Complex Temperature and Concentration Dependent Self-Assembly of Poly(2-oxazoline) Block Copolymers



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
3. Results and Discussion
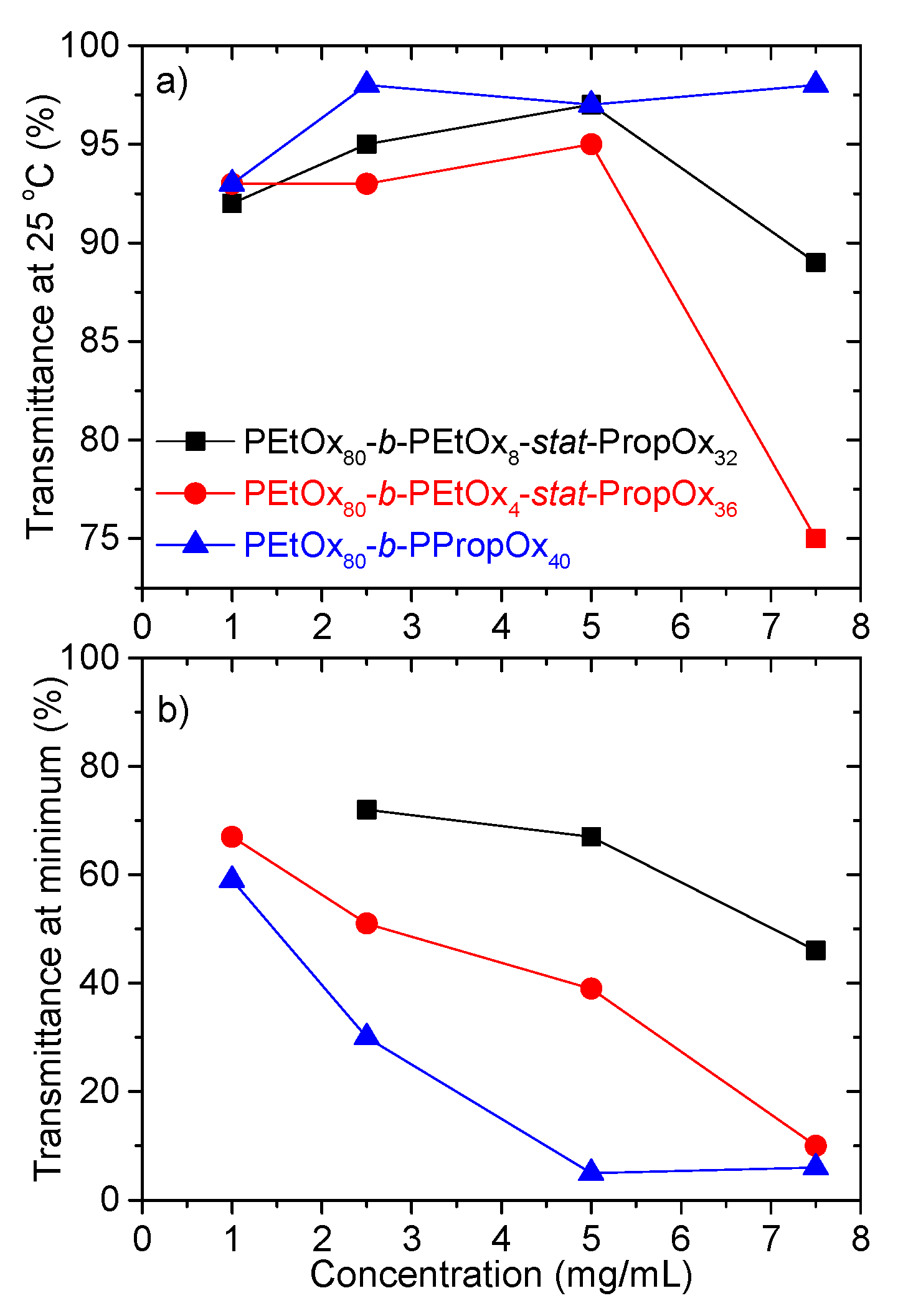
3.1. Turbidimetry
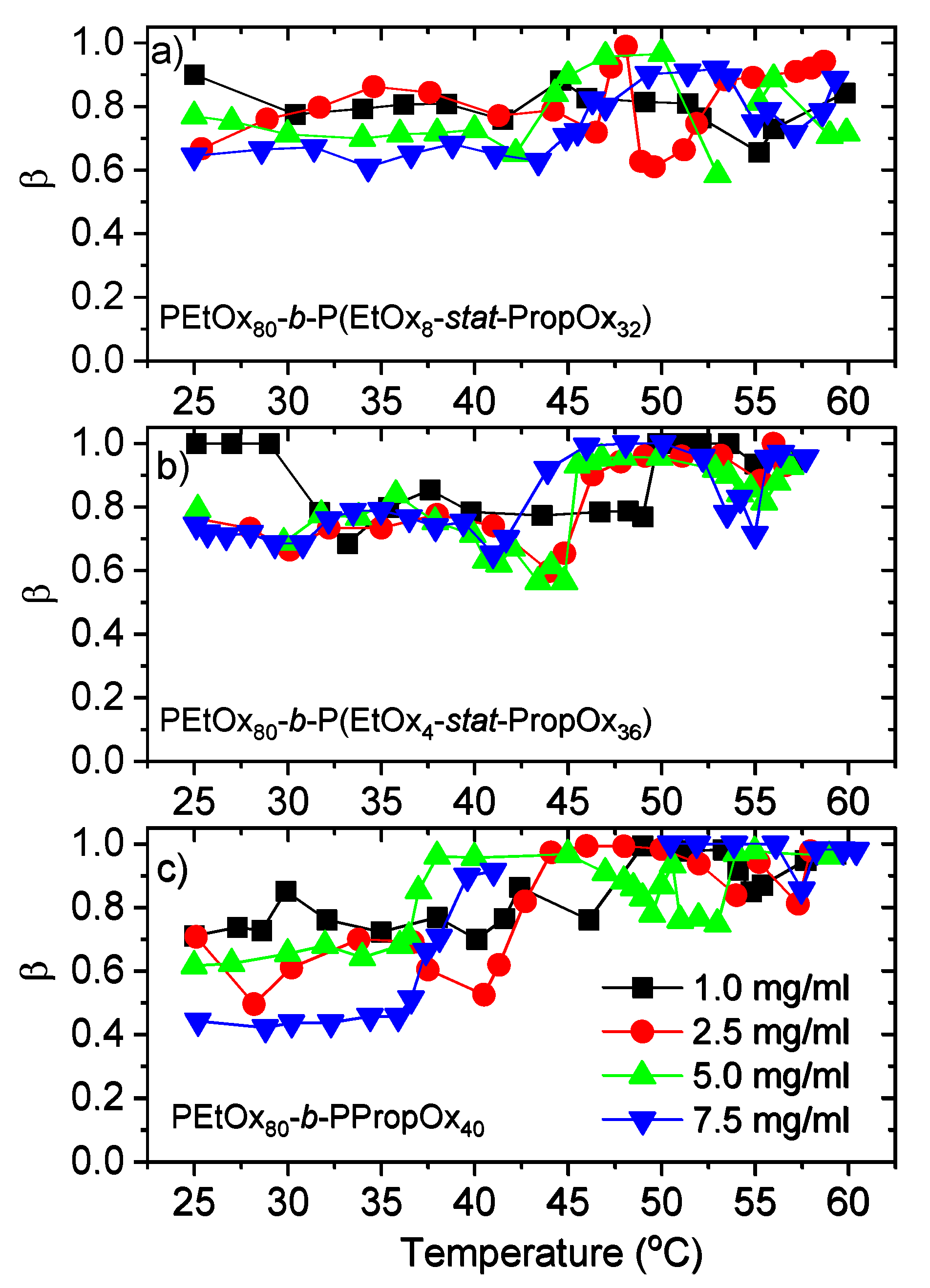
3.2. Dynamic Light Scattering
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Findlay, P.H.; Rannard, S.P.; Royles, B.J.L.; Weaver, J.V.M. Amphiphilic Branched Polymers and Their Use as Emulsifiers. U.S. Patent 20110172314A1, 29 May 2008. [Google Scholar]
- Bobin, M.-F.; Michel, V.; Martini, M.-C. Study of formulation and stability of emulsions with polymeric emulsifiers. Colloids Surfaces A Physicochem. Eng. Asp. 1999, 152, 53–58. [Google Scholar] [CrossRef]
- Mathur, A.M.; Drescher, B.; Scranton, A.B.; Klier, J. Polymeric emulsifiers based on reversible formation of hydrophobic units. Nature 1998, 392, 367–370. [Google Scholar] [CrossRef]
- Alexandridis, P. Amphiphilic copolymers and their applications. Curr. Opin. Colloid Interface Sci. 1996, 1, 490–501. [Google Scholar] [CrossRef]
- Winnik, M.A.; Yekta, A. Associative polymers in aqueous solution. Curr. Opin. Colloid Interface Sci. 1997, 2, 424–436. [Google Scholar] [CrossRef]
- Talelli, M.; Barz, M.; Rijcken, C.J.; Kiessling, F.; Hennink, W.E.; Lammers, T. Core-crosslinked polymeric micelles: Principles, preparation, biomedical applications and clinical translation. Nano Today 2015, 10, 93–117. [Google Scholar] [CrossRef] [PubMed]
- Mura, S.; Nicolas, J.; Couvreur, P.; Patrick, C. Stimuli-responsive nanocarriers for drug delivery. Nat. Mater. 2013, 12, 991–1003. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-Y.; Kim, T.H.; Wu, W.-C.; Huang, C.-M.; Wei, H.; Mount, C.; Tian, Y.; Jang, S.-H.; Pun, S.H.; Jen, A.K.-Y. pH-dependent, thermosensitive polymeric nanocarriers for drug delivery to solid tumors. Biomaterials 2013, 34, 4501–4509. [Google Scholar] [CrossRef]
- Wei, H.; Cheng, S.-X.; Zhang, X.-Z.; Zhuo, R.-X. Thermo-sensitive polymeric micelles based on poly(N-isopropylacrylamide) as drug carriers. Prog. Polym. Sci. 2009, 34, 893–910. [Google Scholar] [CrossRef]
- Hu, J.; Liu, S. Responsive Polymers for Detection and Sensing Applications: Current Status and Future Developments. Macromolecules 2010, 43, 8315–8330. [Google Scholar] [CrossRef]
- Talelli, M.; Hennink, W.E. Thermosensitive polymeric micelles for targeted drug delivery. Nanomedicine 2011, 6, 1245–1255. [Google Scholar] [CrossRef]
- Shao, P.; Wang, B.; Wang, Y.; Li, J.; Zhang, Y. The Application of Thermosensitive Nanocarriers in Controlled Drug Delivery. J. Nanomater. 2011, 2011, 1–12. [Google Scholar] [CrossRef]
- Zhang, Q.; Weber, C.; Schubert, U.S.; Hoogenboom, R. Thermoresponsive polymers with lower critical solution temperature: From fundamental aspects and measuring techniques to recommended turbidimetry conditions. Mater. Horizons 2017, 4, 109–116. [Google Scholar] [CrossRef]
- Gil, E.; Hudson, S.M. Stimuli-reponsive polymers and their bioconjugates. Prog. Polym. Sci. 2004, 29, 1173–1222. [Google Scholar] [CrossRef]
- Ward, M.A.; Georgiou, T.K. Thermoresponsive Polymers for Biomedical Applications. Polymer 2011, 3, 1215–1242. [Google Scholar] [CrossRef]
- Viegas, T.X.; Bentley, M.D.; Harris, J.M.; Fang, Z.; Yoon, K.; Dizman, B.; Weimer, R.; Mero, A.; Pasut, G.; Veronese, F.M. Polyoxazoline: Chemistry, Properties, and Applications in Drug Delivery. Bioconjug. Chem. 2011, 22, 976–986. [Google Scholar] [CrossRef]
- Qiu, X.; Hu, S. “Smart” Materials Based on Cellulose: A Review of the Preparations, Properties, and Applications. Materials 2013, 6, 738–781. [Google Scholar] [CrossRef]
- Luxenhofer, R.; Han, Y.; Schulz, A.; Tong, J.; He, Z.; Kabanov, A.V.; Jordan, R. Poly(2-oxazoline)s as Polymer Therapeutics. Macromol. Rapid Commun. 2012, 33, 1613–1631. [Google Scholar] [CrossRef]
- Schild, H. Poly(N-isopropylacrylamide): Experiment, theory and application. Prog. Polym. Sci. 1992, 17, 163–249. [Google Scholar] [CrossRef]
- Aoshima, S.; Kanaoka, S. Synthesis of Stimuli-Responsive Polymers by Living Polymerization: Poly(N-Isopropylacrylamide) and Poly(Vinyl Ether)s. In Anionic Polymerization; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2007; Volume 210, pp. 169–208. [Google Scholar]
- Lee, J.-S.; Feijen, J. Polymersomes for drug delivery: Design, formation and characterization. J. Control. Release 2012, 161, 473–483. [Google Scholar] [CrossRef]
- Lutz, J.-F. Polymerization of oligo(ethylene glycol) (meth)acrylates: Toward new generations of smart biocompatible materials. J. Polym. Sci. Part A: Polym. Chem. 2008, 46, 3459–3470. [Google Scholar] [CrossRef]
- Lutz, J. Thermo-Switchable Materials Prepared Using the OEGMA-Platform. Adv. Mater. 2011, 23, 2237–2243. [Google Scholar] [CrossRef]
- Hoogenboom, R.; Schlaad, H. Bioinspired Poly(2-oxazoline)s. Polymer 2011, 3, 467–488. [Google Scholar] [CrossRef]
- Schlaad, H.; Diehl, C.; Gress, A.; Meyer, M.; Demirel, A.L.; Nur, Y.; Bertin, A. Poly(2-oxazoline)s as Smart Bioinspired Polymers. Macromol. Rapid Commun. 2010, 31, 511–525. [Google Scholar] [CrossRef] [PubMed]
- Luxenhofer, R.; Schulz, A.; Roques, C.; Li, S.; Bronich, T.K.; Batrakova, E.V.; Jordan, R.; Kabanov, A.V. Doubly amphiphilic poly(2-oxazoline)s as high-capacity delivery systems for hydrophobic drugs. Biomaterial 2010, 31, 4972–4979. [Google Scholar] [CrossRef] [PubMed]
- Hoogenboom, R. Poly(2-oxazoline)s: A Polymer Class with Numerous Potential Applications. Angew. Chem. Int. Ed. 2009, 48, 7978–7994. [Google Scholar] [CrossRef]
- Adams, N.; Schubert, U.S. Poly(2-oxazolines) in biological and biomedical application contexts. Adv. Drug Deliv. Rev. 2007, 59, 1504–1520. [Google Scholar] [CrossRef]
- Knop, K.; Hoogenboom, R.; Fischer, D.; Schubert, U.S. Poly(ethylene glycol) in Drug Delivery: Pros and Cons as Well as Potential Alternatives. Angew. Chem. Int. Ed. 2010, 49, 6288–6308. [Google Scholar] [CrossRef]
- Allen, T.M.; Cullis, P.R. Drug Delivery Systems: Entering the Mainstream. Science 2004, 303, 1818–1822. [Google Scholar] [CrossRef]
- Langer, R. Drug delivery and targeting. Nature 1998, 392, 5. [Google Scholar]
- Panyam, J.; Labhasetwar, V. Biodegradable nanoparticles for drug and gene delivery to cells and tissue. Adv. Drug Deliv. Rev. 2003, 55, 329–347. [Google Scholar] [CrossRef]
- Uhrich, K.E.; Cannizzaro, S.M.; Langer, R.S.; Shakesheff, K.M. Polymeric Systems for Controlled Drug Release. Chem. Rev. 1999, 99, 3181–3198. [Google Scholar] [CrossRef] [PubMed]
- Sedlacek, O.; Monnery, B.D.; Filippov, S.K.; Hoogenboom, R.; Hruby, M. Poly(2-Oxazoline)s—Are They More Advantageous for Biomedical Applications Than Other Polymers? Macromol. Rapid Commun. 2012, 33, 1648–1662. [Google Scholar] [CrossRef] [PubMed]
- De La Rosa, V.R. Poly(2-oxazoline)s as materials for biomedical applications. J. Mater. Sci. Mater. Electron. 2013, 25, 1211–1225. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.; Lautenschlaeger, C.; Kempe, K.; Tauhardt, L.; Schubert, U.S.; Fischer, D. Poly(2-ethyl-2-oxazoline) as Alternative for the Stealth Polymer Poly(ethylene glycol): Comparison of in vitro Cytotoxicity and Hemocompatibility. Macromol. Biosci. 2012, 12, 986–998. [Google Scholar] [CrossRef] [PubMed]
- Frechet, J.J.M.; Yui, K. Polymerizable Macromonomers and Polymers Prepared Therefrom. U.S. Patent 58,309,48A, 30 June 1995. [Google Scholar]
- Warchol, J.F.; Walton, C.D. Creping Adhesives Containing Oxazoline Polymer Blends and Use in Paper Product Applications. U.S. Patent 97-795911, 24 August 1998. [Google Scholar]
- Brinkhuis, R. Hyperbranched Esteroxazoline Polymers. WO2003054060A1, 21 December 2001. [Google Scholar]
- Davis, R.A.; Madison, N.L. Polyoxazoline-Modified, Paper Coating. U.S. Patent 44,367,89A, 24 August 1998. [Google Scholar]
- Warchol, J.F.; Walton, C.D. Creping Adhesives Containing Oxazoline Polymers and Methods of Use Thereof. U.S. Patent 58,377,68A, 8 December 1994. [Google Scholar]
- Hamidi, M.; Shahbazi, M.-A.; Rostamizadeh, K. Copolymers: Efficient Carriers for Intelligent Nanoparticulate Drug Targeting and Gene Therapy. Macromol. Biosci. 2011, 12, 144–164. [Google Scholar] [CrossRef]
- Tomalia, D.A.; Sheetz, D.P. Homopolymerization of 2-alkyl- and 2-aryl-2-oxazolines. J. Polym. Sci. Part A-1: Polym. Chem. 1966, 4, 2253–2265. [Google Scholar] [CrossRef]
- Litt, M.; Herz, J. Polymerization of cyclic imino ethers. J. Colloid Interface Sci. 1969, 31, 248–252. [Google Scholar] [CrossRef]
- Kagiya, T.; Narisawa, S.; Maeda, T.; Fukui, K. Ring-opening polymerization of 2-substituted 2-oxazolines. J. Polym. Sci. Part B Polym. Lett. 1966, 4, 441–445. [Google Scholar] [CrossRef]
- Levy, A.; Litt, M. Polymerization of cyclic iminoethers. III. Effect of ring substituents. J. Polym. Sci. Part A-1 Polym. Chem. 1968, 6, 57–62. [Google Scholar] [CrossRef]
- Levy, A.; Litt, M. Polymerization of cyclic iminoethers. V. 1,3-oxazolines with hydroxy-, acetoxy-, and carboxymethyl-alkyl groups in the 2 position and their polymers. J. Polym. Sci. Part A-1: Polym. Chem. 1968, 6, 1883–1894. [Google Scholar] [CrossRef]
- Moreadith, R.W.; Viegas, T.X.; Bentley, M.D.; Harris, J.M.; Fang, Z.; Yoon, K.; Dizman, B.; Weimer, R.; Rae, B.P.; Li, X.; et al. Clinical development of a poly(2-oxazoline) (POZ) polymer therapeutic for the treatment of Parkinson’s disease – Proof of concept of POZ as a versatile polymer platform for drug development in multiple therapeutic indications. Eur. Polym. J. 2017, 88, 524–552. [Google Scholar] [CrossRef]
- Aoi, K.; Suzuki, H.; Okada, M. Architectural control of sugar-containing polymers by living polymerization: Ring-opening polymerization of 2-oxazolines initiated with carbohydrate derivatives. Macromolecules 1992, 25, 7073–7075. [Google Scholar] [CrossRef]
- Verbraeken, B.; Monnery, B.D.; Lava, K.; Hoogenboom, R. The chemistry of poly(2-oxazoline)s. Eur. Polym. J. 2017, 88, 451–469. [Google Scholar] [CrossRef]
- Fox, M.E.; Szoka, F.C.; Fréchet, J.M.J. Soluble Polymer Carriers for the Treatment of Cancer: The Importance of Molecular Architecture. Accounts Chem. Res. 2009, 42, 1141–1151. [Google Scholar] [CrossRef]
- Glassner, M.; Vergaelen, M.; Hoogenboom, R. Poly(2-oxazoline)s: A comprehensive overview of polymer structures and their physical properties. Polym. Int. 2017, 67, 32–45. [Google Scholar] [CrossRef]
- Zalipsky, S.; Hansen, C.B.; Oaks, J.M.; Allen, T.M. Evaluation of Blood Clearance Rates and Biodistribution of Poly(2-oxazoline)-Grafted Liposomes§. J. Pharm. Sci. 1996, 85, 133–137. [Google Scholar] [CrossRef]
- Gaertner, F.C.; Luxenhofer, R.; Blechert, B.; Jordan, R.; Essler, M. Synthesis, biodistribution and excretion of radiolabeled poly(2-alkyl-2-oxazoline)s. J. Control. Release 2007, 119, 291–300. [Google Scholar] [CrossRef]
- Woodle, M.C.; Engbers, C.M.; Zalipsky, S. New Amphipatic Polymer-Lipid Conjugates Forming Long-Circulating Reticuloendothelial System-Evading Liposomes. Bioconjug. Chem. 1994, 5, 493–496. [Google Scholar] [CrossRef]
- He, Z.; Schulz, A.; Wan, X.; Seitz, J.; Bludau, H.; Alakhova, D.; Darr, D.B.; Perou, C.M.; Jordan, R.; Ojima, I.; et al. Poly(2-oxazoline) based micelles with high capacity for 3rd generation taxoids: Preparation, in vitro and in vivo evaluation. J. Control. Release 2015, 208, 67–75. [Google Scholar] [CrossRef]
- Lin, P.; Clash, C.; Pearce, E.M.; Kwei, T.K.; Aponte, M.A. Solubility and miscibility of poly(ethyl oxazoline). J. Polym. Sci. Part B: Polym. Phys. 1988, 26, 603–619. [Google Scholar] [CrossRef]
- Uyama, H.; Kobayashi, S. A Novel Thermo-Sensitive Polymer. Poly(2-iso-propyl-2-oxazoline). Chem. Lett. 1992, 21, 1643–1646. [Google Scholar] [CrossRef]
- Park, J.-S.; Kataoka, K. Precise Control of Lower Critical Solution Temperature of Thermosensitive Poly(2-isopropyl-2-oxazoline) via Gradient Copolymerization with 2-Ethyl-2-oxazoline as a Hydrophilic Comonomer. Macromolecules 2006, 39, 6622–6630. [Google Scholar] [CrossRef]
- Hoogenboom, R.; Thijs, H.M.L.; Jochems, M.J.H.C.; Van Lankvelt, B.M.; Fijten, M.W.M.; Schubert, U.S. Tuning the LCST of poly(2-oxazoline)s by varying composition and molecular weight: Alternatives to poly(N-isopropylacrylamide)? Chem. Commun. 2008, 5758–5760. [Google Scholar] [CrossRef] [PubMed]
- Trinh, L.T.T.; Lambermont-Thijs, H.M.L.; Schubert, U.S.; Hoogenboom, R.; Kjøniksen, A.-L. Thermoresponsive Poly(2-oxazoline) Block Copolymers Exhibiting Two Cloud Points: Complex Multistep Assembly Behavior. Macromolecules 2012, 45, 4337–4345. [Google Scholar] [CrossRef]
- Siegert, A.J.F. On the Fluctuations in Signals Returned by Many Independently Moving Scatterers; Radiation Laboratory Report; Report 465; Massachusetts Institute of Technology: Cambridge, MA, USA, 1943. [Google Scholar]
- Ngai, K. Dynamics of semidilute solutions of polymers and associating polymers. Adv. Colloid Interface Sci. 1996, 64, 1–43. [Google Scholar] [CrossRef]
- Tirado-Miranda, M.; Haro-Pérez, C.; Quesada-Pérez, M.; Callejas-Fernández, J.; Hidalgo-Alvarez, R. Effective charges of colloidal particles obtained from collective diffusion experiments. J. Colloid Interface Sci. 2003, 263, 74–79. [Google Scholar] [CrossRef]
- Ellis, A.R.; Schaller, J.K.; McKiernan, M.L.; Selser, J.C. Evidence for coupling between hydrodynamic interactions and excluded volume effects in polymer coils. J. Chem. Phys. 1990, 92, 5731–5743. [Google Scholar] [CrossRef]
- Zimm, B.H. Dynamics of Polymer Molecules in Dilute Solution: Viscoelasticity, Flow Birefringence and Dielectric Loss. J. Chem. Phys. 1956, 24, 269. [Google Scholar] [CrossRef]
- Rouse, P.E. A Theory of the Linear Viscoelastic Properties of Dilute Solutions of Coiling Polymers. J. Chem. Phys. 1953, 21, 1272. [Google Scholar] [CrossRef]
- Jia, Y.-G.; Zhu, X.X. Complex thermoresponsive behavior of diblock polyacrylamides. Polym. Chem. 2014, 5, 4358–4364. [Google Scholar] [CrossRef]
- Hua, F.; Jiang, X.; Zhao, B. Temperature-Induced Self-Association of Doubly Thermosensitive Diblock Copolymers with Pendant Methoxytris(oxyethylene) Groups in Dilute Aqueous Solutions. Macromolecules 2006, 39, 3476–3479. [Google Scholar] [CrossRef]
- Käfer, F.; Liu, F.; Stahlschmidt, U.; Jérôme, V.; Freitag, R.; Karg, M.; Agarwal, S. LCST and UCST in One: Double Thermoresponsive Behavior of Block Copolymers of Poly(ethylene glycol) and Poly(acrylamide-co-acrylonitrile). Langmuir 2015, 31, 8940–8946. [Google Scholar] [CrossRef] [PubMed]
- Kermagoret, A.; Fustin, C.-A.; Bourguignon, M.; Detrembleur, C.; Jérôme, C.; Debuigne, A. One-pot controlled synthesis of double thermoresponsive N-vinylcaprolactam-based copolymers with tunable LCSTs. Polym. Chem. 2013, 4, 2575–2583. [Google Scholar] [CrossRef]
- Zhang, B.; Tang, H.; Wu, P. In Depth Analysis on the Unusual Multistep Aggregation Process of Oligo(ethylene glycol) Methacrylate-Based Polymers in Water. Macromolecules 2014, 47, 4728–4737. [Google Scholar] [CrossRef]
- Zhu, K.; Pamies, R.; Al-Manasir, N.; Cifre, J.G.H.; De La Torre, J.G.; Nyström, B.; Kjøniksen, A.-L. The Effect of Number of Arms on the Aggregation Behavior of Thermoresponsive Poly( N -isopropylacrylamide) Star Polymers. ChemPhysChem 2020, 21, 1258–1271. [Google Scholar] [CrossRef]
- Lechner, M. Influence of Mie scattering on nanoparticles with different particle sizes and shapes: Photometry and analytical ultracentrifugation with absorption optics. J. Serbian Chem. Soc. 2005, 70, 361–369. [Google Scholar] [CrossRef]
- Jonassen, H.; Kjøniksen, A.-L. Optical-scattering method for the determination of the local polymer concentration inside nanoparticles. Phys. Rev. E 2011, 84, 022401. [Google Scholar] [CrossRef]
- Bai, Y.; Ma, X.; Wang, W.; Yin, Q.; Du, Z.; Wang, G. Synthesis, aggregation and dispersity properties of novels amphiphilic comb-like terpolymers. Colloids Surfaces A: Physicochem. Eng. Asp. 2017, 526, 40–47. [Google Scholar] [CrossRef]
- Zhu, K.; Pamies, R.; Kjøniksen, A.-L.; Nyström, B. Temperature-Induced Intermicellization of “Hairy” and “Crew-Cut” Micelles in an Aqueous Solution of a Thermoresponsive Copolymer. Langmuir 2008, 24, 14227–14233. [Google Scholar] [CrossRef]
- Motokawa, R.; Morishita, K.; Koizumi, S.; Nakahira, T.; Annaka, M. Thermosensitive Diblock Copolymer of Poly(N-isopropylacrylamide) and Poly(ethylene glycol) in Water: Polymer Preparation and Solution Behavior. Macromolecules 2005, 38, 5748–5760. [Google Scholar] [CrossRef]
- Lutz, J.; Weichenhan, K.; Akdemir, O.; Hoth, A. About the Phase Transitions in Aqueous Solutions of Thermoresponsive Copolymers and Hydrogels Based on 2-(2-methoxyethoxy)ethyl Methacrylate and Oligo(ethylene glycol) Methacrylate. Macromolecules 2007, 40, 2503–2508. [Google Scholar] [CrossRef]
- Casse, O.; Shkilnyy, A.; Linders, J.; Mayer, C.; Häussinger, D.; Völkel, A.; Thünemann, A.F.; Dimova, R.; Cölfen, H.; Meier, W.P.; et al. Solution Behavior of Double-Hydrophilic Block Copolymers in Dilute Aqueous Solution. Macromolecules 2012, 45, 4772–4777. [Google Scholar] [CrossRef]
- Schmidt, B.V.K.J. Double Hydrophilic Block Copolymer Self-Assembly in Aqueous Solution. Macromol. Chem. Phys. 2018, 219, 1700494. [Google Scholar] [CrossRef]
- Willersinn, J.; Schmidt, B.V.K.J. Self-Assembly of Double Hydrophilic Poly(2-ethyl-2-oxazoline)-b-poly(N-vinylpyrrolidone) Block Copolymers in Aqueous Solution. Polym. 2017, 9, 293. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Fernández, J.; Pérez-Juste, J.; Liz-Marzán, L.M.; Lang, P. Dynamic Light Scattering of Short Au Rods with Low Aspect Ratios. J. Phys. Chem. C 2007, 111, 5020–5025. [Google Scholar] [CrossRef]
- Jenkins, P.; Snowden, M. Depletion flocculation in colloidal dispersions. Adv. Colloid Interface Sci. 1996, 68, 57–96. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trinh Che, L.; Hiorth, M.; Hoogenboom, R.; Kjøniksen, A.-L. Complex Temperature and Concentration Dependent Self-Assembly of Poly(2-oxazoline) Block Copolymers. Polymers 2020, 12, 1495. https://doi.org/10.3390/polym12071495
Trinh Che L, Hiorth M, Hoogenboom R, Kjøniksen A-L. Complex Temperature and Concentration Dependent Self-Assembly of Poly(2-oxazoline) Block Copolymers. Polymers. 2020; 12(7):1495. https://doi.org/10.3390/polym12071495
Chicago/Turabian StyleTrinh Che, Loan, Marianne Hiorth, Richard Hoogenboom, and Anna-Lena Kjøniksen. 2020. "Complex Temperature and Concentration Dependent Self-Assembly of Poly(2-oxazoline) Block Copolymers" Polymers 12, no. 7: 1495. https://doi.org/10.3390/polym12071495
APA StyleTrinh Che, L., Hiorth, M., Hoogenboom, R., & Kjøniksen, A.-L. (2020). Complex Temperature and Concentration Dependent Self-Assembly of Poly(2-oxazoline) Block Copolymers. Polymers, 12(7), 1495. https://doi.org/10.3390/polym12071495