The Impact of Vitrimers on the Industry of the Future: Chemistry, Properties and Sustainable Forward-Looking Applications



Abstract
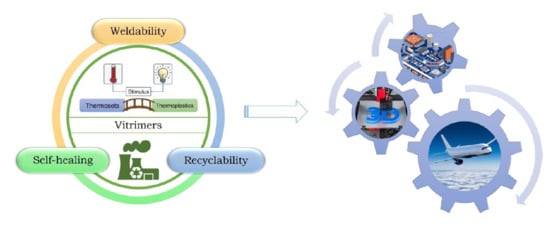
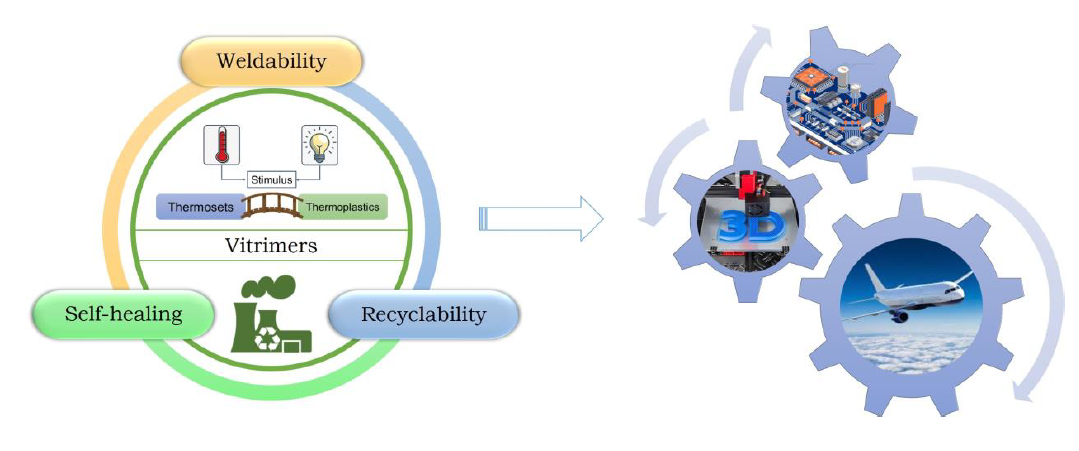{kind=link}
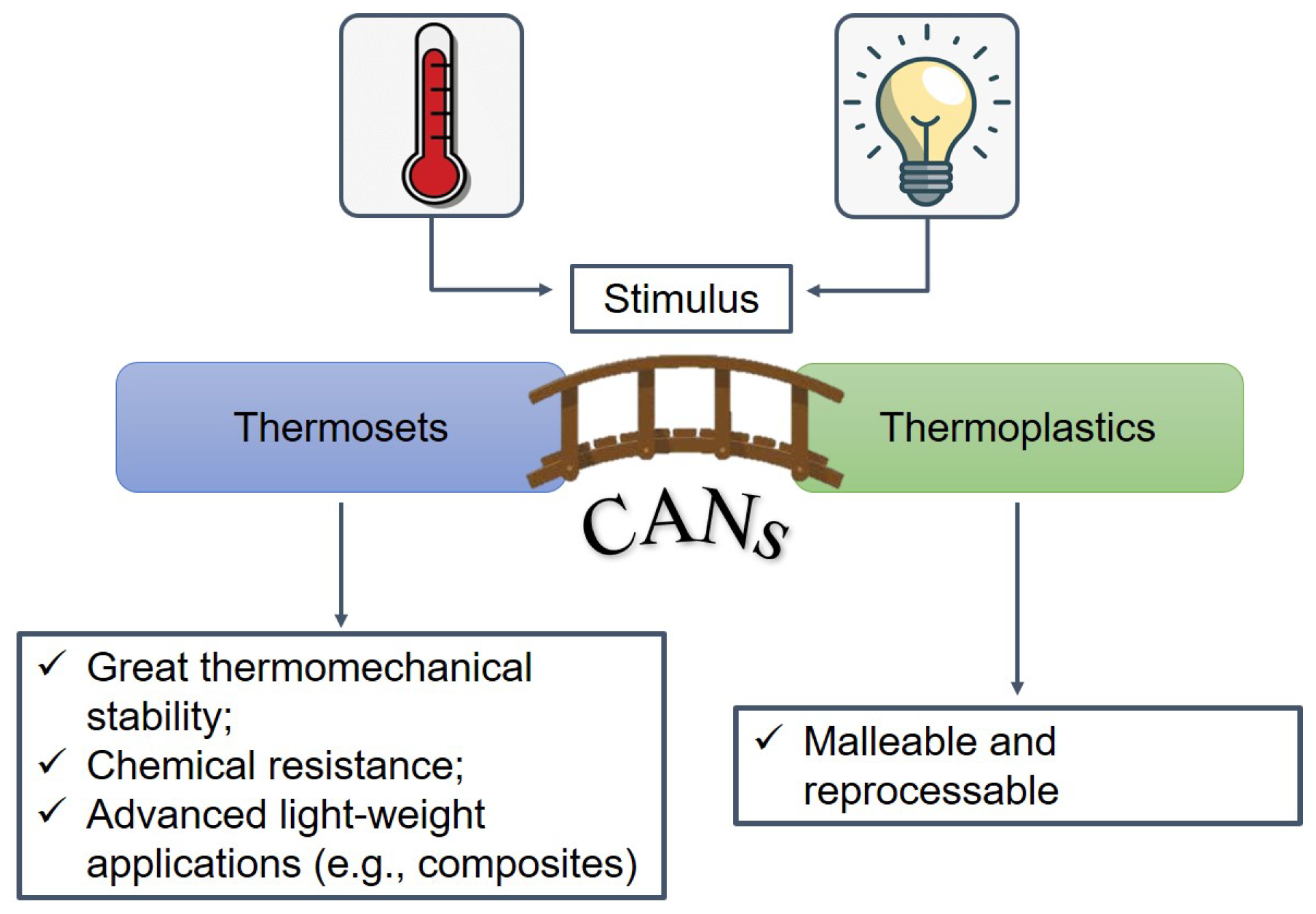{kind=link}
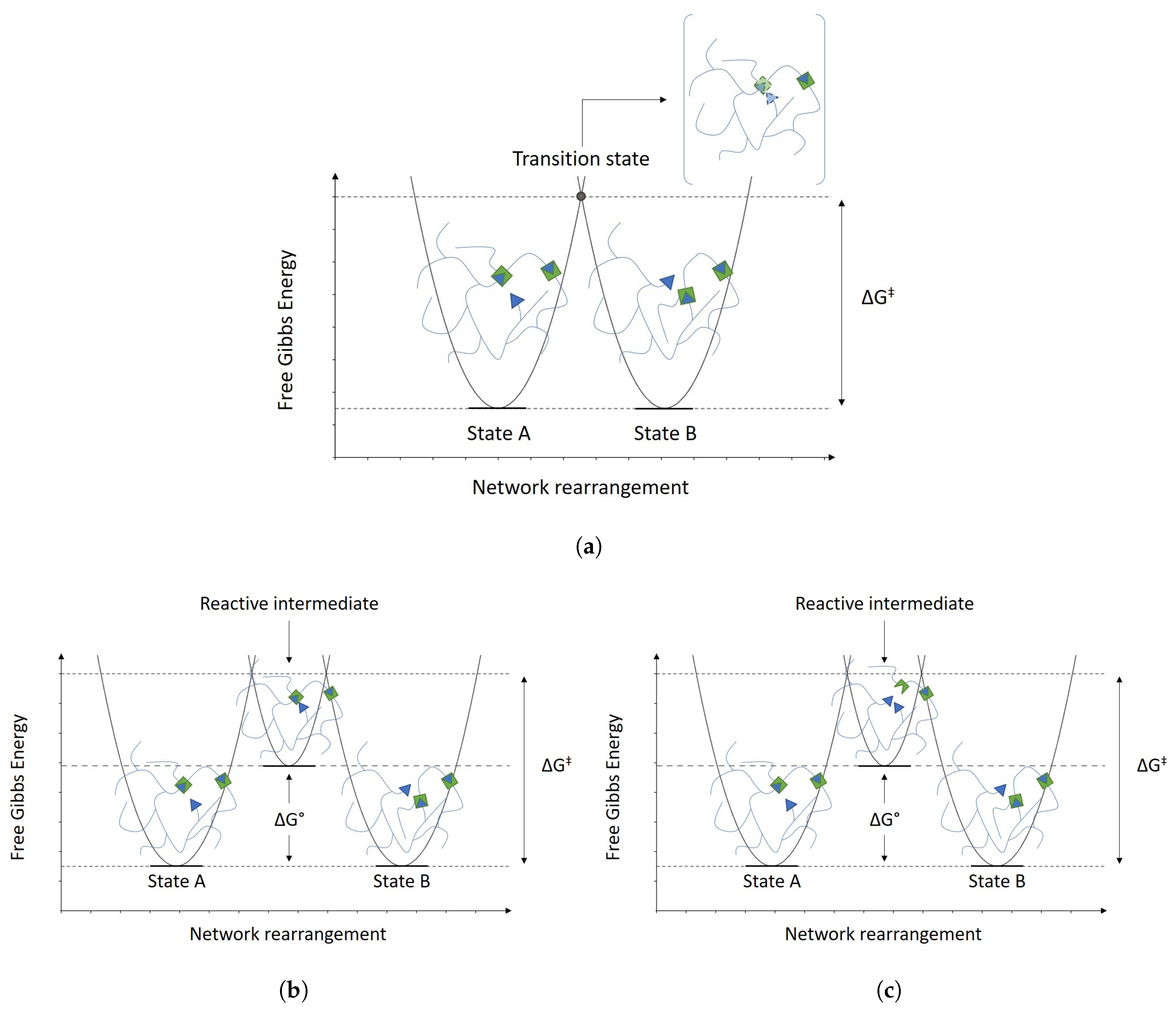{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
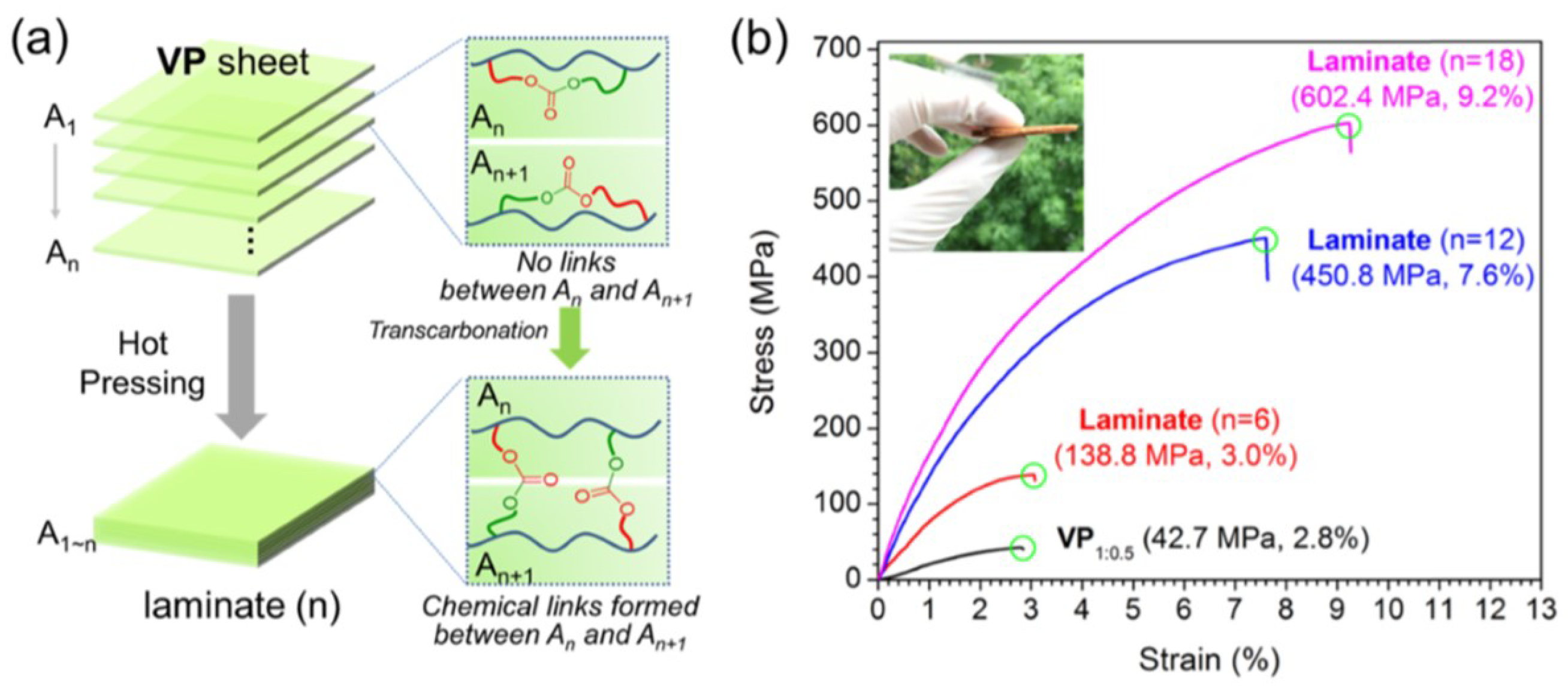{kind=link}
{kind=link}
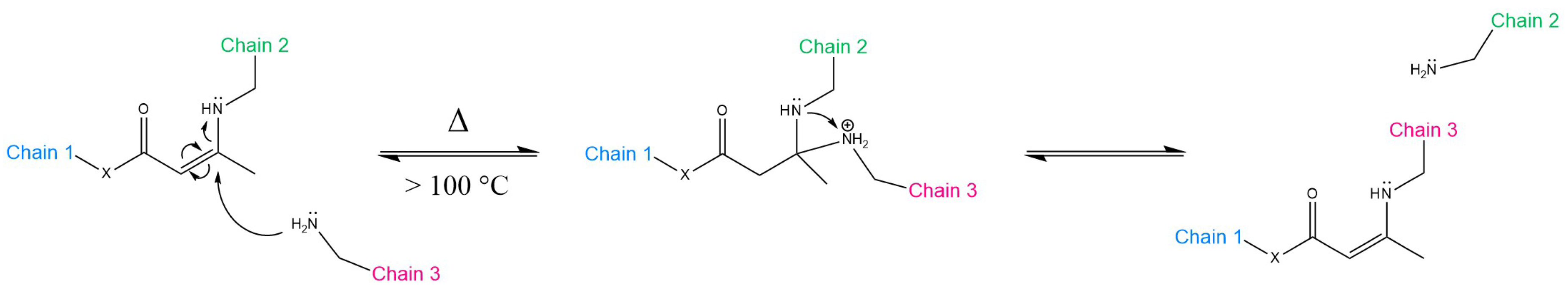{kind=link}
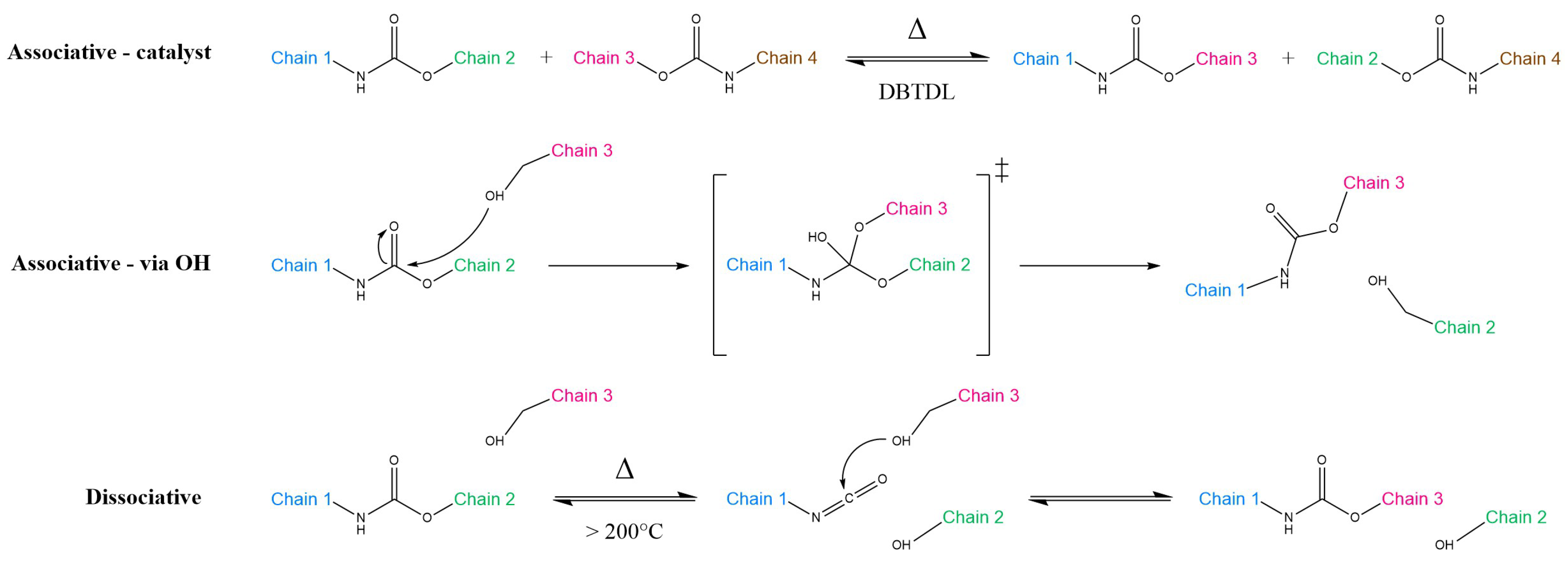{kind=link}
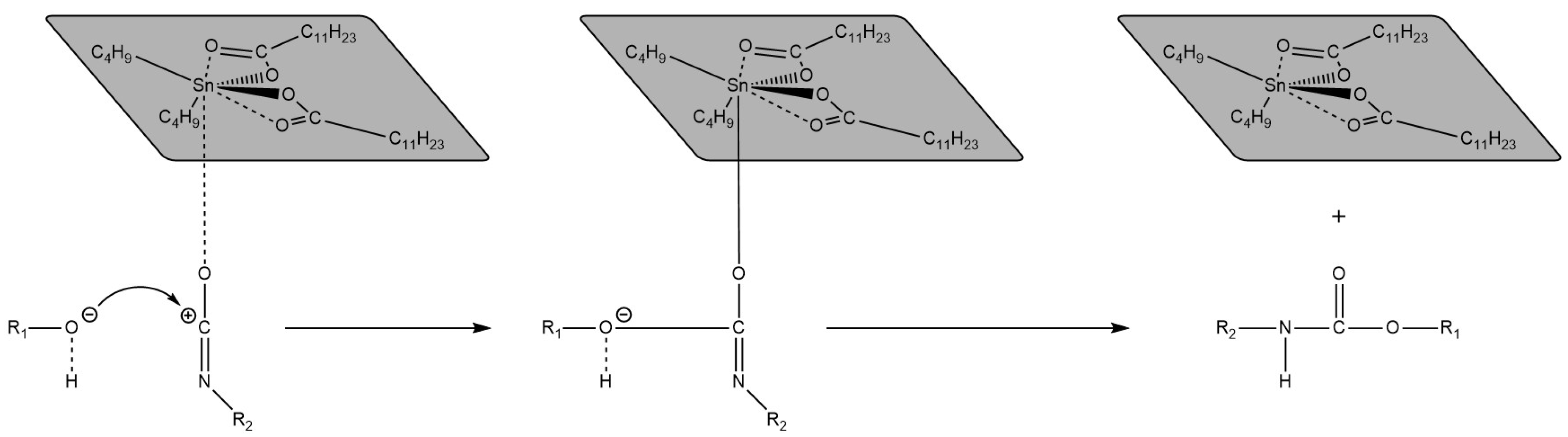{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Theoretical Background of Dynamic Covalent Networks and Vitrimers
2.1. Associative and Dissociative Covalent Adaptable Networks
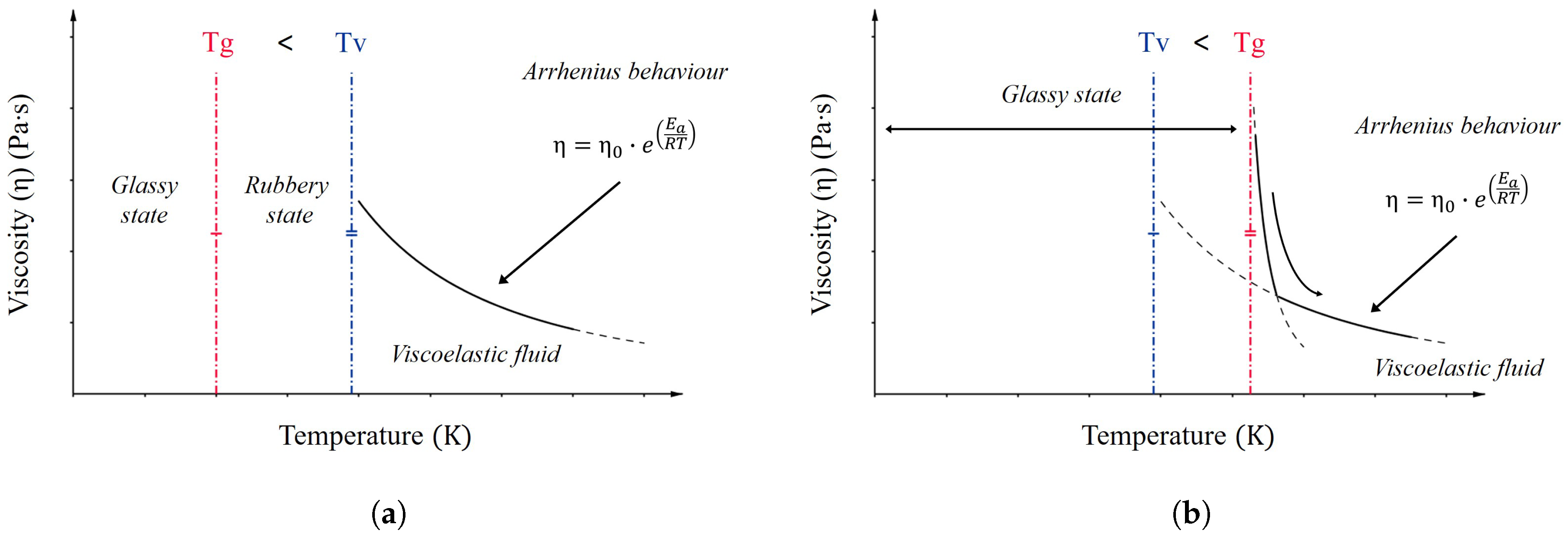
2.2. Vitrimers
- Organic network of covalently bound chains;
- Thermally-induced topology rearrangement via associative exchange reactions (dynamic covalent network);
- Above Tv, viscosity follows the Arrhenius law and decreases as a consequence of the dominant kinetics of chemical exchanges.
- Insolubility and cross-link density kept constant at all temperatures up to degradation.
3. Chemistry of Associative CANs and Vitrimers
3.1. Transesterification
3.2. Transamination of Vinylogous Acyls
3.3. Transcarbamoylation
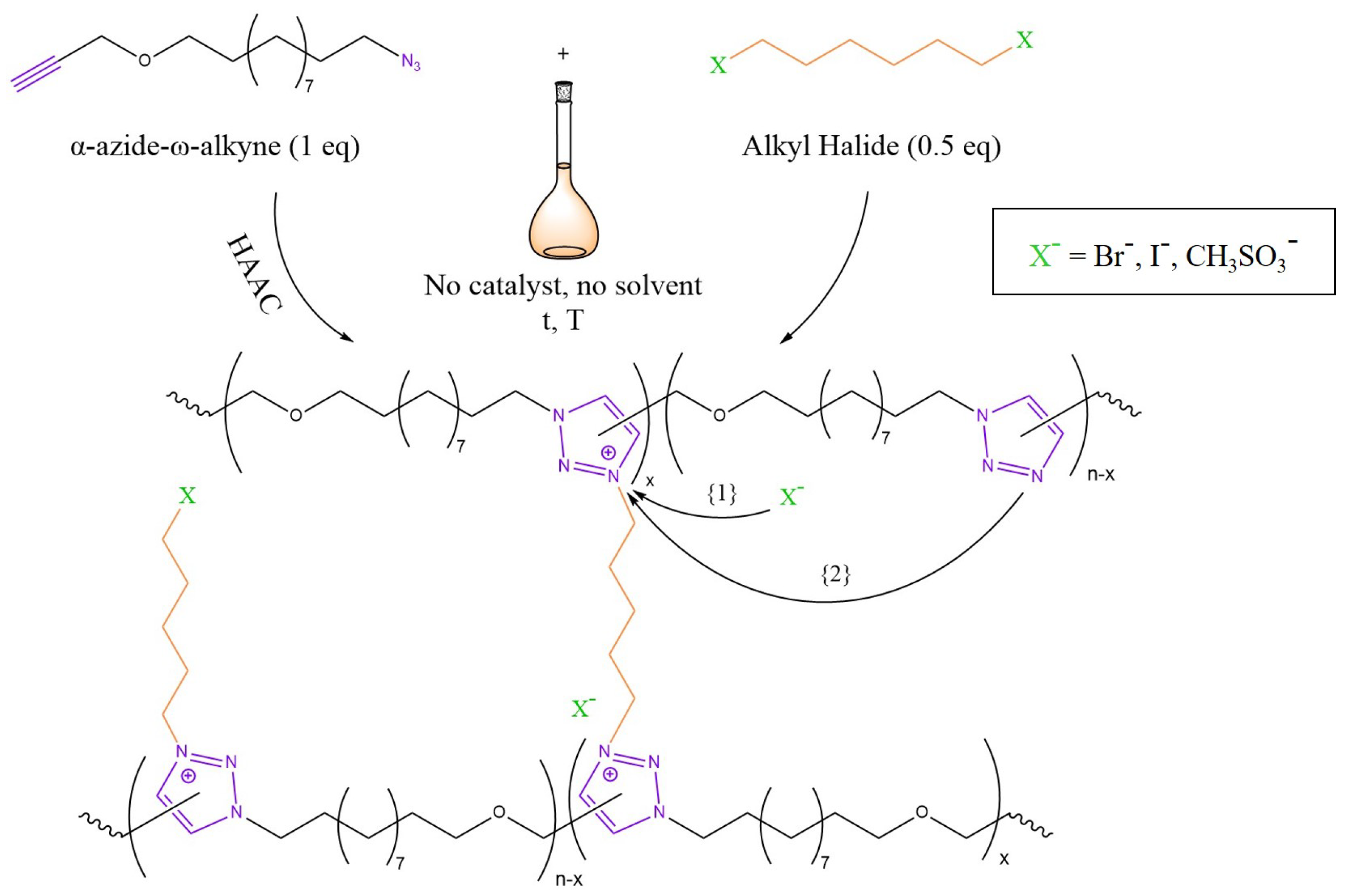
3.4. Transalkylation
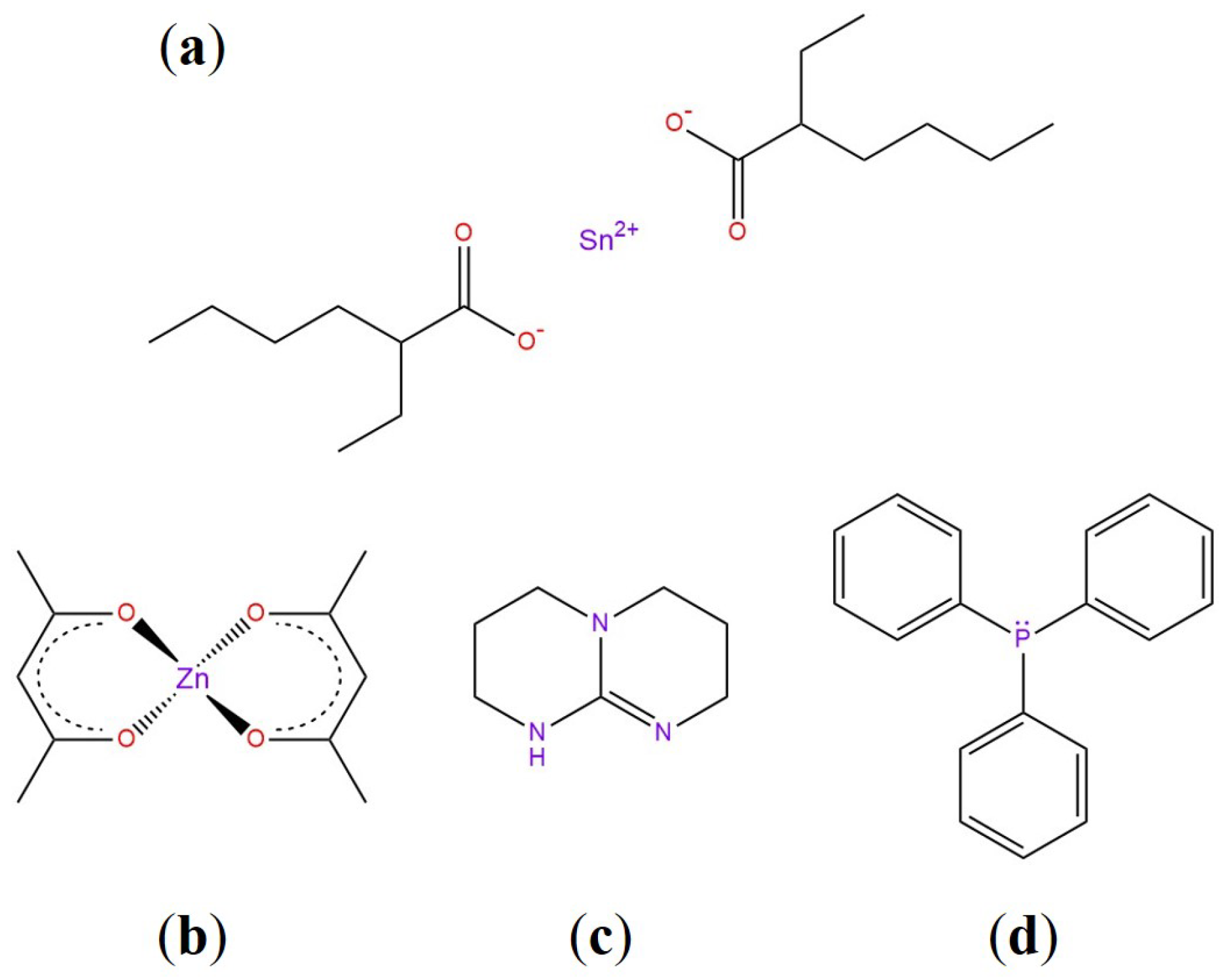
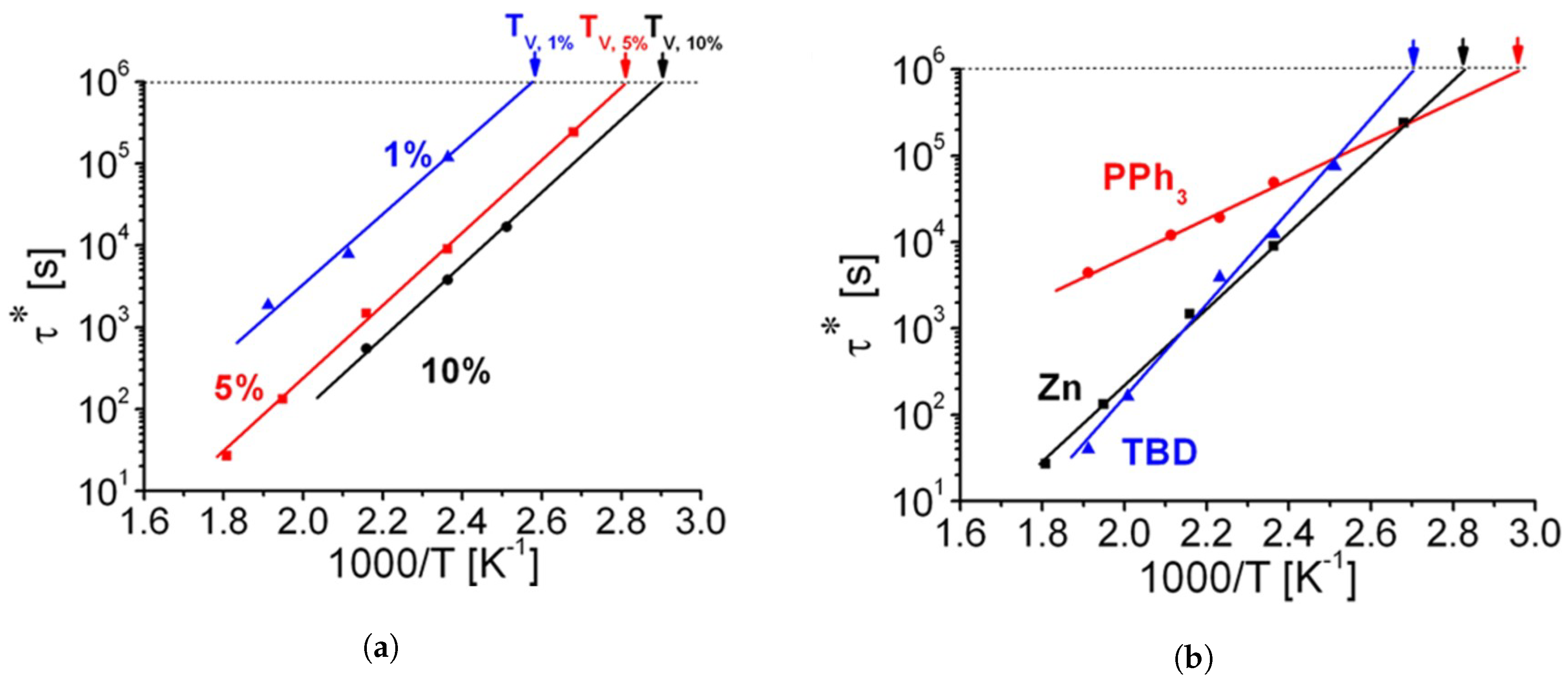
3.5. Common Catalysts and the Role Thereof
4. Unique Properties of CANs
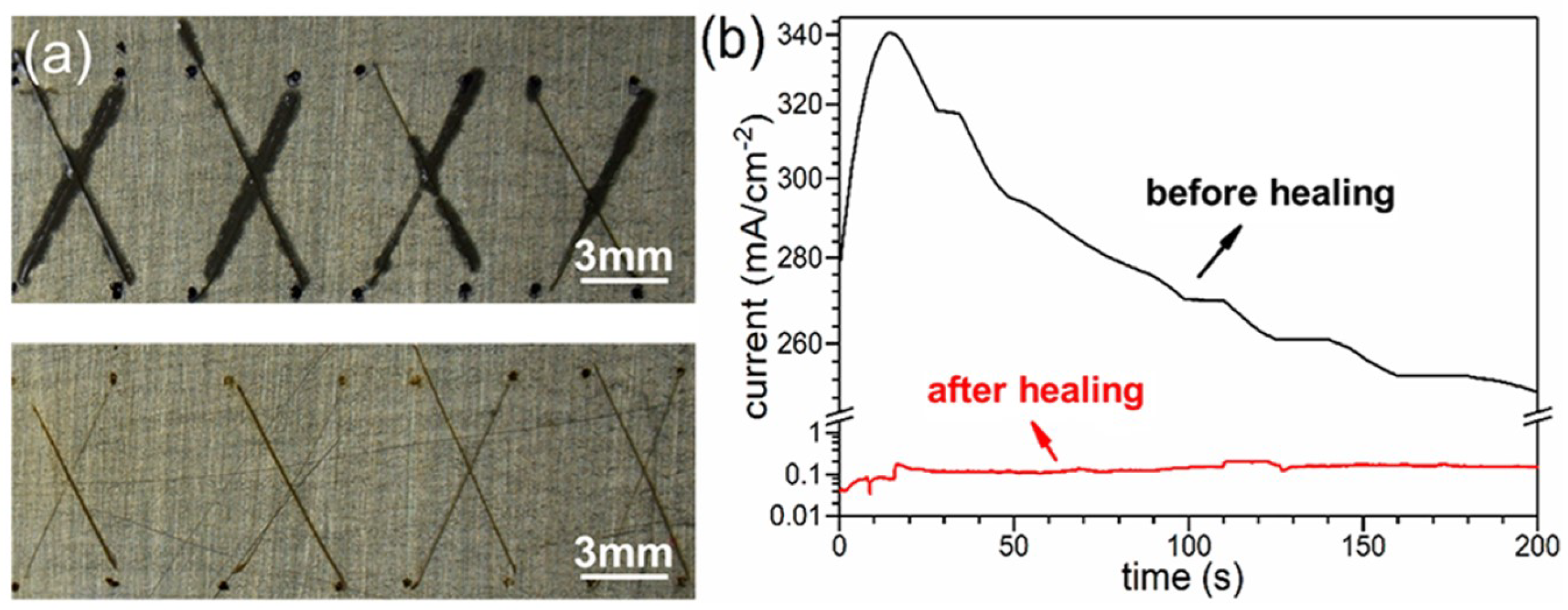
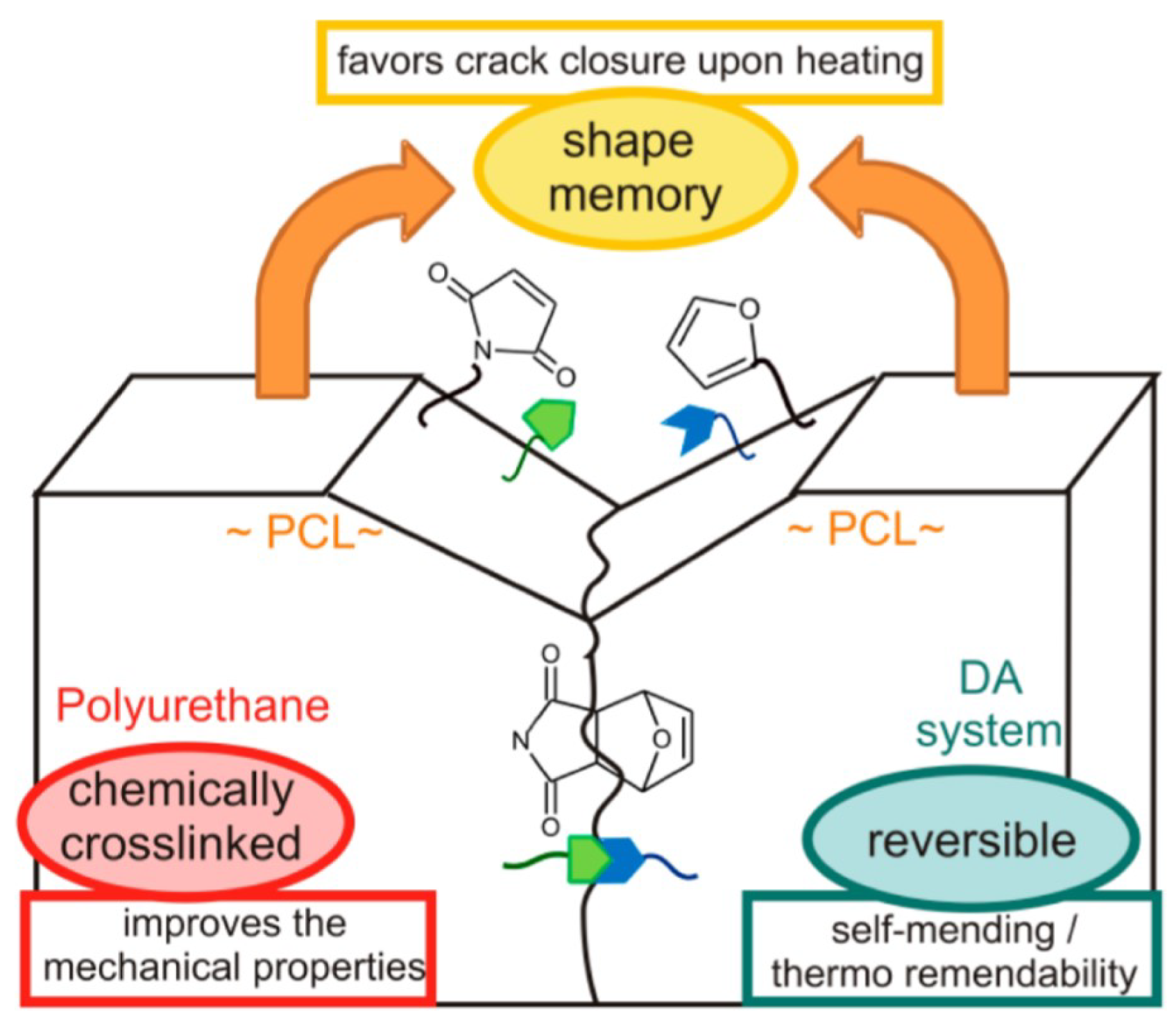
4.1. Self-Healing
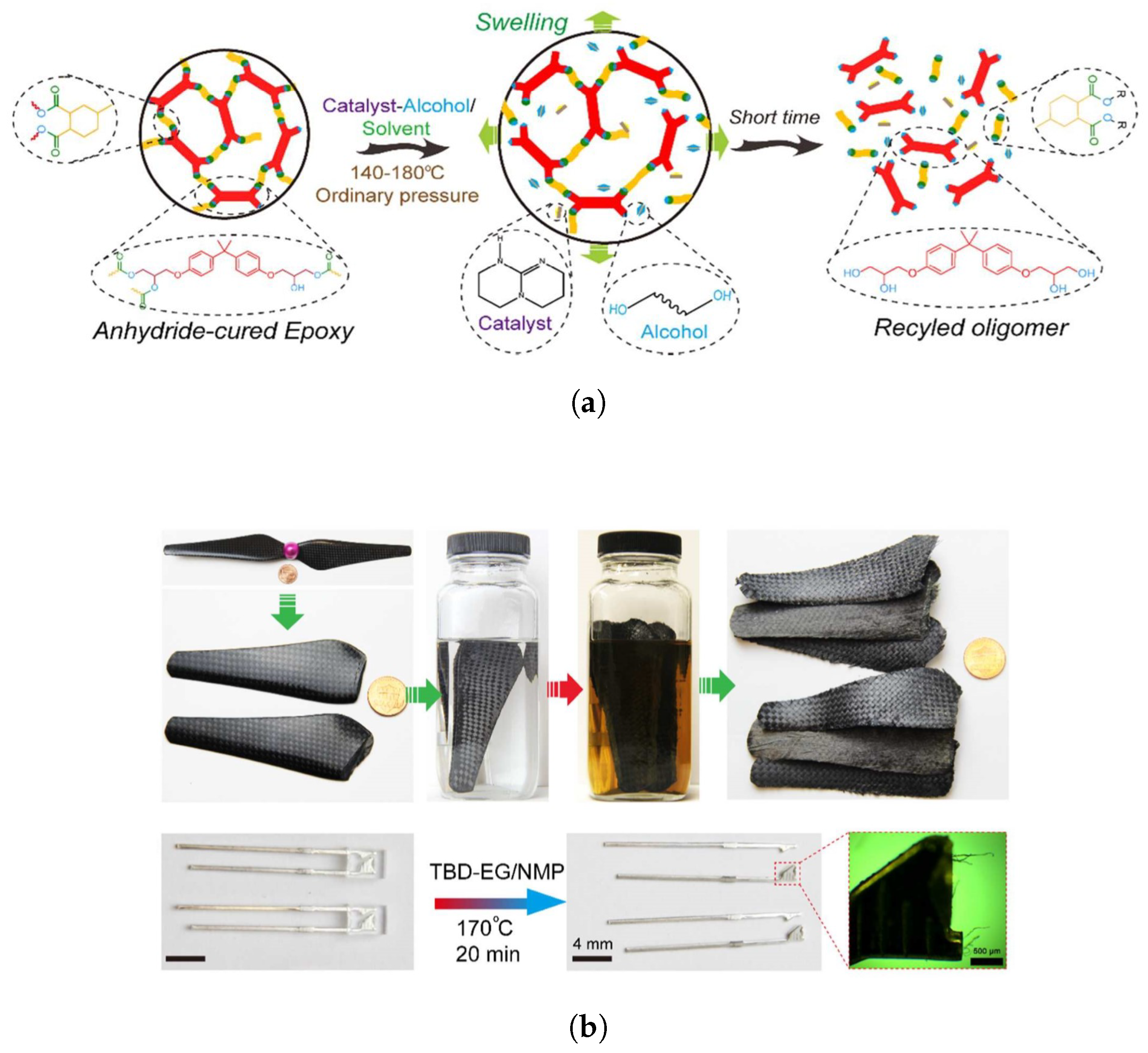
4.2. Recyclability
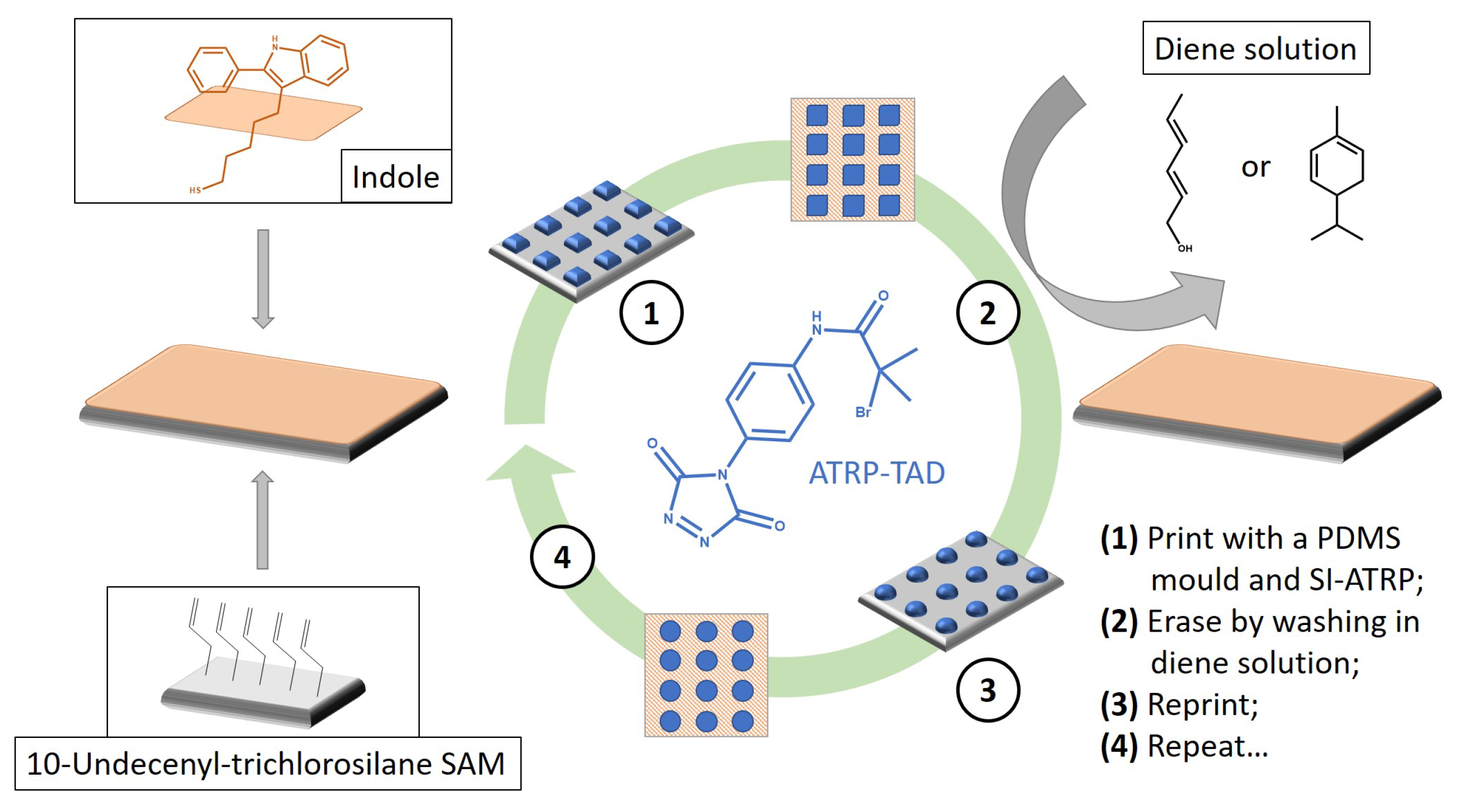
4.3. Weldability
5. State-of-the-Art of Industrially-Relevant Applications
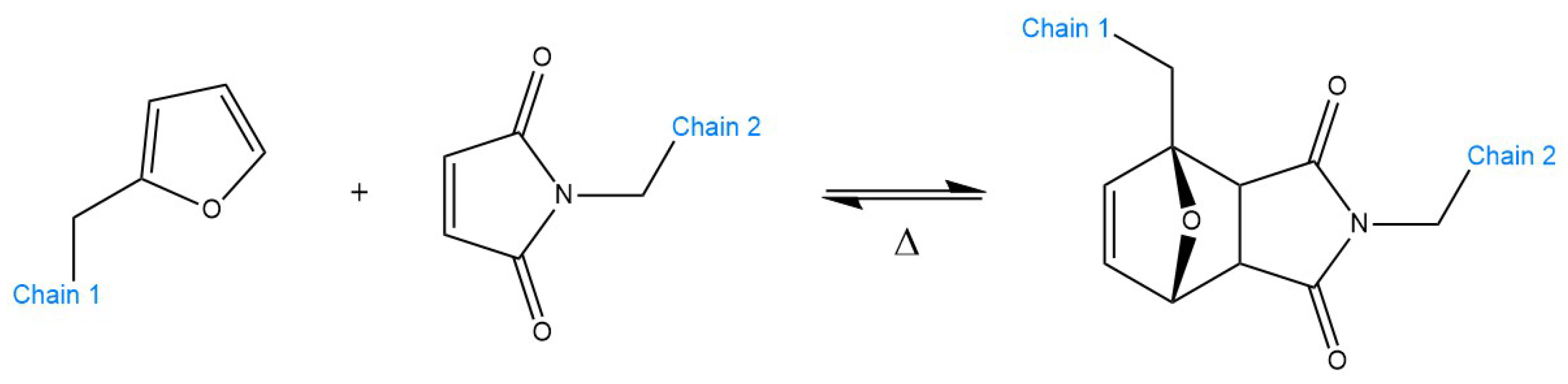
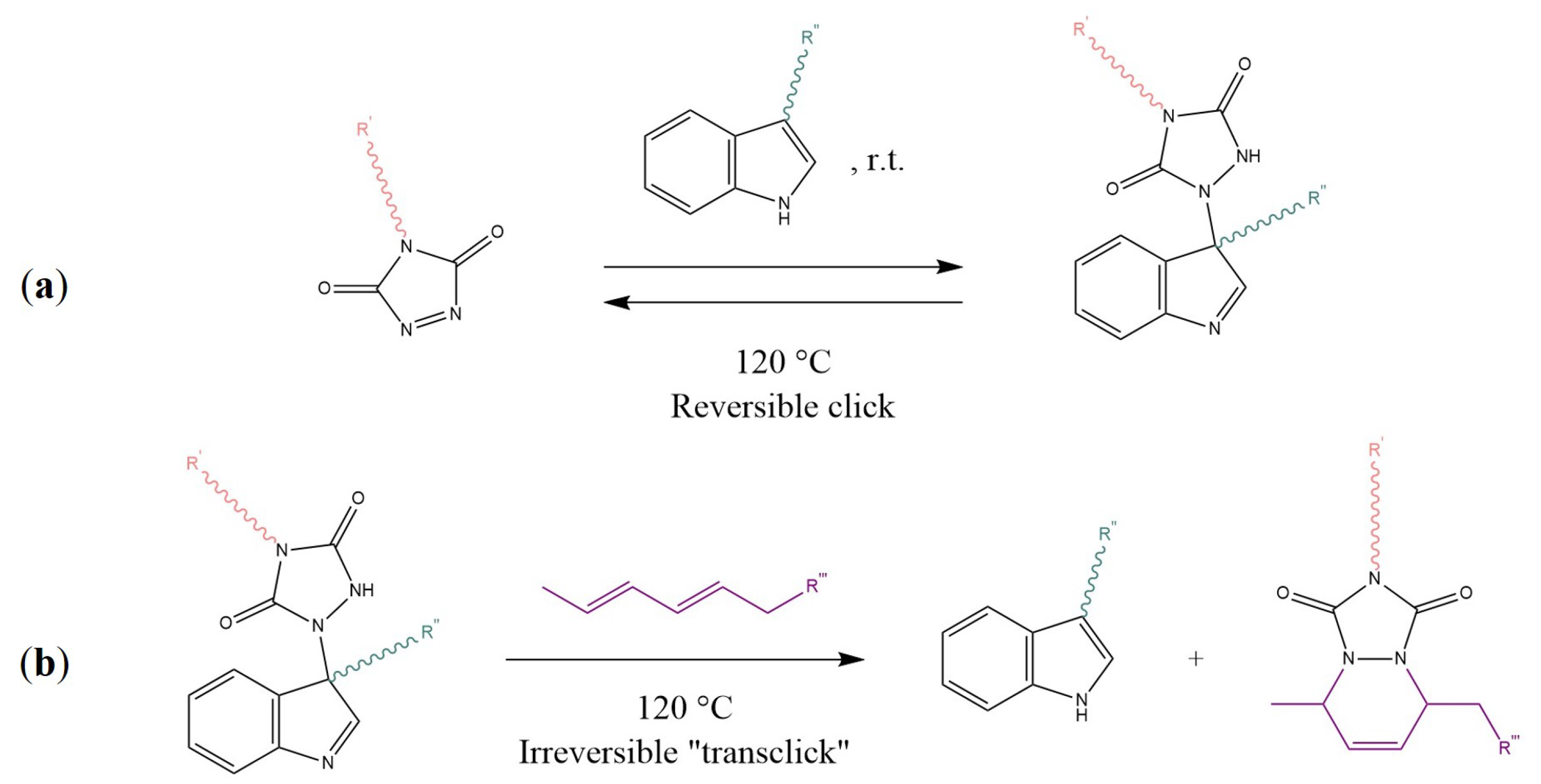
5.1. Examples and Applications of dissociative CANs—Diels–Alder Reaction
5.2. Applications of Associative CANs and Vitrimers in the Industrial World
5.2.1. Sustainable Vitrimers
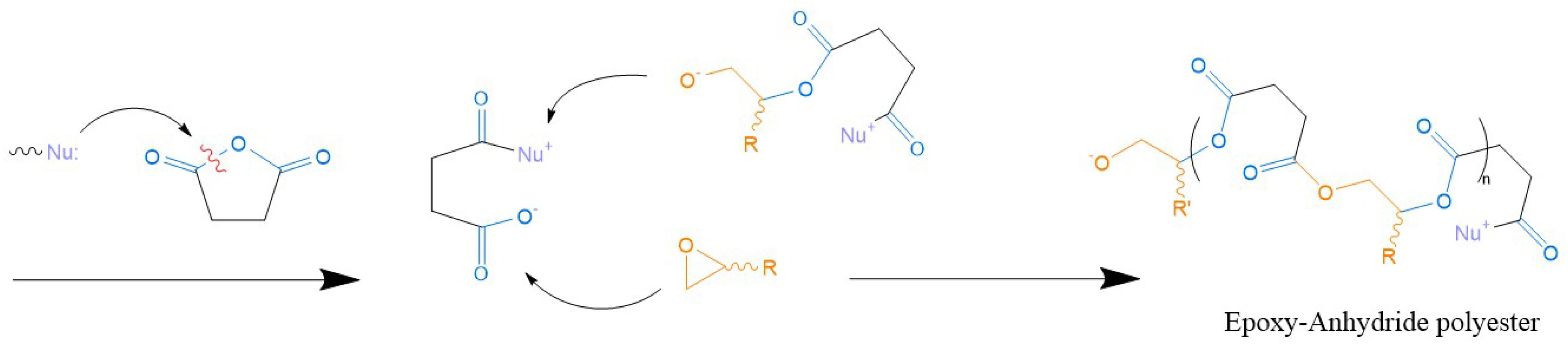
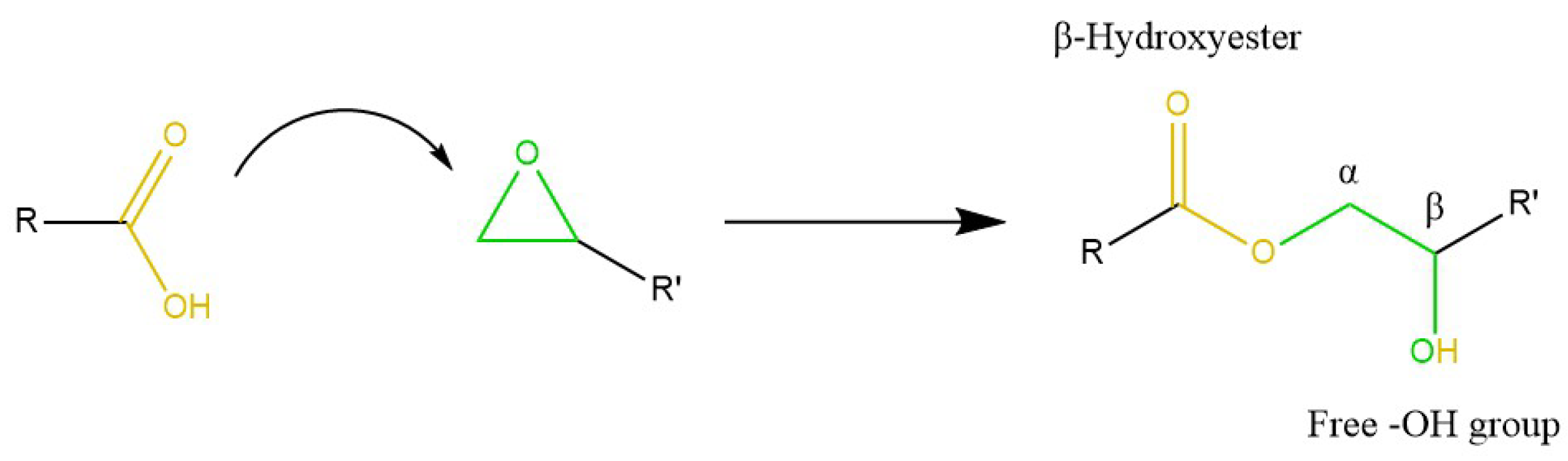
5.2.2. Epoxy Resins
5.2.3. Elastomers
5.2.4. Composites and Nanocomposites
6. Outlook and Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DCN | Dynamic Covalent Network |
| CAN | Covalent Adaptable Network |
| MNR | Molecular Network Rearrangement |
| Tg | Glass transition temperature |
| Tv | Topology freezing transition temperature |
| TE | Transesterification |
| TBD | Triazabicyclodecene |
| TPP | Triphenylphosphine |
| Zn(Ac)2 | Zinc Acetate |
| Zn(Acac)2 | Zinc Acetylacetonate |
| Sn(Oct)2 | Stannous Octoate |
| Ti(Oi-Pr)4 | Titanium (IV) Isopropoxide |
| DBTDL | Dibutyltin Dilaurate |
| DGEBA | Diglycidyl Ether of Bisphenol A |
| FRPC | Fibre-Reinforced Polymer Composite |
| RTM | Resin Transfer Moulding |
| RIM | Reaction Injection Moulding |
| SMASH | Shape Memory Assisted Self-Healing |
| DA | Diels–Alder (reaction) |
| PDMS | Polydimethylsiloxane |
| RAFT | Reversible Addition-Fragmentation chain Transfer |
| WLF | Williams–Landel–Ferry |
| PTIL | Poly(1,2,3-Triazolium Ionic Liquid) |
| GA | Glutaric Anhydride |
| EG | Ethylene Glycol |
| NMP | N-Methyl-2-Pyrrolidone |
| MHHPA | Methylhexahydrophthalic Anhydride |
| NIR | Near-Infrared Radiation |
References
- Pleşa, I.; Noţingher, P.V.; Schlögl, S.; Sumereder, C.; Muhr, M. Properties of Polymer Composites Used in High-Voltage Applications. Polymers 2016, 8, 173. [Google Scholar] [CrossRef] [PubMed]
- Pleşa, I.; Noţingher, P.V.; Stancu, C.; Wiesbrock, F.; Schlögl, S. Polyethylene Nanocomposites for Power Cable Insulations. Polymers 2018, 11, 24. [Google Scholar] [CrossRef] [PubMed]
- Winne, J.M.; Leibler, L.; Du Prez, F.E. Dynamic covalent chemistry in polymer networks: A mechanistic perspective. Polym. Chem. 2019, 10, 6091–6108. [Google Scholar] [CrossRef]
- Zou, W.; Dong, J.; Luo, Y.; Zhao, Q.; Xie, T. Dynamic Covalent Polymer Networks: From Old Chemistry to Modern Day Innovations. Adv. Mater. 2017, 29, 1606100. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Hao, C.; Wang, L.; Li, Y.; Liu, W.; Xin, J.; Zhang, J. Eugenol-Derived Biobased Epoxy: Shape Memory, Repairing, and Recyclability. Macromolecules 2017, 50, 8588–8597. [Google Scholar] [CrossRef]
- Denissen, W.; Winne, J.M.; Du Prez, F.E. Vitrimers: Permanent organic networks with glass-like fluidity. Chem. Sci. 2016, 7, 30–38. [Google Scholar] [CrossRef]
- Kloxin, C.J.; Bowman, C.N. Covalent adaptable networks: Smart, reconfigurable and responsive network systems. Chem. Soc. Rev. 2013, 42, 7161–7173. [Google Scholar] [CrossRef]
- Lu, L.; Fan, J.; Li, G. Intrinsic healable and recyclable thermoset epoxy based on shape memory effect and transesterification reaction. Polymer 2016, 105, 10–18. [Google Scholar] [CrossRef]
- Byrne, J.P. Rubber Elasticity: A Simple Method for Measurement of Thermodynamic Properties. J. Chem. Educ. 1994, 71, 531. [Google Scholar] [CrossRef]
- Imbernon, L.; Oikonomou, E.K.; Norvez, S.; Leibler, L. Chemically cross-linked yet reprocessable epoxidized natural rubber via thermo-activated disulfide rearrangements. Polym. Chem. 2015, 6, 4271–4278. [Google Scholar] [CrossRef]
- Schmolke, W.; Perner, N.; Seiffert, S. Dynamically Cross-Linked Polydimethylsiloxane Networks with Ambient-Temperature Self-Healing. Macromolecules 2015, 48, 8781–8788. [Google Scholar] [CrossRef]
- Montarnal, D.; Capelot, M.; Tournilhac, F.; Leibler, L. Silica-like malleable materials from permanent organic networks. Science 2011, 334, 965–968. [Google Scholar] [CrossRef] [PubMed]
- Chakma, P.; Konkolewicz, D. Dynamic Covalent Bonds in Polymeric Materials. Angew. Chem. Int. Ed. 2019, 58, 9682–9695. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhao, B.; Zhang, J. Recent development of repairable, malleable and recyclable thermosetting polymers through dynamic transesterification. Polymer 2020, 194, 122392. [Google Scholar] [CrossRef]
- Van Zee, N.J.; Nicolaÿ, R. Vitrimers: Permanently cross-linked polymers with dynamic network topology. Prog. Polym. Sci. 2020, 104, 101233. [Google Scholar] [CrossRef]
- Amamoto, Y.; Kamada, J.; Otsuka, H.; Takahara, A.; Matyjaszewski, K. Repeatable photoinduced self-healing of covalently cross-linked polymers through reshuffling of trithiocarbonate units. Angew. Chem. 2011, 50, 1660–1663. [Google Scholar] [CrossRef]
- Amamoto, Y.; Otsuka, H.; Takahara, A.; Matyjaszewski, K. Self-healing of covalently cross-linked polymers by reshuffling thiuram disulfide moieties in air under visible light. Adv. Mater. 2012, 24, 3975–3980. [Google Scholar] [CrossRef]
- Billiet, S.; van Camp, W.; Hillewaere, X.K.; Rahier, H.; Du Prez, F.E. Development of optimized autonomous self-healing systems for epoxy materials based on maleimide chemistry. Polymer 2012, 53, 2320–2326. [Google Scholar] [CrossRef]
- Li, Q.; Liu, C.; Wen, J.; Wu, Y.; Shan, Y.; Liao, J. The design, mechanism and biomedical application of self-healing hydrogels. Chin. Chem. Lett. 2017, 28, 1857–1874. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, L.; Zhao, X.; Hou, S.; Guo, B.; Ma, P.X. Self-healing supramolecular bioelastomers with shape memory property as a multifunctional platform for biomedical applications via modular assembly. Biomaterials 2016, 104, 18–31. [Google Scholar] [CrossRef]
- Zhang, F.; Ju, P.; Pan, M.; Zhang, D.; Huang, Y.; Li, G.; Li, X. Self-healing mechanisms in smart protective coatings: A review. Corros. Sci. 2018, 144, 74–88. [Google Scholar] [CrossRef]
- Liu, T.; Hao, C.; Zhang, S.; Yang, X.; Wang, L.; Han, J.; Li, Y.; Xin, J.; Zhang, J. A Self-Healable High Glass Transition Temperature Bioepoxy Material Based on Vitrimer Chemistry. Macromolecules 2018, 51, 5577–5585. [Google Scholar] [CrossRef]
- Zheng, P.; McCarthy, T.J. A surprise from 1954: Siloxane equilibration is a simple, robust, and obvious polymer self-healing mechanism. J. Am. Chem. Soc. 2012, 134, 2024–2027. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Mather, P.T. Shape Memory Assisted Self-Healing Coating. ACS Macro Lett. 2013, 2, 152–156. [Google Scholar] [CrossRef]
- Han, J.; Liu, T.; Hao, C.; Zhang, S.; Guo, B.; Zhang, J. A Catalyst-Free Epoxy Vitrimer System Based on Multifunctional Hyperbranched Polymer. Macromolecules 2018, 51, 6789–6799. [Google Scholar] [CrossRef]
- Han, J.; Liu, T.; Zhang, S.; Hao, C.; Xin, J.; Guo, B.; Zhang, J. Hyperbranched Polymer Assisted Curing and Repairing of an Epoxy Coating. Ind. Eng. Chem. Res. 2019, 58, 6466–6475. [Google Scholar] [CrossRef]
- Kuang, X.; Zhou, Y.; Shi, Q.; Wang, T.; Qi, H.J. Recycling of Epoxy Thermoset and Composites via Good Solvent Assisted and Small Molecules Participated Exchange Reactions. ACS Sustain. Chem. Eng. 2018, 6, 9189–9197. [Google Scholar] [CrossRef]
- Ruiz de Luzuriaga, A.; Martin, R.; Markaide, N.; Rekondo, A.; Cabañero, G.; Rodríguez, J.; Odriozola, I. Epoxy resin with exchangeable disulfide cross-links to obtain reprocessable, repairable and recyclable fiber-reinforced thermoset composites. Mater. Horizons 2016, 3, 241–247. [Google Scholar] [CrossRef]
- Takahashi, A.; Ohishi, T.; Goseki, R.; Otsuka, H. Degradable epoxy resins prepared from diepoxide monomer with dynamic covalent disulfide linkage. Polymer 2016, 82, 319–326. [Google Scholar] [CrossRef]
- Yu, K.; Shi, Q.; Dunn, M.L.; Wang, T.; Qi, H.J. Carbon Fiber Reinforced Thermoset Composite with Near 100% Recyclability. Adv. Funct. Mater. 2016, 26, 6098–6106. [Google Scholar] [CrossRef]
- Shi, Q.; Yu, K.; Kuang, X.; Mu, X.; Dunn, C.K.; Dunn, M.L.; Wang, T.; Jerry Qi, H. Recyclable 3D printing of vitrimer epoxy. Mater. Horizons 2017, 4, 598–607. [Google Scholar] [CrossRef]
- Zhang, B.; Kowsari, K.; Serjouei, A.; Dunn, M.L.; Ge, Q. Reprocessable thermosets for sustainable three-dimensional printing. Nat. Commun. 2018, 9, 1831. [Google Scholar] [CrossRef]
- Capelot, M.; Montarnal, D.; Tournilhac, F.; Leibler, L. Metal-catalyzed transesterification for healing and assembling of thermosets. J. Am. Chem. Soc. 2012, 134, 7664–7667. [Google Scholar] [CrossRef]
- Chabert, E.; Vial, J.; Cauchois, J.P.; Mihaluta, M.; Tournilhac, F. Multiple welding of long fiber epoxy vitrimer composites. Soft Matter 2016, 12, 4838–4845. [Google Scholar] [CrossRef]
- Yang, Y.; Pei, Z.; Zhang, X.; Tao, L.; Wei, Y.; Ji, Y. Carbon nanotube–vitrimer composite for facile and efficient photo-welding of epoxy. Chem. Sci. 2014, 5, 3486–3492. [Google Scholar] [CrossRef]
- Duquenne, C.; Melas, M.; Beaudrais, S. Composition for Manufacturing Vitrimer Resins of Epoxy/anhydride Type Comprising a Polyol. U.S. Patent 2017/0044361 A1, 16 February 2017. [Google Scholar]
- Duquenne, C.; Melas, M.; Gentilhomme, P.; Disson, J.-P. Composition for Manufacturing Epoxy/Anhydride Vitrimer Resins including an Organic Catalyst. US 20170044305A1, 16 February 2017. [Google Scholar]
- Shi, Q.; Yu, K.; Dunn, M.L.; Wang, T.; Qi, H.J. Solvent Assisted Pressure-Free Surface Welding and Reprocessing of Malleable Epoxy Polymers. Macromolecules 2016, 49, 5527–5537. [Google Scholar] [CrossRef]
- Yu, K.; Shi, Q.; Li, H.; Jabour, J.; Yang, H.; Dunn, M.L.; Wang, T.; Qi, H.J. Interfacial welding of dynamic covalent network polymers. J. Mech. Phys. Solids 2016, 94, 1–17. [Google Scholar] [CrossRef]
- Fouquey, C.; Lehn, J.M.; Levelut, A.M. Molecular recognition directed self-assembly of supramolecular liquid crystalline polymers from complementary chiral components. Adv. Mater. 1990, 2, 254–257. [Google Scholar] [CrossRef]
- Brunsveld, L.; Folmer, B.J.; Meijer, E.W.; Sijbesma, R.P. Supramolecular polymers. Chem. Rev. 2001, 101, 4071–4098. [Google Scholar] [CrossRef]
- Dhers, S.; Vantomme, G.; Avérous, L. A fully bio-based polyimine vitrimer derived from fructose. Green Chem. 2019, 21, 1596–1601. [Google Scholar] [CrossRef]
- Michal, B.T.; Jaye, C.A.; Spencer, E.J.; Rowan, S.J. Inherently Photohealable and Thermal Shape-Memory Polydisulfide Networks. ACS Macro Lett. 2013, 2, 694–699. [Google Scholar] [CrossRef]
- Jourdain, A.; Asbai, R.; Anaya, O.; Chehimi, M.M.; Drockenmuller, E.; Montarnal, D. Rheological Properties of Covalent Adaptable Networks with 1,2,3-Triazolium Cross-Links: The Missing Link between Vitrimers and Dissociative Networks. Macromolecules 2020, 53, 1884–1900. [Google Scholar] [CrossRef]
- Capelot, M.; Unterlass, M.M.; Tournilhac, F.; Leibler, L. Catalytic Control of the Vitrimer Glass Transition. ACS Macro Lett. 2012, 1, 789–792. [Google Scholar] [CrossRef]
- Denissen, W.; Rivero, G.; Nicolaÿ, R.; Leibler, L.; Winne, J.M.; Du Prez, F.E. Vinylogous Urethane Vitrimers. Adv. Funct. Mater. 2015, 25, 2451–2457. [Google Scholar] [CrossRef]
- Denissen, W.; de Baere, I.; van Paepegem, W.; Leibler, L.; Winne, J.; Du Prez, F.E. Vinylogous Urea Vitrimers and Their Application in Fiber Reinforced Composites. Macromolecules 2018, 51, 2054–2064. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, S.; Zhang, X.; Gao, L.; Wei, Y.; Ji, Y. Detecting topology freezing transition temperature of vitrimers by AIE luminogens. Nat. Commun. 2019, 10, 3165. [Google Scholar] [CrossRef]
- Kaiser, S.; Novak, P.; Giebler, M.; Gschwandl, M.; Novak, P.; Pilz, G.; Morak, M.; Schlögl, S. The crucial role of external force in the estimation of the topology freezing transition temperature of vitrimers by elongational creep measurements. Polymer 2020, in press. [Google Scholar] [CrossRef]
- Breuillac, A.; Kassalias, A.; Nicolaÿ, R. Polybutadiene Vitrimers Based on Dioxaborolane Chemistry and Dual Networks with Static and Dynamic Cross-links. Macromolecules 2019, 52, 7102–7113. [Google Scholar] [CrossRef]
- Röttger, M.; Domenech, T.; van der Weegen, R.; Breuillac, A.; Nicolaÿ, R.; Leibler, L. High-performance vitrimers from commodity thermoplastics through dioxaborolane metathesis. Science 2017, 356, 62–65. [Google Scholar] [CrossRef]
- Legrand, A.; Soulié-Ziakovic, C. Silica–Epoxy Vitrimer Nanocomposites. Macromolecules 2016, 49, 5893–5902. [Google Scholar] [CrossRef]
- Obadia, M.M.; Mudraboyina, B.P.; Serghei, A.; Montarnal, D.; Drockenmuller, E. Reprocessing and Recycling of Highly Cross-Linked Ion-Conducting Networks through Transalkylation Exchanges of C-N Bonds. J. Am. Chem. Soc. 2015, 137, 6078–6083. [Google Scholar] [CrossRef]
- Self, J.L.; Dolinski, N.D.; Zayas, M.S.; Read de Alaniz, J.; Bates, C.M. Brønsted-Acid-Catalyzed Exchange in Polyester Dynamic Covalent Networks. ACS Macro Lett. 2018, 7, 817–821. [Google Scholar] [CrossRef]
- Otera, J. Transesterification. Chem. Rev. 1993, 93, 1449–1470. [Google Scholar] [CrossRef]
- Kaiser, S.; Wurzer, S.; Pilz, G.; Kern, W.; Schlögl, S. Stress relaxation and thermally adaptable properties in vitrimer-like elastomers from HXNBR rubber with covalent bonds. Soft Matter 2019, 15, 6062–6072. [Google Scholar] [CrossRef]
- Taplan, C.; Guerre, M.; Winne, J.M.; Du Prez, F.E. Fast processing of highly cross-linked, low-viscosity vitrimers. Mater. Horizons 2020, 7, 104–110. [Google Scholar] [CrossRef]
- Fortman, D.J.; Brutman, J.P.; Cramer, C.J.; Hillmyer, M.A.; Dichtel, W.R. Mechanically activated, catalyst-free polyhydroxyurethane vitrimers. J. Am. Chem. Soc. 2015, 137, 14019–14022. [Google Scholar] [CrossRef]
- Zheng, N.; Fang, Z.; Zou, W.; Zhao, Q.; Xie, T. Thermoset Shape-Memory Polyurethane with Intrinsic Plasticity Enabled by Transcarbamoylation. Angew. Chem. 2016, 55, 11421–11425. [Google Scholar] [CrossRef]
- Zheng, N.; Hou, J.; Xu, Y.; Fang, Z.; Zou, W.; Zhao, Q.; Xie, T. Catalyst-Free Thermoset Polyurethane with Permanent Shape Reconfigurability and Highly Tunable Triple-Shape Memory Performance. ACS Macro Lett. 2017, 6, 326–330. [Google Scholar] [CrossRef]
- Chen, X.; Li, L.; Jin, K.; Torkelson, J.M. Reprocessable polyhydroxyurethane networks exhibiting full property recovery and concurrent associative and dissociative dynamic chemistry via transcarbamoylation and reversible cyclic carbonate aminolysis. Polym. Chem. 2017, 8, 6349–6355. [Google Scholar] [CrossRef]
- Fortman, D.J.; Brutman, J.P.; Hillmyer, M.A.; Dichtel, W.R. Structural effects on the reprocessability and stress relaxation of cross-linked polyhydroxyurethanes. J. Appl. Polym. Sci. 2017, 134, 44984. [Google Scholar] [CrossRef]
- Tang, Z.; Liu, Y.; Huang, Q.; Zhao, J.; Guo, B.; Zhang, L. A real recycling loop of sulphur-cured rubber through transalkylation exchange of C–S bonds. Green Chem. 2018, 20, 5454–5458. [Google Scholar] [CrossRef]
- Lossada, F.; Guo, J.; Jiao, D.; Groeer, S.; Bourgeat-Lami, E.; Montarnal, D.; Walther, A. Vitrimer Chemistry Meets Cellulose Nanofibrils: Bioinspired Nanopapers with High Water Resistance and Strong Adhesion. Biomacromolecules 2019, 20, 1045–1055. [Google Scholar] [CrossRef]
- Altuna, F.I.; Hoppe, C.E.; Williams, R.J.J. Shape memory epoxy vitrimers based on DGEBA cross-linked with dicarboxylic acids and their blends with citric acid. RSC Adv. 2016, 6, 88647–88655. [Google Scholar] [CrossRef]
- DelDonno, T.A. Polyurethane Preparation Using Organo-Zinc Catalyst and Time-Lapse Modifier. U.S. Patent 4,426,510, 17 January 1984. [Google Scholar]
- Zhang, G.; Zhou, X.; Liang, K.; Guo, B.; Li, X.; Wang, Z.; Zhang, L. Mechanically Robust and Recyclable EPDM Rubber Composites by a Green Cross-Linking Strategy. ACS Sustain. Chem. Eng. 2019, 7, 11712–11720. [Google Scholar] [CrossRef]
- Demongeot, A.; Mougnier, S.J.; Okada, S.; Soulié-Ziakovic, C.; Tournilhac, F. Coordination and catalysis of Zn2+ in epoxy-based vitrimers. Polym. Chem. 2016, 7, 4486–4493. [Google Scholar] [CrossRef]
- Tran, T.N.; Rawstron, E.; Bourgeat-Lami, E.; Montarnal, D. Formation of Cross-Linked Films from Immiscible Precursors through Sintering of Vitrimer Nanoparticles. ACS Macro Lett. 2018, 7, 376–380. [Google Scholar] [CrossRef]
- Niu, X.; Wang, F.; Li, X.; Zhang, R.; Wu, Q.; Sun, P. Using Zn2+ Ionomer To Catalyze Transesterification Reaction in Epoxy Vitrimer. Ind. Eng. Chem. Res. 2019, 58, 5698–5706. [Google Scholar] [CrossRef]
- Yang, Z.; Wang, Q.; Wang, T. Dual-Triggered and Thermally Reconfigurable Shape Memory Graphene-Vitrimer Composites. ACS Appl. Mater. Interfaces 2016, 8, 21691–21699. [Google Scholar] [CrossRef]
- Kiesewetter, M.K.; Scholten, M.D.; Kirn, N.; Weber, R.L.; Hedrick, J.L.; Waymouth, R.M. Cyclic guanidine organic Catalysts: What is magic about triazabicyclodecene? J. Org. Chem. 2009, 74, 9490–9496. [Google Scholar] [CrossRef]
- Yadav, J.S.; Reddy, B.; Krishna, A.D.; Reddy, C.S.; Narsaiah, A.V. Triphenylphosphine: An efficient catalyst for transesterification of β-ketoesters. J. Mol. Catal. A Chem. 2007, 261, 93–97. [Google Scholar] [CrossRef]
- Brutman, J.P.; Delgado, P.A.; Hillmyer, M.A. Polylactide Vitrimers. ACS Macro Lett. 2014, 3, 607–610. [Google Scholar] [CrossRef]
- Lou, C.; Gu, J.; Di, M.; Ma, L.; Wang, Y.; Liu, X. Synthesis and Characterization of Trichlorophenol-blocked Polyaryl Polyisocyanate. Iran. Polym. J. 2011, 20, 247–255. [Google Scholar]
- Liu, W.; Schmidt, D.F.; Reynaud, E. Catalyst Selection, Creep, and Stress Relaxation in High-Performance Epoxy Vitrimers. Ind. Eng. Chem. Res. 2017, 56, 2667–2672. [Google Scholar] [CrossRef]
- Snyder, R.L.; Fortman, D.J.; de Hoe, G.X.; Hillmyer, M.A.; Dichtel, W.R. Reprocessable Acid-Degradable Polycarbonate Vitrimers. Macromolecules 2018, 51, 389–397. [Google Scholar] [CrossRef]
- Zhao, W.; Feng, Z.; Liang, Z.; Lv, Y.; Xiang, F.; Xiong, C.; Duan, C.; Dai, L.; Ni, Y. Vitrimer-Cellulose Paper Composites: A New Class of Strong, Smart, Green, and Sustainable Materials. ACS Appl. Mater. Interfaces 2019, 11, 36090–36099. [Google Scholar] [CrossRef] [PubMed]
- Hatano, M.; Tabata, Y.; Yoshida, Y.; Toh, K.; Yamashita, K.; Ogura, Y.; Ishihara, K. Metal-free transesterification catalyzed by tetramethylammonium methyl carbonate. Green Chem. 2018, 20, 1193–1198. [Google Scholar] [CrossRef]
- Fairbanks, B.D.; Singh, S.P.; Bowman, C.N.; Anseth, K.S. Photodegradable, Photoadaptable Hydrogels via Radical-Mediated Disulfide Fragmentation Reaction. Macromolecules 2011, 44, 2444–2450. [Google Scholar] [CrossRef]
- Worrell, B.T.; McBride, M.K.; Lyon, G.B.; Cox, L.M.; Wang, C.; Mavila, S.; Lim, C.H.; Coley, H.M.; Musgrave, C.B.; Ding, Y.; et al. Bistable and photoswitchable states of matter. Nat. Commun. 2018, 9, 2804. [Google Scholar] [CrossRef]
- Zhang, G.; Zhao, Q.; Yang, L.; Zou, W.; Xi, X.; Xie, T. Exploring Dynamic Equilibrium of Diels–Alder Reaction for Solid State Plasticity in Remoldable Shape Memory Polymer Network. ACS Macro Lett. 2016, 5, 805–808. [Google Scholar] [CrossRef]
- Rivero, G.; Nguyen, L.T.T.; Hillewaere, X.K.D.; Du Prez, F.E. One-Pot Thermo-Remendable Shape Memory Polyurethanes. Macromolecules 2014, 47, 2010–2018. [Google Scholar] [CrossRef]
- Billiet, S.; de Bruycker, K.; Driessen, F.; Goossens, H.; van Speybroeck, V.; Winne, J.M.; Du Prez, F.E. Triazolinediones enable ultrafast and reversible click chemistry for the design of dynamic polymer systems. Nat. Chem. 2014, 6, 815–821. [Google Scholar] [CrossRef] [PubMed]
- Roling, O.; de Bruycker, K.; Vonhören, B.; Stricker, L.; Körsgen, M.; Arlinghaus, H.F.; Ravoo, B.J.; Du Prez, F.E. Rewritable Polymer Brush Micropatterns Grafted by Triazolinedione Click Chemistry. Angew. Chem. Int. Ed. 2015, 54, 13126–13129. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Liu, T.; Hao, C.; Wang, L.; Han, J.; Liu, H.; Zhang, J. Preparation of a lignin-based vitrimer material and its potential use for recoverable adhesives. Green Chem. 2018, 20, 2995–3000. [Google Scholar] [CrossRef]
- Zhang, W.; Ma, Y.; Wang, C.; Li, S.; Zhang, M.; Chu, F. Preparation and properties of lignin–phenol– formaldehyde resins based on different biorefinery residues of agricultural biomass. Ind. Crops Prod. 2013, 43, 326–333. [Google Scholar] [CrossRef]
- Hernandez, E.D.; Bassett, A.W.; Sadler, J.M.; La Scala, J.J.; Stanzione, J.F. Synthesis and Characterization of Bio-based Epoxy Resins Derived from Vanillyl Alcohol. ACS Sustain. Chem. Eng. 2016, 4, 4328–4339. [Google Scholar] [CrossRef]
- Effendi, A.; Gerhauser, H.; Bridgwater, A.V. Production of renewable phenolic resins by thermochemical conversion of biomass: A review. Renew. Sustain. Energy Rev. 2008, 12, 2092–2116. [Google Scholar] [CrossRef]
- Foyer, G.; Chanfi, B.H.; Boutevin, B.; Caillol, S.; David, G. New method for the synthesis of formaldehyde-free phenolic resins from lignin-based aldehyde precursors. Eur. Polym. J. 2016, 74, 296–309. [Google Scholar] [CrossRef]
- Xin, J.; Li, M.; Li, R.; Wolcott, M.P.; Zhang, J. Green Epoxy Resin System Based on Lignin and Tung Oil and Its Application in Epoxy Asphalt. ACS Sustain. Chem. Eng. 2016, 4, 2754–2761. [Google Scholar] [CrossRef]
- Cantarutti, C.; Dinu, R.; Mija, A. Biorefinery Byproducts and Epoxy Biorenewable Monomers: A Structural Elucidation of Humins and Triglycidyl Ether of Phloroglucinol Cross-Linking. Biomacromolecules 2020, 21, 517–533. [Google Scholar] [CrossRef]
- Geng, H.; Wang, Y.; Yu, Q.; Gu, S.; Zhou, Y.; Xu, W.; Zhang, X.; Ye, D. Vanillin-Based Polyschiff Vitrimers: Reprocessability and Chemical Recyclability. ACS Sustain. Chem. Eng. 2018, 6, 15463–15470. [Google Scholar] [CrossRef]
- Yan, N.; Chen, X. Sustainability: Don’t waste seafood waste. Nature 2015, 524, 155–157. [Google Scholar] [CrossRef]
- Zhang, J.; Feng, M.; Lu, X.; Shi, C.; Li, X.; Xin, J.; Yue, G.; Zhang, S. Base-free preparation of low molecular weight chitin from crab shell. Carbohydr. Polym. 2018, 190, 148–155. [Google Scholar] [CrossRef]
- Anitha, A.; Sowmya, S.; Kumar, P.S.; Deepthi, S.; Chennazhi, K.P.; Ehrlich, H.; Tsurkan, M.; Jayakumar, R. Chitin and chitosan in selected biomedical applications. Prog. Polym. Sci. 2014, 39, 1644–1667. [Google Scholar] [CrossRef]
- Ghosh, B.; Urban, M.W. Self-repairing oxetane-substituted chitosan polyurethane networks. Science 2009, 323, 1458–1460. [Google Scholar] [CrossRef]
- Boey, F.; Qiang, W. Experimental modeling of the cure kinetics of an epoxy-hexaanhydro-4-methylphthalicanhydride (MHHPA) system. Polymer 2000, 41, 2081–2094. [Google Scholar] [CrossRef]
- Paramarta, A.; Webster, D.C. Curing kinetics of bio-based epoxy-anhydride thermosets with zinc catalyst. J. Therm. Anal. Calorim. 2017, 130, 2133–2144. [Google Scholar] [CrossRef]
- Giebler, M.; Sperling, C.; Kaiser, S.; Duretek, I.; Schlögl, S. Epoxy-Anhydride Vitrimers from Aminoglycidyl Resins with High Glass Transition Temperature and Efficient Stress Relaxation. Polymers 2020, 12, 1148. [Google Scholar] [CrossRef]
- Lu, L.; Pan, J.; Li, G. Recyclable high-performance epoxy based on transesterification reaction. J. Mater. Chem. A 2017, 5, 21505–21513. [Google Scholar] [CrossRef]
- Altuna, F.I.; Hoppe, C.E.; Williams, R.J.J. Epoxy Vitrimers: The Effect of Transesterification Reactions on the Network Structure. Polymers 2018, 10, 43. [Google Scholar] [CrossRef]
- Han, H.; Xu, X. Poly(methyl methacrylate)-epoxy vitrimer composites. J. Appl. Polym. Sci. 2018, 135, 46307. [Google Scholar] [CrossRef]
- Yu, L.; Zhu, C.; Sun, X.; Salter, J.; Wu, H.; Jin, Y.; Zhang, W.; Long, R. Rapid Fabrication of Malleable Fiber Reinforced Composites with Vitrimer Powder. ACS Appl. Polym. Mater. 2019, 1, 2535–2542. [Google Scholar] [CrossRef]
- Tesoro, G.C.; Sastri, V. Reversible cross-linking in epoxy resins. I. Feasibility studies. J. Appl. Polym. Sci. 1990, 39, 1425–1437. [Google Scholar] [CrossRef]
- Chen, M.; Zhou, L.; Wu, Y.; Zhao, X.; Zhang, Y. Rapid Stress Relaxation and Moderate Temperature of Malleability Enabled by the Synergy of Disulfide Metathesis and Carboxylate Transesterification in Epoxy Vitrimers. ACS Macro Lett. 2019, 8, 255–260. [Google Scholar] [CrossRef]
- Deng, J.; Kuang, X.; Liu, R.; Ding, W.; Wang, A.C.; Lai, Y.C.; Dong, K.; Wen, Z.; Wang, Y.; Wang, L.; et al. Vitrimer Elastomer-Based Jigsaw Puzzle-Like Healable Triboelectric Nanogenerator for Self-Powered Wearable Electronics. Adv. Mater. 2018, 30, e1705918. [Google Scholar] [CrossRef]
- Lötters, J.C.; Olthuis, W.; Veltink, P.H.; Bergveld, P. The mechanical properties of the rubber elastic polymer polydimethylsiloxane for sensor applications. J. Micromech. Microeng. 1997, 7, 145–147. [Google Scholar] [CrossRef]
- Stukenbroeker, T.; Wang, W.; Winne, J.M.; Du Prez, F.E.; Nicolaÿ, R.; Leibler, L. Polydimethylsiloxane quenchable vitrimers. Polym. Chem. 2017, 8, 6590–6593. [Google Scholar] [CrossRef]
- Krishnakumar, B.; Prasanna Sanka, R.; Binder, W.H.; Park, C.; Jung, J.; Parthasarthy, V.; Rana, S.; Yun, G.J. Catalyst free self-healable vitrimer/graphene oxide nanocomposites. Compos. Part B Eng. 2020, 184, 107647. [Google Scholar] [CrossRef]
- Yan, P.; Zhao, W.; Jiang, L.; Wu, B.; Hu, K.; Yuan, Y.; Lei, J. Reconfiguration and shape memory triggered by heat and light of carbon nanotube-polyurethane vitrimer composites. J. Appl. Polym. Sci. 2018, 135, 45784. [Google Scholar] [CrossRef]




















© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alabiso, W.; Schlögl, S. The Impact of Vitrimers on the Industry of the Future: Chemistry, Properties and Sustainable Forward-Looking Applications. Polymers 2020, 12, 1660. https://doi.org/10.3390/polym12081660
Alabiso W, Schlögl S. The Impact of Vitrimers on the Industry of the Future: Chemistry, Properties and Sustainable Forward-Looking Applications. Polymers. 2020; 12(8):1660. https://doi.org/10.3390/polym12081660
Chicago/Turabian StyleAlabiso, Walter, and Sandra Schlögl. 2020. "The Impact of Vitrimers on the Industry of the Future: Chemistry, Properties and Sustainable Forward-Looking Applications" Polymers 12, no. 8: 1660. https://doi.org/10.3390/polym12081660
APA StyleAlabiso, W., & Schlögl, S. (2020). The Impact of Vitrimers on the Industry of the Future: Chemistry, Properties and Sustainable Forward-Looking Applications. Polymers, 12(8), 1660. https://doi.org/10.3390/polym12081660