The DOE was performed to study the influence of
S, VTMS co-precursor amount and KP amount on three key properties, the bulk density (
ρb), thermal conductivity (
k) and Young’s modulus (
YM), in order to identify guidelines for the synthesis conditions. The systems were also characterized by other techniques to obtain additional information about their chemical, structural-morphological, thermal and mechanical features. The prepared aerogels with different combinations of parameters are listed in
Table 1, together with their main properties.
3.1. Design of Experiments (DOE)
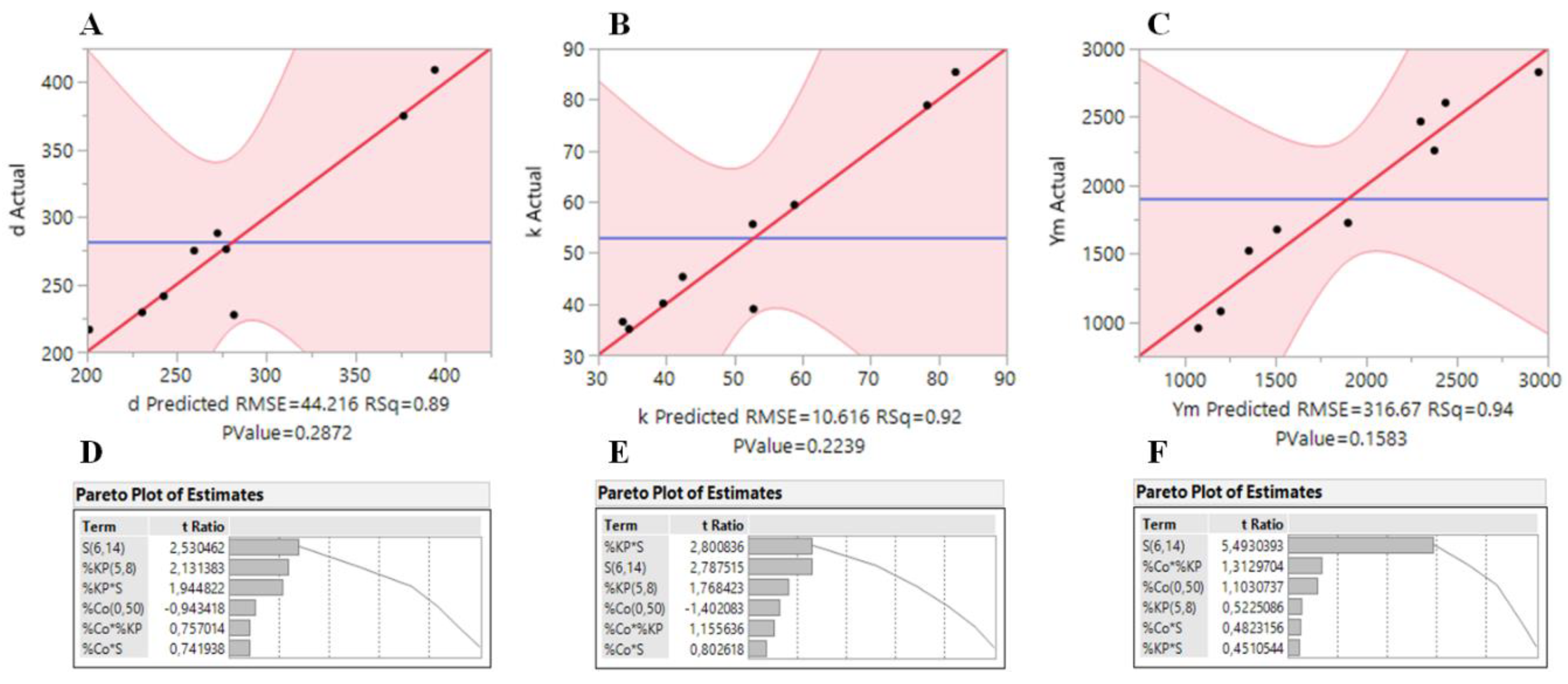
In order to confirm the accuracy of the models, plots of the real responses (bulk density, thermal conductivity and Young’s modulus) of the samples versus the predicted responses are presented in
Figure 1A–C. In general, the models fit well to the data, with
R2 values of 0.89, 0.92 and 0.94 for
ρb,
k and
YM, respectively. Pareto plots of the estimates obtained for the responses are also shown in
Figure 1D–F, where it can be seen that the
S factor has the highest influence in the responses. However, all the factors appear to have a non-negligible effect in the obtained responses, which makes the system much more complex.
For a deeper understanding of the system, surface responses were obtained using a central composite test design model. The surface responses for bulk density, thermal conductivity and Young’s modulus are represented in
Figure 2, but taking only the most influent factors.
The factors that show major influence on the bulk density are % CoP and
S, in agreement with previous studies [
23,
24]. The surface response indicates that the lowest values for bulk density are obtained for the following most favorable conditions: % CoP = 20, % KP = 8 and
S = 6.
A similar trend was observed for thermal conductivity, which is also mostly influenced by the % CoP and S. The response surface graph of thermal conductivity showed that for the lowest value of k, the optimum values for % CoP, % KP and S are 24, 8 and 6, respectively.
Regarding the mechanical behavior, the most dominant factor for the YM is S. In fact, the model indicates that, with an increase in S value, the YM also increases. The lowest value was achieved using % CoP = 19, % KP = 6.5 and S = 6.
Therefore, an optimum aerogel composite is likely to be obtained in the following range of conditions: % CoP = 19–24, % KP = 6.5–8 and
S = 6. In fact, the results in
Table 1 are in good agreement with this trend.
The desirability function method, already used for aerogel samples [
21], was applied here in order to obtain the global minimum value for density, thermal conductivity and Young’s modulus simultaneously, and the weights of 0.4, 0.4 and 0.2 for these responses were adopted, respectively (giving less significance to the mechanical properties). Under these circumstances, the optimum preparation conditions were found to be % CoP = 18, % KP = 7.2 and
S = 6, corresponding to the following optimum response values:
ρb = 0.173 g·cm
−3,
k = 27.6 mW·m
−1·K
−1 and
YM = 1.1 MPa.
3.2. Chemical Characterisation
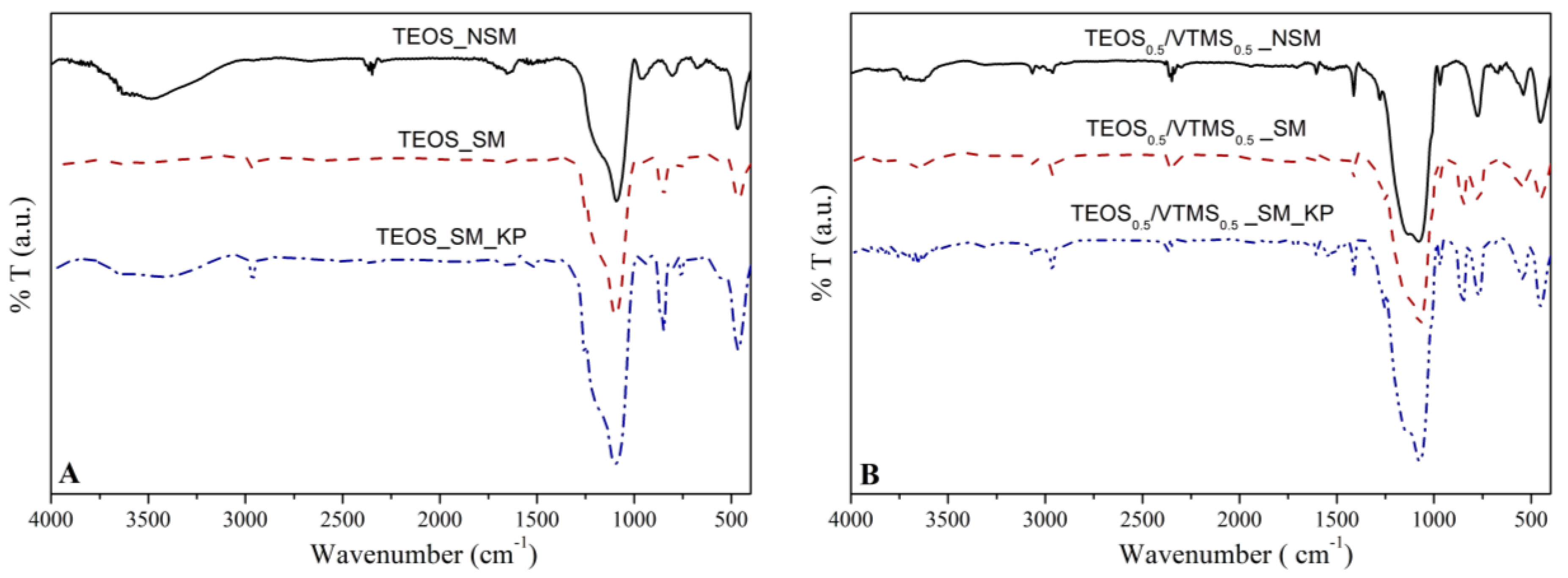
FTIR analysis. This analysis allowed to observe the constituents of the network, as well as to evaluate the success of surface modification. For all spectra (
Figure 3), the strong Si–O–Si vibration bands near 460 cm
−1 (deformation), 788 cm
−1 (symmetric stretching) and 1084 cm
−1 (asymmetric stretching) confirm the SiO
2 network [
25]. In
Figure 3B, the presence of VTMS was confirmed by the appearance of the following bands: (i) at 540 cm
−1, due to the twisting vibration of H derived from vinyl groups, (ii) at 1412 cm
−1, which corresponds to the deformation vibration of =C–H groups, (iii) at 1605 cm
−1 from the stretching vibration of C=C, and (iv) between 3000 to 3070 cm
−1, attributed to the stretching of =C–H groups [
7,
25].
After surface modification with HMDZ, the bands related to –OH groups became weaker, indicating an increase in the hydrophobic character of the nanocomposites. It is also perceptible a higher intensity of the bands at 2800–3000 cm−1 and at ca. 1250–1280 cm−1 (stretching and deformation vibrations of –C–H groups, respectively) and the appearance of the absorption band at 848 cm−1, which results from the stretching of the Si−CH3 bonds. This indicates that the aerogel nanocomposites have been efficiently modified.
Three main bands are observed due to the presence of KP at: ca. 3330 cm
−1, which is assigned as the –NH– stretching vibration; 1650 cm
−1, representing the stretching of –C=O bonds; and 1542 cm
−1, which is characteristic of the –NH– bending [
26].
Contact angle measurements. To evaluate the wettability of the prepared composites, the water contact angles (WCA) were measured for samples in different conditions. The obtained WCA are reported in
Table 1. It is well known that the hydrophobicity of a sample is strongly dependent on the chemical properties of the surface [
27,
28]. Since the samples were subjected to a surface modification step with a silylation agent (HMDZ), the modified materials are hydrophobic or even superhydrophobic in some cases, with contact angles ranging from 137° to 156°. After silylation, the hydroxyl groups were replaced by methyl groups in the silica surface, which leads to an increase in the hydrophobic character, by reducing the surface free energy. The surface modification was also confirmed by FTIR analysis (
Figure 3), as already discussed. The hydrophobic character ensures a high durability of the materials when subjected to atmospheric moisture [
1].
3.3. Structural and Morphological Features
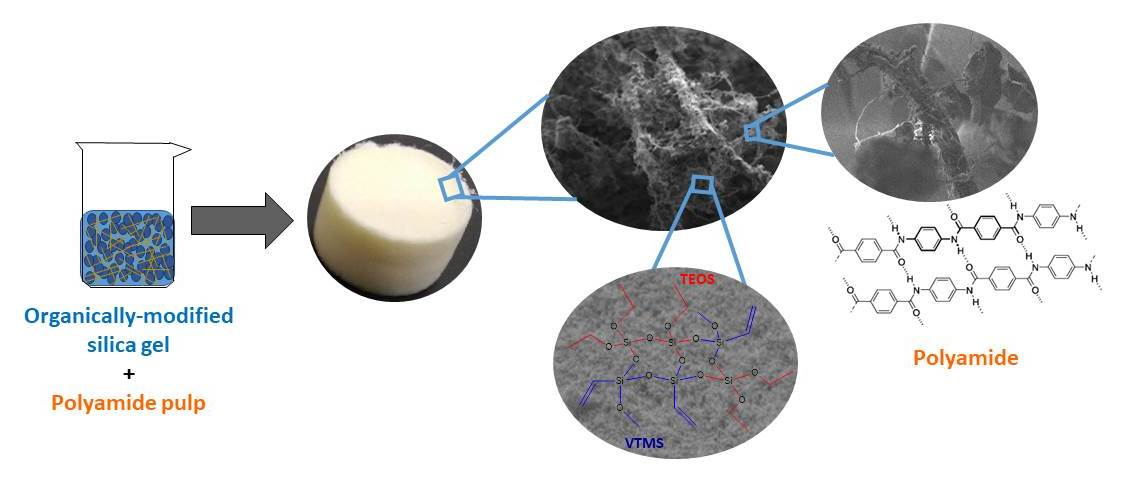
SEM analysis. The morphology and microstructure of the aerogel nanocomposites were analyzed by SEM. The micrographs of different configurations are shown in
Figure 4. All samples exhibited an interconnected three-dimensional matrix (
Figure 4A–D). In
Figure 4A,B, a comparison between TEOS-based aerogel (
S = 10) before and after surface modification is presented. After silylation (
Figure 4B), the structure is more porous and less agglomerates were formed, due to the Si–(CH
3)
3 groups attached on the surface of the nanocomposites. By decreasing the
S value (
S = 6,
Figure 4C), a less porous network was obtained, as expected. The introduction of VTMS as a co-precursor in the matrix (
Figure 4D) led to a smooth and regular network, revealing less heterogeneity of the medium during gel formation, which may be due to a higher affinity of this matrix to KP fibers, compared to the system only with TEOS. Thus, the type of precursor, the solvent/Si molar ratio (
S) and surface modification have a significant influence on the microstructure of the samples.
In
Figure 4E,F, it is possible to observe that KP fibers form a reinforcing network that is covered by the silica matrix, in this way providing monolithicity to the aerogels. In samples prepared with lower
S value (
Figure 4F), a better coverage of the fibers by the silica phase is observed, as expected.
Linear shrinkage. Shrinkage of the samples occurred both in diameter and height, since the pulp in the matrix has a random orientation. The diameter of the samples was measured after ageing/washing/surface modification and after drying. The total shrinkage values varied between 14.4% and 39.7% (
Table 1). However, in general, main part of shrinkage (10%–20%) was observed after solvent exchange and surface modification steps (
Table S1). This happens due to a swelling/de-swelling phenomenon that KP suffers during the synthesis protocol of aerogels. First, when dispersed in ethanol, the KP swells. After solvent exchange, when ethanol is replaced by heptane, the phenomenon reverses. The swelling occurs because ethanol may easily form hydrogen bonds with amine groups of the Kevlar
® pulp and the apolar tail of ethanol increases the space between the fibers and decreases their interconnection. During the washing step, ethanol is removed and exchanged by heptane, a non-polar solvent that is not able to maintain the hydrogen bridges. Thus, de-swelling of pulp occurs, leading to a decrease in the gel volume.
The linear shrinkage after drying decreased with the increase in KP amount from 5 to 8%wt., in the case of TEOS-based aerogels. With the introduction of VTMS, the shrinkage is almost independent of KP amount. In all cases, the linear shrinkage increases with increase in the
S value, as well as with increase in the VTMS content (
Table 1). This may be explained by the less interconnected silica network with higher
S, which results in lower strength of the gel to withstand the drying stresses. The effect of VTMS in the shrinkage may also be explained by the less connectivity of the network with the increase in VTMS/TEOS ratio, since VTMS has one non-hydrolysable group.
Bulk density. The influence of the Kevlar
® pulp concentration,
S ratio and the ratio of co-precursors on the bulk density was investigated, as mentioned in the DOE, being the results presented in
Table 1. Generally, a lower bulk density was obtained for the systems with
S = 6, reaching a value of 0.208 g·m
−3 (the lowest) for the TEOS
0.75/VTMS
0.25 aerogel with 6.5% KP. By increasing the
S from 6 to 10,
ρb slightly increases in the case of the aerogels containing only TEOS and those with smaller amount of VTMS (TEOS
0.75/VTMS
0.25). A further increase in
S led to additional increase in bulk density values for all systems. Moreover, by introduction of a higher amount of VTMS in the aerogel matrix (TEOS
0.5/VTMS
0.5), an increase in the bulk density value was observed. The highest bulk density value was obtained for the TEOS
0.5/VTMS
0.5 system with
S = 14.
Hence, it can be concluded that the increase in KP concentration leads to a decrease in the bulk density, certainly due to the higher support provided by the fibers. On the other hand, it was observed that the increase in S value, as well as the increase in VTMS amount, led to higher bulk density values, in agreement with shrinkage results.
Skeletal density, specific surface area and porosity. These properties were studied for the samples used for the initial DOE analysis, but only for KP = 8% (due to lower bulk density observed).
The obtained values of the skeletal density of the selected aerogel samples are shown in
Table 2; they varied between 1.13 and 1.48 g·cm
−3 and are in agreement with those in literature for similar organically-modified aerogel systems [
7]. The porosities, obtained from Equation (3), are in the interval 81%–84.5%, with only one exception of a sample with 2-fold higher bulk density (
Table 2; sample TEOS
0.5/VTMS
0.5,
S = 14 and KP = 8%).
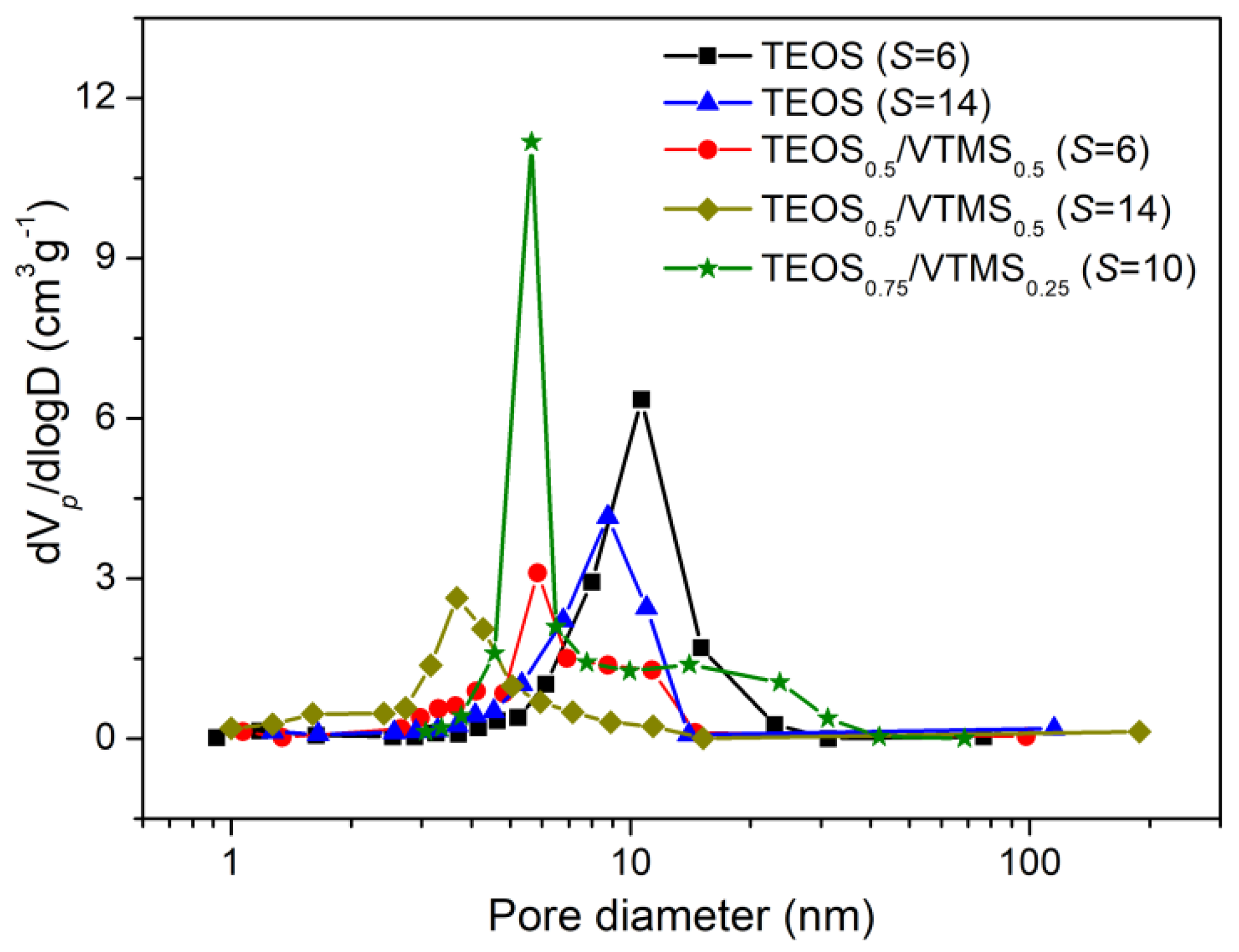
The specific surface areas and the pore size distributions of the prepared aerogels were evaluated from the nitrogen adsorption/desorption isotherms (
Figure S3), and their values are presented in
Table 2 and
Figure 5. Notice that the average pore sizes in
Table 2 were obtained from Equation (5) and not from N
2 adsorption/desorption, in order to include all pore sizes.
As observed from
Figure S3, the shape of the adsorption/desorption isotherms followed the type IV of IUPAC classification, characteristic of mesoporous materials, recognized by their hysteresis loop. Adsorption/desorption hysteresis shape of the aerogels corresponds approximately to H2 type of pores, indicating bottleneck-like pores. This is more evident in the samples with higher pore volume. The H2 type of pores is typical of disordered porous networks, with irregular pore shapes and complex interconnectivity.
The specific surface areas of the TEOS-based aerogels (~450 m
2·g
−1) were lower than those of TEOS-based aerogels synthesized in methanol [
29], or synthesized in ethanol but that were silylated with TMCS [
13,
17,
18,
30], which is due to the more favorable synthesis compounds in the latter. However, these compounds (methanol and TMCS) represent more hazard for the operators and environment. The introduction of VTMS as a co-precursor leads to a slight increase in the surface area for
S = 6 (
Table 2), and for
S = 14 the values are much higher (~615 m
2·g
−1), comparable with those for a similar TEOS/VTMS system [
7]. The increase in surface area for the TEOS/VTMS-based aerogels may be explained by the higher amount of mesopores, since when vinyl groups were introduced in the aerogels, a reduction in the average pore diameter was observed (
Table 2,
Figure 5), probably due to more interaction between the KP and the organically modified matrix and to the higher shrinkage already discussed.
3.4. Thermal Characterisation
Thermal conductivity. The evolution of the thermal conductivity is in agreement with the trend of bulk density (
Table 1)—generally both values decrease when using higher aramid pulp amount and lower
S value. By increasing the
S to 10, for the same KP amount, 6.5%, it is clear that the introduction of a small amount of VTMS in the aerogel matrix led to a decrease in 13.8% of the thermal conductivity, probably due to the reduction in pore size; a further increase in VTMS content contributes to almost double the
k value for the TEOS
0.5/VTMS
0.5 sample, justified by an obvious dominant effect of the increase in bulk density.
The larger samples were measured with a sensor with higher radius, and the obtained thermal conductivities are significantly lower than the ones obtained by the smaller sensor (
Table 1). The results of the larger sensor (5501) are more representative of the thermal conductivity of these samples, since they are assessed considering a larger volume for heat transfer. It is expected that for samples even larger, with size compatible with Guarded Hot Plate measurements (negligible boundary effect), the thermal conductivity may decrease further. Future work of scale-up of these aerogels will be performed to confirm this hypothesis.
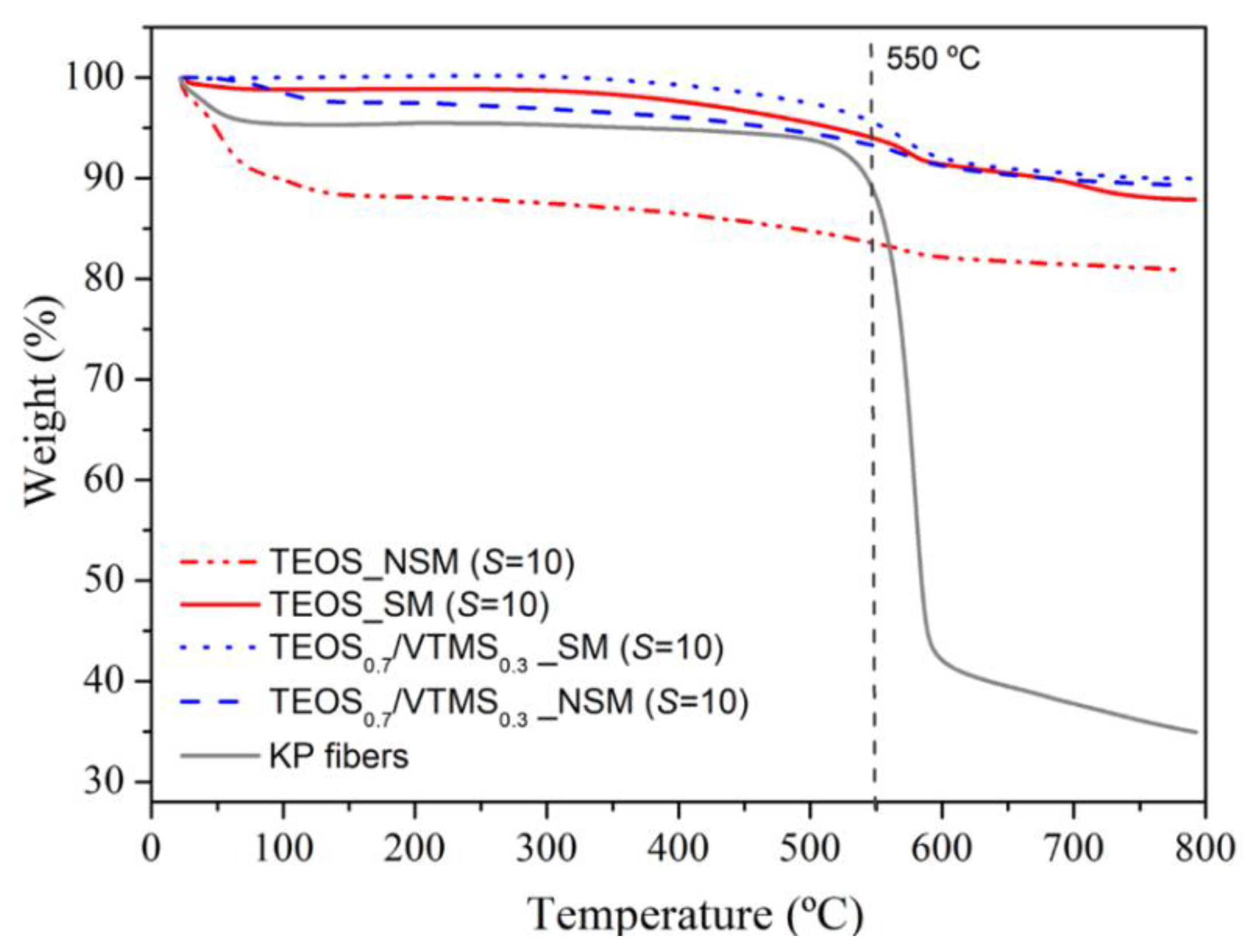
Thermal stability. In order to assess the thermal stability of the KP-reinforced silica aerogel composites, a thermogravimetric analysis from room temperature to 800 °C was carried out under nitrogen atmosphere (
Figure 6;
Table S2). The first weight loss (~9%) of the non-silylated sample starts at room temperature and is due to adsorbed water and residual solvents/by-products (
Tonset = 23.0 °C). It is noteworthy that in the case of samples containing VTMS this loss is much smaller (~2.3%) due to the greater hydrophobicity provided by the vinyl groups, even without silylation. The second mass loss is observed from ca. 350 up to 700 °C (
Tonset = 450 °C), in an earlier stage attributed to the loss of structural hydroxyl groups and, in a later stage, to the thermal decomposition of the vinyl groups derived from VTMS co-precursor, decomposition of methyl groups attached to the silica surface after silylation, and decomposition of KP. After this phenomenon, it is expected the loss of the hydrophobic nature of the modified aerogel, as a result of the decomposition of methyl/vinyl groups. The final sample mass percentage was high (80%–90%) and is attributed to carbon and silica that remain in the crucible. In general, the prepared aerogel composites can be used at high temperature, up to 450–550 °C, for considerable periods of time, without significant thermal degradation and maintaining their size/monolithicity (
Figure S4). In the case of TEOS/VTMS samples with surface modification, the thermal stability is higher, up to 550 °C (
Table S2).
3.5. Mechanical Characterisation
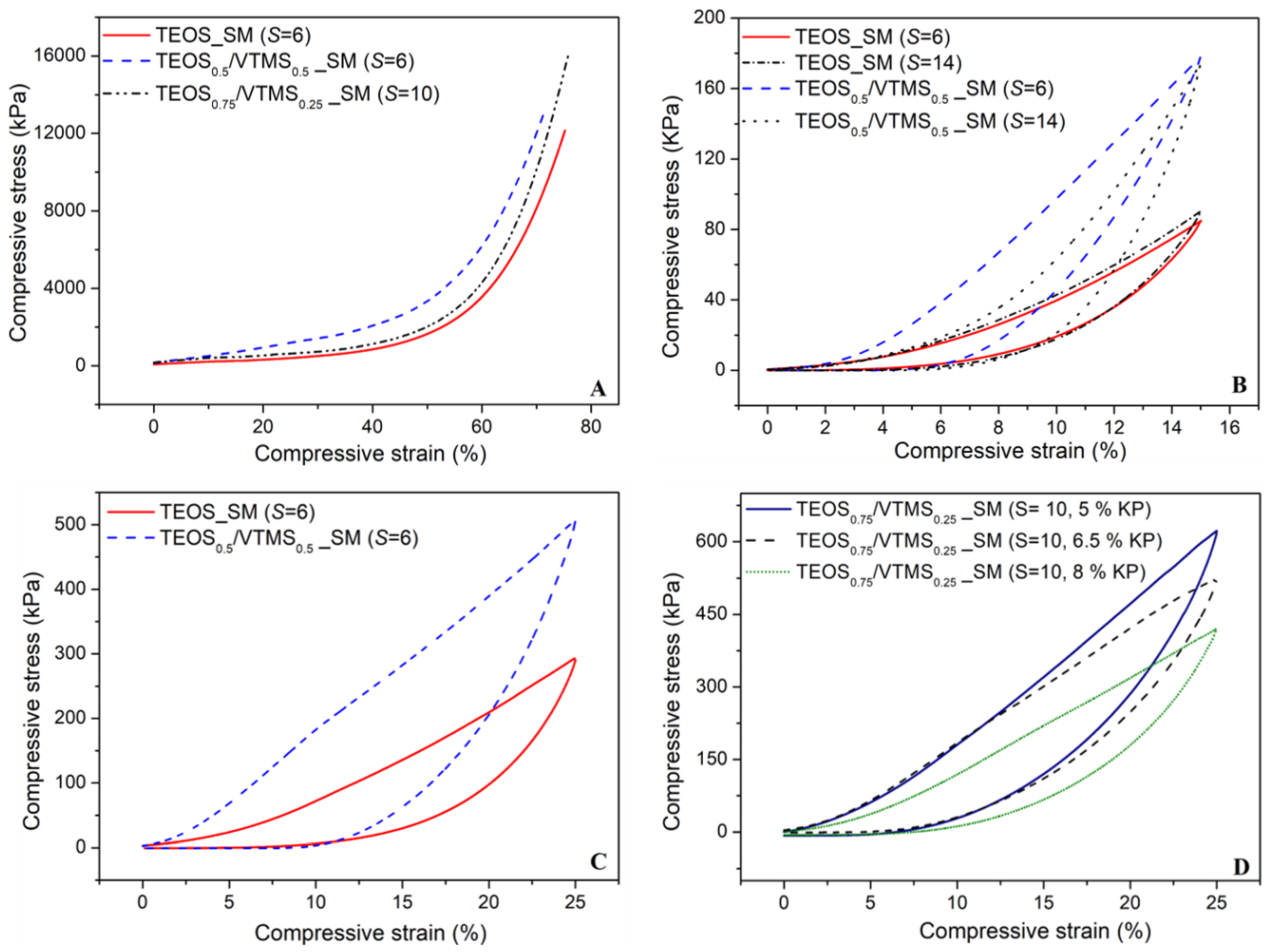
The mechanical behavior of the silica aerogel composites reinforced with KP was assessed by uniaxial compression-decompression tests, consisting in loading and unloading of the samples with strain between 0% and 15% with a load cell of 50 N and between 0% and 25% with a load cell of 3 kN. A destructive test was also performed with the cell of 3 kN, up to the maximum allowed force in order to describe the mechanical behavior of the samples. The results of compressive stress versus strain obtained from the latter referred test is illustrated in
Figure 7A for different samples. The curves can be divided essentially in three regions: linear stage, yielding stage and densification stage (elastic–plastic stage). In the beginning of linear stage (until ~6%), the compression curve slope is almost unchanged and the main bearing support during this phase corresponds to the open pores and the elastic bending of the KP fibers and pore walls. During the yielding stage (6%–40% strain), an increase in the stress is observed. The fibers in the aerogel matrix play the role of supporting skeleton, dispersing and transferring the external force to the whole sample and avoiding the collapse of the structure. This increase can also result from the elastic buckling of the pore walls [
7,
17]. In the final stage (>50% strain), the slope of the stress–strain curve increases significantly, due to the densification of the porous structure and correspondent increase in stiffness of the compressed specimen. Despite there being no expected chemical bonds between fibers and the silica matrix, the large interfacial adhesion increases the van der Waals’ force between the two phases [
13]. The dispersion of KP fibers in the silica matrix is multidirectional and uniform, thus the external stress is transferred to the whole composite, avoiding stress concentration and being able to stand a larger external stress. As a consequence, the KP-reinforced aerogel composites can support higher compressive stresses when compared with composites reinforced with larger fibers [
17].
The Young’s modulus was evaluated from the yielding stress–strain region and the values are presented in
Table 1. Generally, the
YM value increased with increase in
S value, thus more flexible aerogel composites can be achieved at lower
S values, which is in agreement with the bulk density decrease. In addition, the increase in VTMS amount in the matrix led to more rigid aerogel samples. Beside the effect of bulk density, this trend comes also probably from the interaction between vinyl group of the silica network with the aromatic ring of aramid via their pi-systems [
31]. Regarding the fibers content, the influence on
YM is not clear, although it has a tendency to decrease with the increase in the wt. % of KP, related to the decrease in bulk density.
The mechanical properties of the obtained aerogels are important to select the most suitable material for large volume applications. For example, good elasticity is required in Space applications (e.g., suits, pipes and panels) for adaptation to large bending. For this reason, the capacity of these materials to recover to their original shapes during unloading is very important. Thus, recovery tests were performed (
Figure 7C,D) and the results indicate that in general, after 25% strain, the samples restore up to 77% their shape.
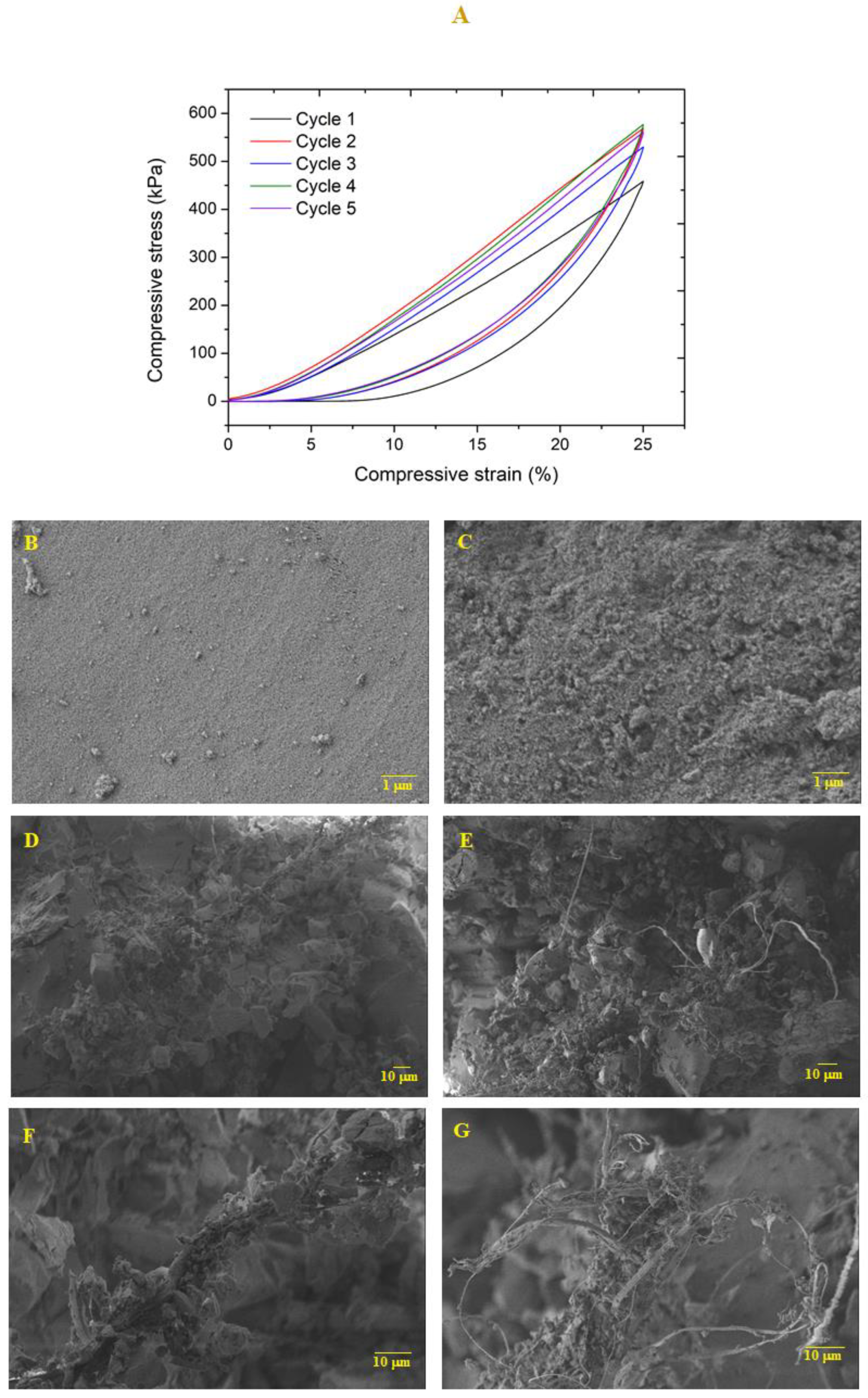
In order to evaluate the capacity of the nanocomposites to withstand dimensional loads, axial cyclic compression tests (5 cycles) were performed until 25% deformation, at a speed compression of 1 mm/min (
Figure 8). After each cycle, the sample height was measured to evaluate its recovery capacity (
Table S3). It featured a small decrease in height during the successive cycles. Regarding the maximum compressive stress, this value increased from ca. 450 to 550 kPa in the second cycle and remained almost unchanged during the next compressive cycles (
Table S3). The sample retained its overall shape without noticeable fissures.
Sample microstructure was analyzed through SEM images (
Figure 8B–G) before and after the cyclic compression-decompression. Surface images shows that, after the compression test, the sample topography became more irregular. By analyzing the cross section, it is possible to verify that after the compressive loads, the sample is more compact and the fibers are apparently less coated by the sol-gel matrix, resulting from the effect of axial compression forces. However, the integrity of the sample was maintained at the microstructural level after five compressive cycles, indicating that these nanocomposites are capable of supporting reasonable loads without fragmentation.















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}