Characterization and Release Behavior of a Thiosemicarbazone from Electrospun Polyvinyl Alcohol Core-Shell Nanofibers




Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Characterization of the Polymer Solutions
2.3. Fabrication of TSC-Loaded Monolithic and Core–Shell PVA Fibers
2.4. Characterization of the Fibers
2.5. In Vitro Release
2.6. In Vitro Antibacterial Assay
2.7. In Vitro Cytotoxicity
3. Results and Discussion
3.1. Preparation and Characterization of the Fibers
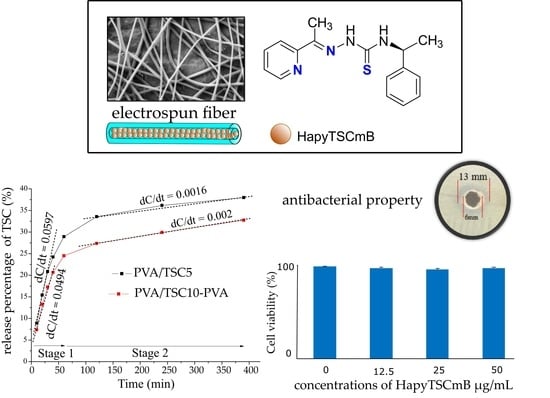
3.2. Release Studies
3.3. Growth Inhibition Studies–Minimal Inhibitory Concentration
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bahojb Noruzi, E.; Kheirkhahi, M.; Shaabani, B.; Geremia, S.; Hickey, N.; Asaro, F.; Nitti, P.; Kafil, H.S. Design of a thiosemicarbazide-functionalized Calix[4]arene ligand and related transition metal complexes: Synthesis, characterization, and biological studies. Front. Chem. 2019, 7, 663. [Google Scholar] [CrossRef]
- Bonaccorso, C.; Marzo, T.; La Mendola, D. Biological applications of thiocarbohydrazones and their metal complexes: A perspective review. Pharmaceuticals 2020, 13, 4. [Google Scholar] [CrossRef]
- Scarim, C.B.; Jornada, D.H.; Machado, M.G.M.; Ferreira, C.M.R.; dos Santos, J.L.; Chung, M.C. Thiazole, thio and semicarbazone derivatives against tropical infective diseases: Chagas disease, human African trypanosomiasis (HAT), leishmaniasis, and malaria. Eur. J. Med. Chem. 2019, 162, 378–395. [Google Scholar] [CrossRef] [PubMed]
- Pelosi, G. Thiosemicarbazone metal complexes: From structure to activity. Open Crystallogr. J. 2010, 3, 16–28. [Google Scholar] [CrossRef]
- de Siqueira, L.R.P.; de Moraes Gomes, P.A.T.; de Lima Ferreira, L.P.; de Melo Rêgo, M.J.B.; Leite, A.C.L. Multi-target compounds acting in cancer progression: Focus on thiosemicarbazone, thiazole and thiazolidinone analogues. Eur. J. Med. Chem. 2019, 170, 237–260. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Qiao, H.; Yang, F.; Zhou, W.; Gong, Y.; Zhang, X.; Wang, H.; Zhao, B.; Ma, L.; Liu, H.-M.; et al. Novel thiosemicarbazone derivatives containing indole fragment as potent and selective anticancer agent. Eur. J. Med. Chem. 2019, 184, 111764. [Google Scholar] [CrossRef]
- Akbari, A.; Ghateazadeh, H.; Takjoo, R.; Sadeghi-Nejad, B.; Mehrvar, M.; Mague, J.T. Synthesis & crystal structures of four new biochemical active Ni(II) complexes of thiosemicarbazone and isothiosemicarbazone-based ligands: In vitro antimicrobial study. J. Mol. Struct. 2019, 1181, 287–294. [Google Scholar] [CrossRef]
- Volynets, G.P.; Tukalo, M.A.; Bdzhola, V.G.; Derkach, N.M.; Gumeniuk, M.I.; Tarnavskiy, S.S.; Starosyla, S.A.; Yarmoluk, S.M. Benzaldehyde thiosemicarbazone derivatives against replicating and nonreplicating Mycobacterium tuberculosis. J. Antibiot. 2019, 72, 218–224. [Google Scholar] [CrossRef]
- Pingaew, R.; Prachayasittikul, S.; Ruchirawat, S. Synthesis, cytotoxic and antimalarial activities of benzoyl thiosemicarbazone analogs of isoquinoline and related compounds. Molecules 2010, 15, 988–996. [Google Scholar] [CrossRef]
- Pelosi, G.; Bisceglie, F.; Bignami, F.; Bignami, P.; Schiavone, P.; Re, M.C.; Casoli, C.; Pilotti, E. Antiretroviral activity of thiosemicarbazone metal complexes. J. Med. Chem. 2010, 53, 8765–8769. [Google Scholar] [CrossRef]
- Carradori, S.; Secci, D.; D’Ascenzio, M.; Chimenti, P.; Bolasco, A. Microwave and ultrasound-assisted synthesis of thiosemicarbazones and their corresponding (4,5-substituted-thiazol-2-yl)hydrazines. J. Heterocycl. Chem. 2014, 51, 1856–1861. [Google Scholar] [CrossRef]
- Klayman, D.L.; Scovill, J.P.; Bartosevich, J.F.; Bruce, J. 2-acetylpyridine thiosemicarbazones. 5. 1-[1-(2-Pyridyl)ethyl]-3-thiosemicarbazides as potential antimalarial agents. J. Med. Chem. 1983, 26, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Katz, E. Thiosemicarbazones: Inhibition of the growth of pox viruses and requirement for the growth of an isatin-β-thiosemicarbazone dependent mutant. J. Basic Clin. Physiol. Pharmacol. 1987, 6, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Kang, I.J.; Wang, L.W.; Hsu, T.A.; Yueh, A.; Lee, C.C.; Lee, Y.C.; Lee, C.Y.; Chao, Y.S.; Shih, S.R.; Chern, J.H. Isatin-β-thiosemicarbazones as potent herpes simplex virus inhibitors. Bioorganic Med. Chem. Lett. 2011, 21, 1948–1952. [Google Scholar] [CrossRef] [PubMed]
- Shipman, C.; Smith, S.H.; Drach, J.C.; Klayman, D.L. Antiviral activity of 2-acetylpyridine thiosemicarbazones against herpes simplex virus. Antimicrob. Agents Chemother. 1981, 19, 682–685. [Google Scholar] [CrossRef]
- Levinson, W.; Coleman, V.; Woodson, B.; Rabson, A.; Lanier, J.; Whitcher, J.; Dawson, C. Inactivation of herpes simplex virus by thiosemicarbazones and certain cations. Antimicrob. Agents Chemother. 1974, 5, 398–402. [Google Scholar] [CrossRef][Green Version]
- Genova, P.; Varadinova, T.; Matesanz, A.I.; Marinova, D.; Souza, P. Toxic effects of bis(thiosemicarbazone) compounds and its palladium(II) complexes on herpes simplex virus growth. Toxicol. Appl. Pharmacol. 2004, 197, 107–112. [Google Scholar] [CrossRef]
- Shipman, C.; Smith, S.H.; Drach, J.C.; Klayman, D.L. Thiosemicarbazones of 2-acetylpyridine, 2-acetylquinoline, 1-acetylisoquinoline, and related compounds as inhibitors of herpes simplex virus in vitro and in a cutaneous herpes guinea pig model. Antivir. Res. 1986, 6, 197–222. [Google Scholar] [CrossRef]
- Turk, S.R.; Shipman, C.; Drach, J.C. Structure-activity relationships among α-(N)-heterocyclic acyl thiosemicarbazones and related compounds as inhibitors of herpes simplex virus type 1-specified ribonucleoside diphosphate reductase. J. Gen. Virol. 1986, 67, 1625–1632. [Google Scholar] [CrossRef]
- Altun, A.; Kumru, M.; Dimoglo, A. The role of conformational and electronic parameters of thiosemicarbazone and thiosemicarbazide derivatives for their dermal toxicity. J. Mol. Struct. THEOCHEM 2001, 572, 121–134. [Google Scholar] [CrossRef]
- Altun, A.; Kumru, M.; Dimoglo, A. Study of electronic and structural features of thiosemicarbazone and thiosemicarbazide derivatives demonstrating anti-HSV-1 activity. J. Mol. Struct. THEOCHEM 2001, 535, 235–246. [Google Scholar] [CrossRef]
- Savjani, K.T.; Gajjar, A.K.; Savjani, J.K. Drug solubility: Importance and enhancement techniques. ISRN Pharm. 2012, 2012, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Xing, H.; Zhao, Y.; Ma, Z. Pharmaceutical dispersion techniques for dissolution and bioavailability enhancement of poorly water-soluble drugs. Pharmaceutics 2018, 10, 74. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, H.; Bisht, S.; Yadav, M.; Singh, V. Bioavailability enhancement: A review. Int. J. Pharma Bio Sci. 2011, 2, 202–216. [Google Scholar]
- Rao, V.M.; Sanghvi, R.; Zhu, H. Solubility of pharmaceutical solids. In Developing Solid Oral Dosage Forms; Elsevier: Amsterdam, The Netherlands, 2017; pp. 3–22. [Google Scholar] [CrossRef]
- Serajuddin, A.T.M. Solid dispersion of poorly water-soluble drugs: Early promises, subsequent problems, and recent breakthroughs. J. Pharm. Sci. 1999, 88, 1058–1066. [Google Scholar] [CrossRef]
- Tran, P.; Pyo, Y.C.; Kim, D.H.; Lee, S.E.; Kim, J.K.; Park, J.S. Overview of the manufacturing methods of solid dispersion technology for improving the solubility of poorly water-soluble drugs and application to anticancer drugs. Pharmaceutics 2019, 11, 132. [Google Scholar] [CrossRef]
- Xue, J.; Wu, T.; Dai, Y.; Xia, Y. Electrospinning and electrospun nanofibers: Methods, materials, and applications. Chem. Rev. 2019, 119, 5298–5415. [Google Scholar] [CrossRef]
- Islam, M.S.; Ang, B.C.; Andriyana, A.; Afifi, A.M. A review on fabrication of nanofibers via electrospinning and their applications. SN Appl. Sci. 2019, 1, 1248. [Google Scholar] [CrossRef]
- Barani, H. Antibacterial continuous nanofibrous hybrid yarn through in situ synthesis of silver nanoparticles: Preparation and characterization. Mater. Sci. Eng. C 2014, 43, 50–57. [Google Scholar] [CrossRef]
- Maleki, H.; Gharehaghaji, A.A.; Toliyat, T.; Dijkstra, P.J. Drug release behavior of electrospun twisted yarns as implantable medical devices. Biofabrication 2016, 8, 035019. [Google Scholar] [CrossRef]
- Singh, A.; Singh, N. Recent review on nanofiber for drug delivery systems. World J. Pharm. Res. 2017, 6, 611–631. [Google Scholar] [CrossRef]
- Pereira, E.D.; Cerruti, R.; Fernandes, E.; Peña, L.; Saez, V.; Pinto, J.C.; Ramón, J.A.; Oliveira, G.E.; de Souza Júnior, F.G. Influence of PLGA and PLGA-PEG on the dissolution profile of oxaliplatin. Polímeros 2016, 26, 137–143. [Google Scholar] [CrossRef]
- Vasita, R.; Mani, G.; Agrawal, C.M.; Katti, D.S. Surface hydrophilization of electrospun PLGA micro-/nano-fibers by blending with Pluronic® F-108. Polymer (Guildford) 2010, 51, 3706–3714. [Google Scholar] [CrossRef]
- Evrova, O.; Hosseini, V.; Milleret, V.; Palazzolo, G.; Zenobi-Wong, M.; Sulser, T.; Buschmann, J.; Eberli, D. Hybrid randomly electrospun poly(lactic-co-glycolic acid):poly(ethylene oxide) (PLGA:PEO) fibrous scaffolds enhancing myoblast differentiation and alignment. ACS Appl. Mater. Interfaces 2016, 8, 31574–31586. [Google Scholar] [CrossRef] [PubMed]
- Teo, W.-E.; Inai, R.; Ramakrishna, S. Technological advances in electrospinning of nanofibers. Sci. Technol. Adv. Mater. 2011, 12, 013002. [Google Scholar] [CrossRef] [PubMed]
- Pillay, V.; Dott, C.; Choonara, Y.E.; Tyagi, C.; Tomar, L.; Kumar, P.; du Toit, L.C.; Ndesendo, V.M.K. A review of the effect of processing variables on the fabrication of electrospun nanofibers for drug delivery applications. J. Nanomater. 2013, 2013, 789289. [Google Scholar] [CrossRef]
- Liechty, W.B.; Kryscio, D.R.; Slaughter, B.V.; Peppas, N.A. Peppas, polymers for drug delivery systems. Annu. Rev. Chem. Biomol. Eng. 2010, 1, 149–173. [Google Scholar] [CrossRef]
- Hu, X.; Liu, S.; Zhou, G.; Huang, Y.; Xie, Z.; Jing, X. Electrospinning of polymeric nanofibers for drug delivery applications. J. Control. Release 2014, 185, 12–21. [Google Scholar] [CrossRef]
- Vlachou, M.; Siamidi, A.; Kyriakou, S. Electrospinning and drug delivery. In Electrospinning and Electrospraying—Techniques and Applications; Intechopen: London, UK, 2019. [Google Scholar] [CrossRef]
- Yu, D.-G.; Li, J.-J.; Williams, G.R.; Zhao, M. Electrospun amorphous solid dispersions of poorly water-soluble drugs: A review. J. Control. Release 2018, 292, 91–110. [Google Scholar] [CrossRef]
- Imani, R.; Yousefzadeh, M.; Nour, S. Functional nanofiber for drug delivery applications. In Handbook of Nanofibers; Springer International Publishing: Cham, Switzerland, 2019. [Google Scholar]
- Haider, A.; Haider, S.; Kang, I.-K. A comprehensive review summarizing the effect of electrospinning parameters and potential applications of nanofibers in biomedical and biotechnology. Arab. J. Chem. 2018, 11, 1165–1188. [Google Scholar] [CrossRef]
- Akhgari, A.; Shakib, Z.; Sanati, S. A review on electrospun nanofibers for oral drug delivery. Nanomed. J. 2017, 4, 197–207. [Google Scholar] [CrossRef]
- Buzgo, M.; Mickova, A.; Rampichova, M.; Doupnik, M. Blend electrospinning, coaxial electrospinning, and emulsion electrospinning techniques. In Core-Shell Nanostructures for Drug Delivery and Theranostics; Woodhead Publishing: Sawston, UK, 2018; pp. 325–347. [Google Scholar] [CrossRef]
- Cornejo Bravo, J.M.; Villarreal Gómez, L.J.; Serrano-Medina, A. Electrospinning for drug delivery systems: Drug incorporation techniques. In Electrospinning—Material, Techniques, and Biomedical Applications; IntechOpen: London, UK, 2016. [Google Scholar] [CrossRef]
- Teixeira, M.A.; Amorim, M.T.P.; Felgueiras, H.P. Poly(vinyl alcohol)-based nanofibrous electrospun scaffolds for tissue engineering applications. Polymers (Basel) 2019, 12, 7. [Google Scholar] [CrossRef] [PubMed]
- Bhattarai, R.S.; Bachu, R.D.; Boddu, S.H.S.; Bhaduri, S. Biomedical applications of electrospun nanofibers: Drug and nanoparticle delivery. Pharmaceutics 2019, 11, 5. [Google Scholar] [CrossRef] [PubMed]
- Teodorescu, M.; Bercea, M.; Morariu, S. Biomaterials of poly(vinyl alcohol) and natural polymers. Polym. Rev. 2018, 58, 247–287. [Google Scholar] [CrossRef]
- Truong, Y.B.; Choi, J.; Mardel, J.; Gao, Y.; Maisch, S.; Musameh, M.; Kyratzis, I.L. Functional cross-linked electrospun polyvinyl alcohol membranes and their potential applications. Macromol. Mater. Eng. 2017, 302, 1700024. [Google Scholar] [CrossRef]
- Tang, X.; Alavi, S. Recent advances in starch, polyvinyl alcohol based polymer blends, nanocomposites and their biodegradability. Carbohydr. Polym. 2011, 85, 7–16. [Google Scholar] [CrossRef]
- Khalf, A.; Madihally, S.V. Recent advances in multiaxial electrospinning for drug delivery. Eur. J. Pharm. Biopharm. 2017, 112, 1–17. [Google Scholar] [CrossRef]
- Han, D.; Steckl, A.J.; Han, D. Coaxial electrospinning formation of complex polymer fibers and their applications. ChemPlusChem 2019, 84, 1453–1497. [Google Scholar] [CrossRef]
- Qu, H.; Wei, S.; Guo, Z. Coaxial electrospun nanostructures and their applications. J. Mater. Chem. A 2013, 38, 11513–11528. [Google Scholar] [CrossRef]
- Lu, Y.; Huang, J.; Yu, G.; Cardenas, R.; Wei, S.; Wujcik, E.K.; Guo, Z. Coaxial electrospun fibers: Applications in drug delivery and tissue engineering. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnology 2016, 8, 654–677. [Google Scholar] [CrossRef]
- Pant, B.; Park, M.; Park, S.-J. Drug Delivery Applications of Core-Sheath Nanofibers Prepared by Coaxial Electrospinning: A Review. Pharmaceutics 2019, 11, 305. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.-F.; Carson, D.; Woodrow, K.A. Current strategies for sustaining drug release from electrospun nanofibers. J. Control. Release 2015, 220, 584–591. [Google Scholar] [CrossRef] [PubMed]
- Komiya, S.; Otsuka, E.; Hirashima, Y.; Suzuki, A. Salt effects on formation of microcrystallites in poly(vinyl alcohol) gels prepared by cast-drying method. Prog. Nat. Sci. Mater. Int. 2011, 21, 375–379. [Google Scholar] [CrossRef]
- Rodríguez-Argüelles, M.C.; Tourón-Touceda, P.; Cao, R.; García-Deibe, A.M.; Pelagatti, P.; Pelizzi, C.; Zani, F. Complexes of 2-acetyl-γ-butyrolactone and 2-furancarbaldehyde thiosemicarbazones: Antibacterial and antifungal activity. J. Inorg. Biochem. 2009, 103, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Barani, H.; Montazer, M.; Samadi, N.; Toliyat, T. Nano silver entrapped in phospholipids membrane: Synthesis, characteristics and antibacterial kinetics. Mol. Membr. Boil. 2011, 28, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, C. Advanced Nanofibrous Materials Manufacturing Technology Based on Electrospinning; CRC Press, Taylor & Francis Group: Boca Raton, FL, USA, 2019. [Google Scholar]
- Angammana, C.J.; Jayaram, S.H. Analysis of the Effects of Solution Conductivity on Electrospinning Process and Fiber Morphology. IEEE Trans. Ind. Appl. 2011, 47, 1109–1117. [Google Scholar] [CrossRef]
- Elkasaby, M.; Hegab, H.A.; Mohany, A.; Rizvi, G.M. Modeling and optimization of electrospinning of polyvinyl alcohol (PVA). Adv. Polym. Tech. 2018, 37, 2114–2122. [Google Scholar] [CrossRef]
- Rwei, S.-P.; Huang, C.-C. Electrospinning PVA solution-rheology and morphology analyses. Fibers Polym. 2012, 13, 44–50. [Google Scholar] [CrossRef]
- Okutan, N.; Terzi, P.; Altay, F. Affecting parameters on electrospinning process and characterization of electrospun gelatin nanofibers. Food Hydrocoll. 2014, 39, 19–26. [Google Scholar] [CrossRef]
- Yoon, Y.I.; Park, K.E.; Lee, S.J.; Park, W.H. Fabrication of microfibrous and nano-/microfibrous scaffolds: Melt and hybrid electrospinning and surface modification of poly(L-lactic acid) with plasticizer. Biomed. Res. Int. 2013, 2013, 309048. [Google Scholar] [CrossRef]
- Dasdemir, M.; Topalbekiroglu, M.; Demir, A. Electrospinning of thermoplastic polyurethane microfibers and nanofibers from polymer solution and melt. J. Appl. Polym. Sci. 2013, 127, 1901–1908. [Google Scholar] [CrossRef]
- Nazari, T.; Garmabi, H. The effects of processing parameters on the morphology of PLA/PEG melt electrospun fibers. Polym. Int. 2018, 67, 178–188. [Google Scholar] [CrossRef]
- Kim, G.-M. Fabrication of bio-nanocomposite nanofibers mimicking the mineralized hard tissues via electrospinning process. In Nanofibers; IntechOpen: London, UK, 2010. [Google Scholar] [CrossRef]
- Wiśniewska, M.; Chibowski, S.; Urban, T.; Sternik, D. Investigation of the alumina properties with adsorbed polyvinyl alcohol. J. Therm. Anal. Calorim. 2011, 103, 329–337. [Google Scholar] [CrossRef]
- Fathi, E.; Atyabi, N.; Imani, M.; Alinejad, Z. Physically crosslinked polyvinyl alcohol–dextran blend xerogels: Morphology and thermal behavior. Carbohydr. Polym. 2011, 84, 145–152. [Google Scholar] [CrossRef]
- Jose, T.; George, S.C.; Maya, M.G.; Maria, H.J.; Wilson, R.; Thomas, S. Effect of bentonite clay on the mechanical, thermal, and pervaporation performance of the poly(vinyl alcohol) nanocomposite membranes. Ind. Eng. Chem. Res. 2014, 53, 16820–16831. [Google Scholar] [CrossRef]
- Thitiwongsawet, P.; Supaphol, P. Carbendazim-loaded electrospun poly(vinyl alcohol) fiber mats and release characteristics of carbendazim therefrom. Polym. Adv. Technol. 2011, 22, 1366–1374. [Google Scholar] [CrossRef]
- Wen, P.; Wen, Y.; Zong, M.-H.; Linhardt, R.J.; Wu, H. Encapsulation of bioactive compound in electrospun fibers and its potential application. J. Agric. Food Chem. 2017, 65, 9161–9179. [Google Scholar] [CrossRef]
- Korbag, I.; Saleh, S.M. Studies on the formation of intermolecular interactions and structural characterization of polyvinyl alcohol/lignin film. Int. J. Environ. Stud. 2016, 73, 226–235. [Google Scholar] [CrossRef]
- Negim, E.-S.; Bekbayeva, L.; Adam, H.; Yeligbayeva, G.; Ganjian, E.; Saleh, M.; Saad, B. The effect of blend ratios on physico-mechanical properties and miscibility of cross-linked poly(vinyl alcohol)/urea blends. J. Phys. Conf. Ser. 2018, 1123, 012066. [Google Scholar] [CrossRef]
- Sakurada, I. Polyvinyl Alcohol Fibers; Marcel Dekker: New York, NY, USA; Basel, Switzerland, 1985; p. 78. [Google Scholar]
- Moritani, T.; Kuruma, I.; Shibatani, K.; Fujiwara, Y. Tacticity of poly(vinyl alcohol) studied by nuclear magnetic resonance of hydroxyl protons. Macromolecules 1972, 5, 577–580. [Google Scholar] [CrossRef]
- Mansur, H.S.; Sadahira, C.M.; Souza, A.N.; Mansur, A.A.P. FTIR spectroscopy characterization of poly (vinyl alcohol) hydrogel with different hydrolysis degree and chemically crosslinked with glutaraldehyde. Mater. Sci. Eng. C 2008, 28, 539–548. [Google Scholar] [CrossRef]
- Usha; Chandra, S. Pd(II), Pt(II), Rh(III), Ir(III) and Ru(III) complexes of n-pentyl and n-hexyl ketone thiosemicarbazones. Synth. React. Inorg. Met. Chem. 1992, 22, 1565–1579. [Google Scholar] [CrossRef]
- Ferraz, K.S.O.; Silva, N.F.; Da Silva, J.G.; Speziali, N.L.; Mendes, I.C.; Beraldo, H. Structural studies on acetophenone- and benzophenone-derived thiosemicarbazones and their zinc(II) complexes. J. Mol. Struct. 2012, 1008, 102–107. [Google Scholar] [CrossRef]
- Offiong, O.E. Synthesis and spectral studies of platinum metal complexes of benzoin thiosemicarbazone. Spectrochim. Acta Part A Mol. Spectrosc. 1994, 50, 2167–2175. [Google Scholar] [CrossRef]
- Huang, X.; Brazel, C.S. On the importance and mechanisms of burst release in matrix-controlled drug delivery systems. J. Control. Release 2001, 73, 121–136. [Google Scholar] [CrossRef]
- Mahmud, M.M.; Zaman, S.; Perveen, A.; Jahan, R.A.; Islam, M.F.; Arafat, M.T. Controlled release of curcumin from electrospun fiber mats with antibacterial activity. J. Drug Deliv. Sci. Technol. 2020, 55, 101386. [Google Scholar] [CrossRef]
- Ritger, P.L.; Peppas, N.A. A simple equation for description of solute release I. Fickian and non-Fickian release from non-swellable devices in the form of slabs, spheres, cylinders or discs. J. Control. Release 1987, 5, 23–36. [Google Scholar] [CrossRef]
- Natu, M.V.; de Sousa, H.C.; Gil, M.H. Effects of drug solubility, state and loading on controlled release in bicomponent electrospun fibers. Int. J. Pharm. 2010, 397, 50–58. [Google Scholar] [CrossRef]
- Sun, Y.; Cheng, S.; Lu, W.; Wang, Y.; Zhang, P.; Yao, Q. Electrospun fibers and their application in drug controlled release, biological dressings, tissue repair, and enzyme immobilization. RSC Adv. 2019, 9, 25712–25729. [Google Scholar] [CrossRef]
- Calatayud, D.G.; López-Torres, E.; Mendiola, M.A. Diphenyllead(IV) chloride complexes with benzilthiosemicarbazones. The first bis(thiosemicarbazone) derivatives. Inorg. Chem. 2007, 46, 10434–10443. [Google Scholar] [CrossRef]
- Khan, S.A.; Asiri, A.M.; Al-Amry, K.; Malik, M.A. Synthesis, characterization, electrochemical studies, and in vitro antibacterial activity of novel thiosemicarbazone and its Cu(II), Ni(II), and Co(II) complexes. Sci. World J. 2014, 2014, 592375. [Google Scholar] [CrossRef]
- Pahontu, E.; Julea, F.; Rosu, T.; Purcarea, V.; Chumakov, Y.; Petrenco, P.; Gulea, A. Antibacterial, antifungal and in vitro antileukaemia activity of metal complexes with thiosemicarbazones. J. Cell. Mol. Med. 2015, 19, 865–878. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Code | Method b | HapyTSCmB Content (wt% Relative to PVA) | |||
|---|---|---|---|---|---|
| Fibers c | Spinning Solutions | ||||
| core | shell | core | shell | ||
| PVA | Method II/monolithic | 0 | 0 | PVA | PVA |
| PVA/TSC5 | Method II/monolithic | 5 | 5 | PVA/TSC-5% | PVA/TSC-5% |
| PVA/TSC10-PVA | Method I/core-shell | 10 | 0 | PVA/TSC-10% | PVA |
| Sample | ||||||||
|---|---|---|---|---|---|---|---|---|
| Phase I R2 | Phase II R2 | Phase I R2 | Phase II R2 | Phase I R2 | Phase II R2 | Phase I R2 | Phase II R2 | |
| PVA/TSC5 | 0.71 | 0.85 | 0.83 | 0.86 | 0.98 | 0.88 | 0.99 | 0.97 |
| n = 0.67 | n = 0.14 | |||||||
| PVA/TSC10-PVA | 0.72 | 0.96 | 0.72 | 0.97 | 0.98 | 0.98 | 0.98 | 0.99 |
| n = 0.68 | n = 0.15 | |||||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barani, H.; Khorashadizadeh, M.; Haseloer, A.; Klein, A. Characterization and Release Behavior of a Thiosemicarbazone from Electrospun Polyvinyl Alcohol Core-Shell Nanofibers. Polymers 2020, 12, 1488. https://doi.org/10.3390/polym12071488
Barani H, Khorashadizadeh M, Haseloer A, Klein A. Characterization and Release Behavior of a Thiosemicarbazone from Electrospun Polyvinyl Alcohol Core-Shell Nanofibers. Polymers. 2020; 12(7):1488. https://doi.org/10.3390/polym12071488
Chicago/Turabian StyleBarani, Hossein, Mohsen Khorashadizadeh, Alexander Haseloer, and Axel Klein. 2020. "Characterization and Release Behavior of a Thiosemicarbazone from Electrospun Polyvinyl Alcohol Core-Shell Nanofibers" Polymers 12, no. 7: 1488. https://doi.org/10.3390/polym12071488
APA StyleBarani, H., Khorashadizadeh, M., Haseloer, A., & Klein, A. (2020). Characterization and Release Behavior of a Thiosemicarbazone from Electrospun Polyvinyl Alcohol Core-Shell Nanofibers. Polymers, 12(7), 1488. https://doi.org/10.3390/polym12071488