Lipofectamine® 2000 was used as a positive control, while transfection with pDNA alone was used as a negative control, and all transfections were performed in duplicate. After incubation, the volume of transfection mixes was adjusted to 365 μL with serum-free medium (Opti-MEM), and the growth medium in the wells was changed to 180 μL of transfection mix. The cells were incubated with the transfection mix for 3 h in the 5% CO2 incubator at 37 °C, and then 0.5 mL of complete medium was added to each well. After transfection, the cells were incubated in the 5% CO2 incubator at 37 °C for 24 h, the medium in the wells was changed to fresh medium, and cells were further incubated for 24 h. After that, cells were washed with 0.5 mL of PBS and then lysed using 100 μL of 1× PLB/well. Prior to the measurement of luciferase activity, the lysates were centrifuged to pellet, and the insoluble debris was removed. The transfection experiments were performed in triplicate to quantify the luciferase activity.
To estimate EGFP expression using fluorescence microscopy and flow cytometry, transfections with histones in N/P 10, Lipofectamine® 2000, and pDNA alone were performed, as described above. Two days after the transfection, cells were imaged using a ZOE™ Fluorescent Cell Imager (Bio-Rad) in a bright field and GFP-fluorescence channel. The images obtained from ZOETM were then processed and combined. After imaging, the cells were washed with 0.5 mL of PBS, trypsinized using 150 μL of Trypsin-EDTA, and resuspended in 350 μL of FACS buffer (0.1% FBS and 2 mM EDTA in PBS) for flow cytometric analysis.
Plasmid DNA internalization was also analyzed using flow cytometry. Toward this end, pCMV-EGFP-P2A-luc2 was labeled with tetramethyl(TM)-rhodamine using Label IT® Nucleic Acid Labeling kit (Mirus Bio LLC, Madison, WI, USA). NIH/3T3 (1.5 × 105/well) and 293T (2 × 105/well) cells were plated in 24-well plates in growth medium and incubated in the 5% CO2 incubator at 37 °C for 24 h. The next day, when the cells reached 60–80% confluence, transfections with histones in N/P 10, Lipofectamine® 2000, pDNA alone, and Opti-MEM (serum-free medium) were performed using TM-rhodamine-labeled pDNA. All the transfections were performed in duplicate. The cells were incubated with the transfection mix for 3 h in the 5% CO2 incubator at 37 °C or for 1 h at 4 °C. After transfection, the plates were placed on ice, and the cells were washed with 0.5 mL of ice-cold heparin solution (1 mg/mL in PBS), followed by a wash in 0.5 mL of ice-cold PBS. Then, the cells were trypsinized using 150 μL of Trypsin-EDTA, resuspended in 350 μL of FACS buffer (0.1% FBS and 2 mM EDTA in PBS), and precipitated by centrifugation at 1000 rpm for 5 min. The cell pellet was washed with ice-cold PBS and resuspended in 0.5 mL of FACS buffer for flow cytometric analysis.
2.9. Luciferase Activity Assay
The transfection efficiency was analyzed using a Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer’s protocol with a GENios Pro luminometer (Tecan, Mannedorf, Switzerland). Luciferase activity was measured in 10 μL of the cell lysates and normalized to the total amount of protein in the lysate. The amount of protein was quantified using the Protein Assay Dye Reagent Concentrate (Bio-Rad) according to the manufacturer’s protocol.
2.10. Flow Cytometry
To evaluate the presence of PDGFRβ on the surface of the cells, CT 26 and NIH/3T3 cell lines were stained with anti-PDGFRβ antibodies conjugated with phycoerythrin (PE) (ab93534, Abcam, Cambridge, MA, USA) and then analyzed by flow cytometry. The cells were washed with PBS, detached with PBS containing 2 mM EDTA, washed with PBS, and stained with antibodies at a 1:150 dilution in FACS buffer (0.1% FBS and 2 mM EDTA in PBS) for 30 min on ice. The cells were then washed with FACS buffer three times and used for flow cytometric analysis. Cell suspensions were analyzed using BD FACSAria™ III (BD Biosciences, Franklin Lakes, NJ, USA). Twenty thousand events were collected for each sample. The acquired data were analyzed using the Flowing Software 2.5.1. (Mr. Perttu Terho, Turku Centre for Biotechnology, Turku, Finland). The debris and dead cells were excluded based on FSC (forward scatter) and SSC (side scatter) coordinates. Histograms for unstained and stained cells in PE (λexcitation = 561 nm, detection at 582/15 nm) channel were acquired.
To analyze the EGFP expression, the cell suspensions obtained after transfections were analyzed using FACScanTM (Becton Dickinson Biosciences, San Jose, CA, USA). Ten thousand events were collected for each sample. The acquired data were analyzed using the Flowing Software 2.5.1. The debris and dead cells were excluded based on FSC and SSC coordinates. Next, based on the dot-plot in FL1 (detection at 530/30 nm)-FL2 (detection at 585/42 nm) coordinates for pDNA-transfected cells, EGFP-positive gate was established (λexcitation = 488 nm). The proportion of EGFP-positive cells and overall EGFP fluorescence (proportion multiplied by mean fluorescence intensity) were analyzed for each transfection point.
Cell suspensions obtained for internalization studies were analyzed using BD FACSAria™ III (BD Biosciences). Twenty thousand events were collected for each sample. The acquired data were analyzed using the Flowing Software 2.5.1. The debris and dead cells were excluded based on FSC and SSC coordinates. Next, based on dot-plot in TM-rhodamine (λexcitation = 561 nm, detection at 582/15 nm) and FSC coordinates for Opti-MEM, TM-rhodamine-positive gate was established. The proportion of TM-rhodamine-positive cells and overall TM-rhodamine fluorescence (proportion multiplied by mean fluorescence intensity) were analyzed for each transfection point.
2.11. Statistical Analysis
To estimate the difference between the different pairs of samples, Student’s t-test (two-sample assuming unequal variances) was used where applicable. Differences were considered statistically significant at p (one-tail) < 0.05.
4. Discussion
The efficiency of delivery into eukaryotic cells remains a major challenge when using cancer gene-therapeutic drugs in medicine [
23]. Viral vectors demonstrate the high efficiency of the delivery of genetic drugs into cells, but they are characterized by low packaging capacity, relatively high production costs, immunogenicity, and toxicity that can provoke inflammation [
24]. Although the first gene therapies in the market were based on viral carriers (Gendicine, Oncorine, Imlygic), the problems mentioned above seem to limit their wider application. Extensive efforts for the development of non-viral carriers for gene delivery are ongoing. Synthetic polycationic compounds, lipids, and peptides are now being increasingly used as alternative carriers. Non-viral vectors are advantageous because of their low immunogenicity (which makes them potentially safer for clinical use), practically unlimited packaging capacity, simplicity, and low-cost production [
25]. It may be safer to use natural polycationic proteins as well as histones because of their natural DNA-binding function and formation of strong complexes with DNA molecules in the cell. Moreover, histones contain intrinsic NLS [
26], which may facilitate the entry of therapeutic DNA-histone complexes into the nucleus. We previously demonstrated the ability of histone H2A to transfect different eukaryotic cells [
5], which corresponded well with the results of Balicki and Beutler [
27].
A common disadvantage of non-viral carriers is the low gene transfer efficiency [
3]. A practical solution to this problem involves the incorporation of a domain capable of binding to the surface of cells, thus enhancing the internalization of gene-therapeutic drugs into cells [
28]. We previously [
5] showed that the linking of histone H2A with HIV TAT-peptide enhanced the transfection efficiency of histone, likely due to the high positive charge of the peptide, allowing efficient binding to negatively charged cell membranes and subsequent internalization of the complexes. TAT-peptide was also used to enhance the efficiency of polyethyleneimine (PEI)-polyethylene glycol (PEG) copolymer and successfully applied for DNA delivery during suicide gene therapy in a murine tumor model [
29]. Ligands of cell surface receptors that induce their internalization are also frequently used as non-viral carriers. These ligands can also target DNA delivery toward certain types of cancer cells [
7,
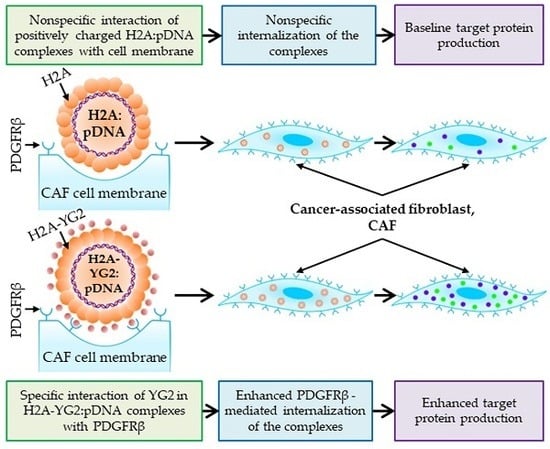
30]. In this work, we constructed a non-viral carrier that targets the cells in the tumor microenvironment, more specifically, the CAFs. To achieve this, we spliced histone H2A with the peptide ligand, YG2, capable of binding to PDGFRβ, a surface marker on CAFs.
Using this approach, we generated a new recombinant protein H2A-YG2 and demonstrated that this histone H2A modification noticeably increased the transfection efficiency of PDGFRβ-positive murine fibroblasts NIH/3T3, but not that of PDGFRβ-negative human embryonic kidney 293T cells. To better understand the mechanism underlying the enhanced transfection efficiency, we studied the internalization of DNA:histone complexes in both the cell lines. The presence of the peptide increased the amount of DNA internalized by NIH/3T3 cells, but not by 293T cells. The observed internalization was energy-dependent because it was significantly inhibited at low temperatures (+4 °C). These results suggest that the H2A-YG2 in the complexes binds to PDGFRβ on the cell surface through the YG2 peptide and thereby induces receptor-mediated endocytosis of the complexes, resulting in greater transfection efficiency compared to histone H2A alone.
The CAF-targeted non-viral DNA carrier developed in this study remains to be tested in vivo. Administration of H2A-YG2:DNA complexes into a tumor, consisting of different types of cells, will provide a better assessment of the stroma specificity of the carrier. The transfection efficiency of the carrier was relatively low compared to that of Lipofectamine
® 2000, despite similar internalization efficiency. These results indicate that the histone complexes may have a lower endosomal escape rate. The endosomal escape could potentially be increased using endosomolytic agents, such as Ca
2+ [
31] and chloroquine [
32], or through endosomolytic domains attached to the carrier [
33]. The successful application of this approach has been previously demonstrated using other non-viral histone-based carriers [
31,
32,
33].

 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}