Selection of Water-Soluble Chitosan by Microwave-Assisted Degradation and pH-Controlled Precipitation



Abstract
1. Introduction
2. Materials and Methods
2.1. General Conditions
2.2. Chitosan Depolymerization
- Heat as fast as possible to 100 °C;
- Hold at 100 °C for 19 min;
- Cool down to 35 °C as fast as possible.
2.3. Evaluation of the Solubility of dCS in Acidic Aqueous Solutions and in DMSO
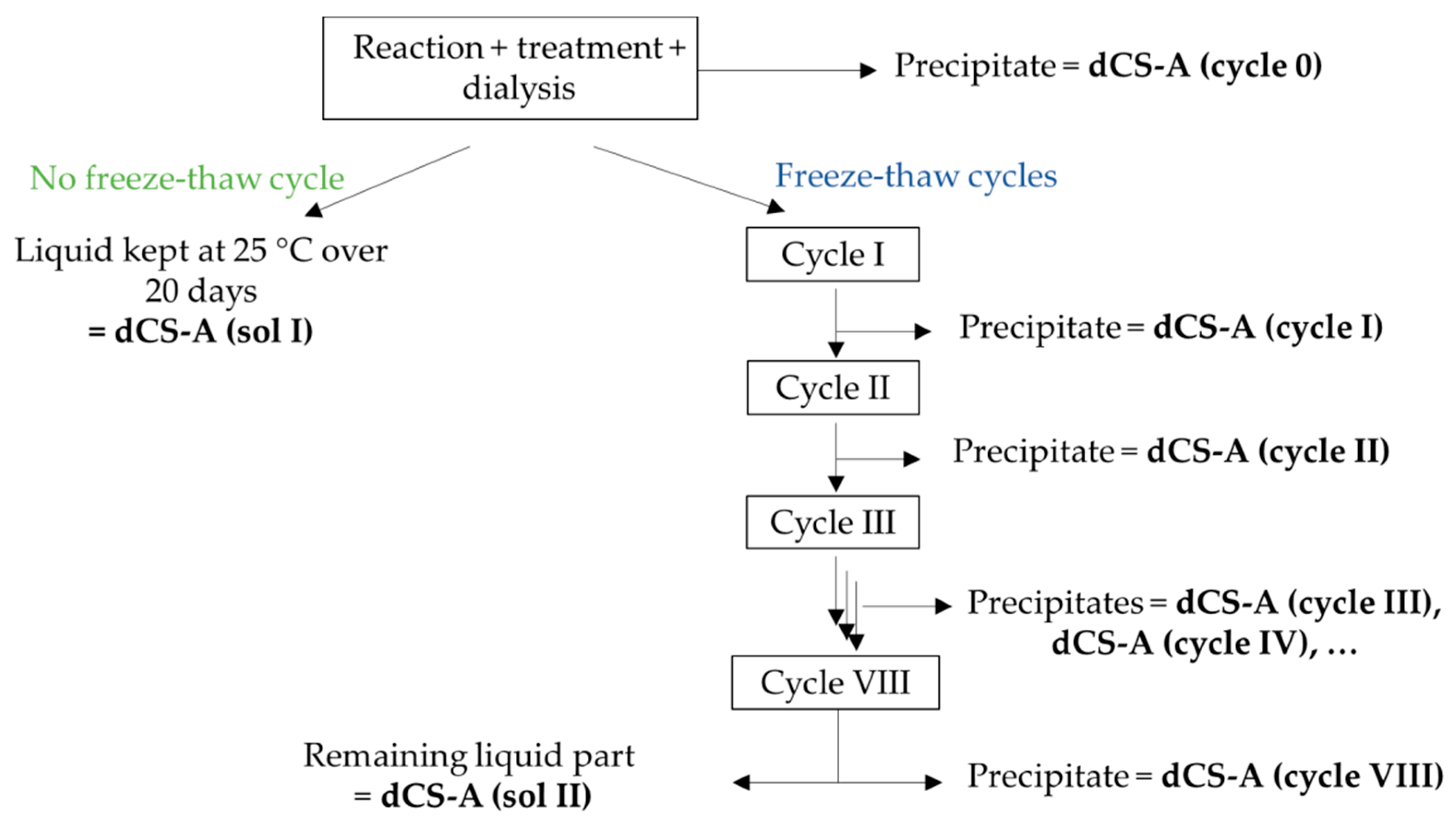
2.4. Protocol for Complete Study of Post-Dialysis dCS Aggregation
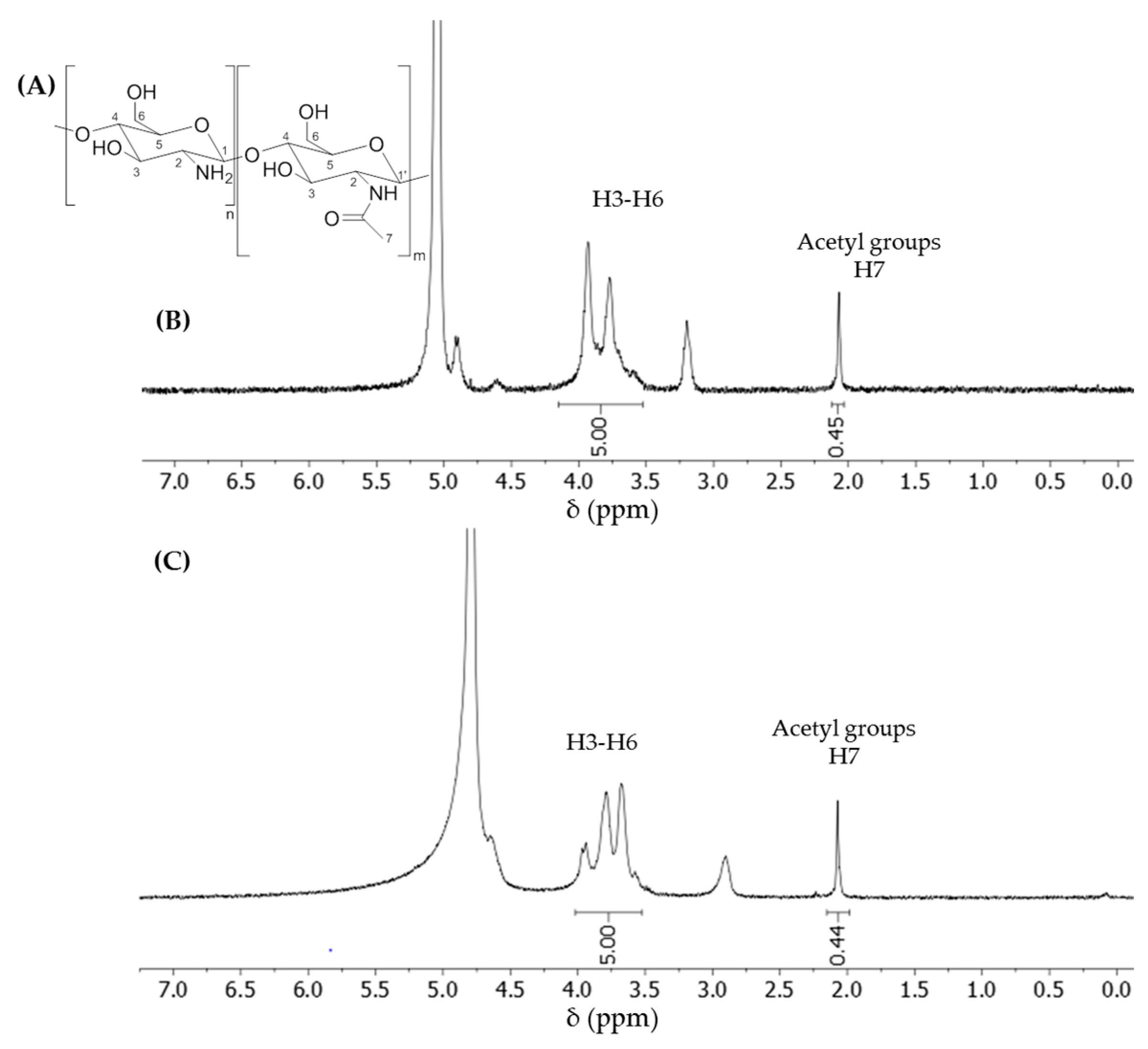
2.5. NMR Analysis
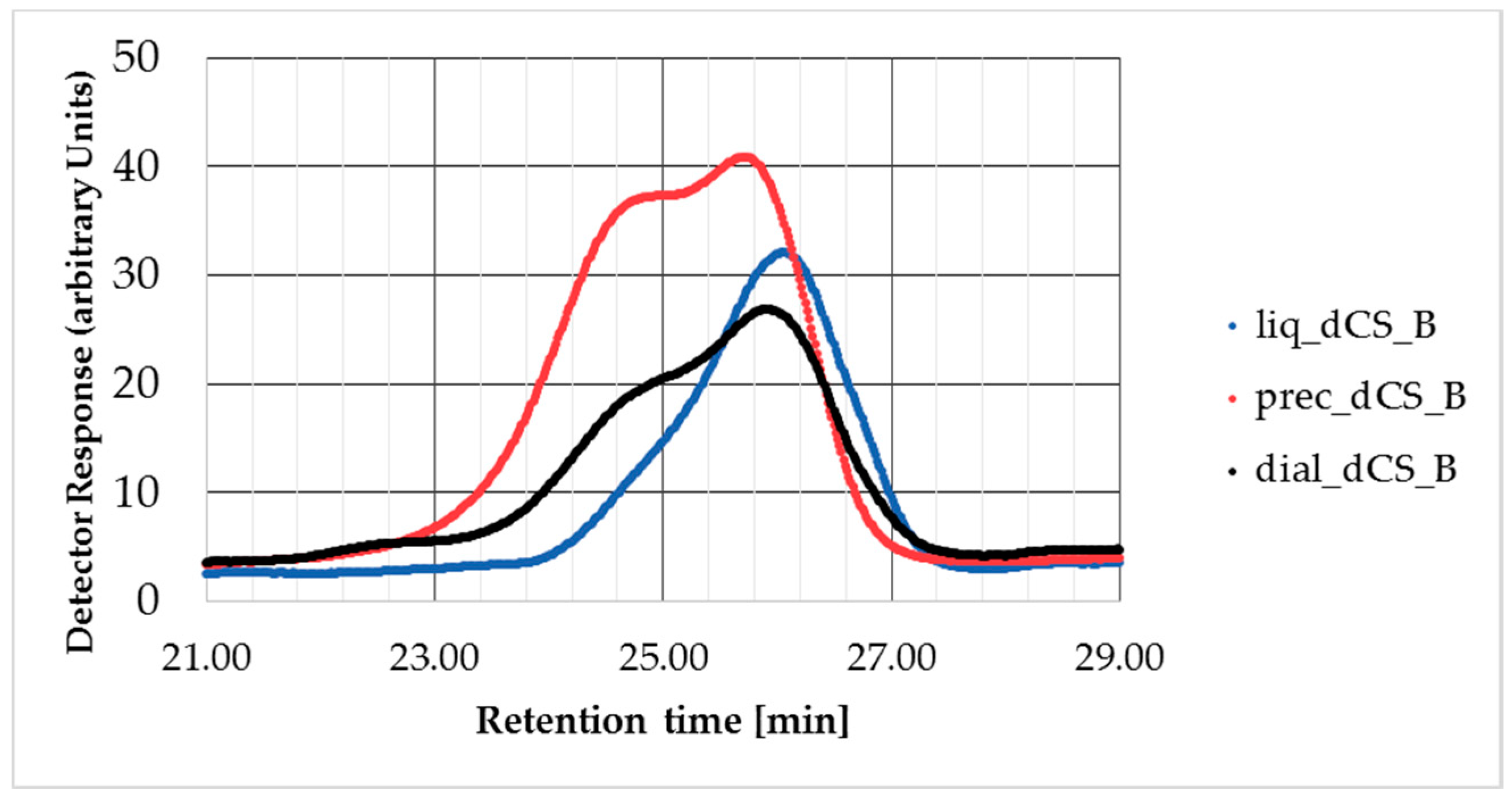
2.6. Gel Permeation Chromatography (GPC) Analysis
3. Results
3.1. Acid-Mediated Depolymerization of Chitosan under Conventional and Micro-Wave Assisted Heating
3.2. Molecular Weight Selectivity
3.3. Solubility of Depolymerized Chitosan
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rajoka, M.S.R.; Zhao, L.; Mehwish, H.M.; Wu, Y.; Mahmood, S. Chitosan and its derivatives: Synthesis, biotechnological applications, and future challenges. Appl. Microbiol. Biotechnol. 2019, 103, 1557–1571. [Google Scholar] [CrossRef]
- Yadav, M.; Goswami, P.; Paritosh, K.; Kumar, M.; Pareek, N.; Vivekanand, V. Seafood waste: A source for preparation of commercially employable chitin/chitosan materials. Bioresour. Bioprocess. 2019, 6, 8. [Google Scholar] [CrossRef]
- LogithKumar, R.; KeshavNarayan, A.; Dhivya, S.; Chawla, A.; Saravanan, S.; Selvamurugan, N. A review of chitosan and its derivatives in bone tissue engineering. Carbohydr. Polym. 2016, 151, 172–188. [Google Scholar] [CrossRef]
- Kean, T.J.; Thanou, M. Biodegradation, biodistribution and toxicity of chitosan. Adv. Drug Deliv. Rev. 2010, 62, 3–11. [Google Scholar] [CrossRef]
- Cheung, R.C.F.; Ng, T.B.; Wong, J.H.; Chan, W.Y. Chitosan: An update on potential biomsdical and pharmaceutical applications. Mar. Drugs 2015, 13, 5156–5186. [Google Scholar] [CrossRef]
- Wang, T.; Zhu, X.-K.; Xue, X.-T.; Wu, D.-Y. Hydrogel sheets of chitosan, honey and gelatin as burn wound dressings. Carbohydr. Polym. 2012, 88, 75–83. [Google Scholar] [CrossRef]
- Saravanan, S.; Leena, R.; Selvamurugan, N. Chitosan based biocomposite scaffolds for bone tissue engineering. Int. J. Biol. Macromol. 2016, 93, 1354–1365. [Google Scholar] [CrossRef]
- Ghormade, V.; Gholap, H.; Kale, S.; Kulkarni, V.; Bhat, S.; Paknikar, K. Fluorescent cadmium telluride quantum dots embedded chitosan nanoparticles: A stable, biocompatible preparation for bio-imaging. J. Biomater. Sci. Polym. Ed. 2014, 26, 42–56. [Google Scholar] [CrossRef]
- Goswami, R.; Subramanian, G.; Silayeva, L.; Newkirk, I.; Doctor, D.; Chawla, K.; Chattopadhyay, S.; Chandra, D.; Chilukuri, N.; Betapudi, V.; et al. Gene therapy leaves a vicious cycle. Front. Oncol. 2019, 9, 297. [Google Scholar] [CrossRef]
- Saranya, N.; Moorthi, A.; Saravanan, S.; Devi, M.P.; Selvamurugan, N. Chitosan and its derivatives for gene delivery. Int. J. Biol. Macromol. 2011, 48, 234–238. [Google Scholar] [CrossRef]
- Wang, W.; Bo, S.; Li, S.; Qin, W. Determination of the Mark-Houwink equation for chitosans with different degrees of deacetylation. Int. J. Biol. Macromol. 1991, 13, 281–285. [Google Scholar] [CrossRef]
- Berkovich, L.; Timofeyeva, G.; Tsyurupa, M.; Davankov, V. Hydrodynamic and conformational parameters of chitosane. Polym. Sci. USSR 1980, 22, 2009–2018. [Google Scholar] [CrossRef]
- Marques, C.; Som, C.; Schmutz, M.; Borges, O.; Borchard, G. How the Lack of Chitosan Characterization Precludes Implementation of the Safe-by-Design Concept. Front. Bioeng. Biotechnol. 2020, 8. [Google Scholar] [CrossRef]
- Davydova, V.N.; Nagorskaya, V.P.; Gorbach, V.I.; Kalitnik, A.A.; Reunov, A.V.; Solov’Eva, T.F.; Ermak, I.M. Chitosan antiviral activity: Dependence on structure and depolymerization method. Appl. Biochem. Microbiol. 2011, 47, 103–108. [Google Scholar] [CrossRef]
- Bellich, B.; D’Agostino, I.; Semeraro, S.; Gamini, A.; Cesàro, A. “The good, the bad and the ugly” of Chitosans. Mar. Drugs 2016, 14, 99. [Google Scholar] [CrossRef]
- Otterlei, M.; Varum, K.; Ryan, L.; Espevik, T. Characterization of binding and TNF-α-inducing ability of chitosans on monocytes: The involvement of CD14. Vaccine 1994, 12, 825–832. [Google Scholar] [CrossRef]
- Prasitsilp, M.; Jenwithisuk, R.; Kongsuwan, K.; Damrongchai, N.; Watts, P. Cellular responses to chitosan in vitro: The importance of deacetylation. J. Mater. Sci. Mater. Electron. 2000, 11, 773–778. [Google Scholar] [CrossRef]
- Firdous, K.; Chakraborty, S. A review: Naturally available sources of chitosan and analysis of chitosan derivatives for its antimicrobial activity. Int. J. Recent Sci. Res. 2017, 8, 15773–15776. [Google Scholar] [CrossRef][Green Version]
- Weinhold, M.X.; Thöming, J. On conformational analysis of chitosan. Carbohydr. Polym. 2011, 84, 1237–1243. [Google Scholar] [CrossRef]
- Aiba, S.-I. Studies on chitosan: 3. Evidence for the presence of random and block copolymer structures in partially N-acetylated chitosans. Int. J. Biol. Macromol. 1991, 13, 40–44. [Google Scholar] [CrossRef]
- Weinhold, M.X.; Sauvageau, J.C.; Kumirska, J.; Thöming, J. Studies on acetylation patterns of different chitosan preparations. Carbohydr. Polym. 2009, 78, 678–684. [Google Scholar] [CrossRef]
- Basa, S.; Nampally, M.; Honorato, T.; Das, S.N.; Podile, A.R.; El Gueddari, N.E.; Moerschbacher, B.M. The pattern of acetylation defines the priming activity of chitosan tetramers. J. Am. Chem. Soc. 2020, 142, 1975–1986. [Google Scholar] [CrossRef]
- Szymańska, E.; Winnicka, K. Stability of chitosan—A challenge for pharmaceutical and biomedical applications. Mar. Drugs 2015, 13, 1819–1846. [Google Scholar] [CrossRef]
- Fiamingo, A.; Delezuk, J.A.D.M.; Trombotto, S.; David, L.; Campana-Filho, S.P. Extensively deacetylated high molecular weight chitosan from the multistep ultrasound-assisted deacetylation of beta-chitin. Ultrason. Sonochem. 2016, 32, 79–85. [Google Scholar] [CrossRef]
- Prajapat, A.L.; Gogate, P.R. Intensification of depolymerization of polyacrylic acid solution using different approaches based on ultrasound and solar irradiation with intensification studies. Ultrason. Sonochem. 2016, 32, 290–299. [Google Scholar] [CrossRef]
- Jung, W.-J.; Park, R.-D. Bioproduction of chitooligosaccharides: Present and perspectives. Mar. Drugs 2014, 12, 5328–5356. [Google Scholar] [CrossRef]
- Gamzazade, A.I.; Šlimak, V.M.; Skljar, A.M.; Štykova, E.V.; Pavlova, S.-S.A.; Rogožin, S.V. Investigation of the hydrodynamic properties of chitosan solutions. Acta Polym. 1985, 36, 420–424. [Google Scholar] [CrossRef]
- Tsao, C.T.; Chang, C.-H.; Lin, Y.Y.; Wu, M.F.; Han, J.L.; Hsieh, K.-H. Kinetic study of acid depolymerization of chitosan and effects of low molecular weight chitosan on erythrocyte rouleaux formation. Carbohydr. Res. 2011, 346, 94–102. [Google Scholar] [CrossRef]
- Chebotok, E.N.; Novikov, V.; Konovalova, I.N. Depolymerization of chitin and chitosan in the course of base deacetylation. Russ. J. Appl. Chem. 2006, 79, 1162–1166. [Google Scholar] [CrossRef]
- Qin, C.; Du, Y.; Xiao, L. Effect of hydrogen peroxide treatment on the molecular weight and structure of chitosan. Polym. Degrad. Stab. 2002, 76, 211–218. [Google Scholar] [CrossRef]
- Dos Santos, D.M.; Bukzem, A.L.; Campana-Filho, S.P. Response surface methodology applied to the study of the microwave-assisted synthesis of quaternized chitosan. Carbohydr. Polym. 2016, 138, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Xing, R.; Liu, S.; Qin, Y.; Meng, X.; Li, P. Microwave-assisted degradation of chitosan for a possible use in inhibiting crop pathogenic fungi. Int. J. Biol. Macromol. 2012, 51, 767–773. [Google Scholar] [CrossRef] [PubMed]
- Nouri, M.; Khodaiyan, F.; Razavi, S.H.; Mousavi, M. Improvement of chitosan production from Persian Gulf shrimp waste by response surface methodology. Food Hydrocoll. 2016, 59, 50–58. [Google Scholar] [CrossRef]
- Allan, G.; Peyron, M. Molecular weight manipulation of chitosan I: Kinetics of depolymerization by nitrous acid. Carbohydr. Res. 1995, 277, 257–272. [Google Scholar] [CrossRef]
- Vårum, K.M.; Ottøy, M.H.; Smidsrød, O. Water-solubility of partially N-acetylated chitosans as a function of pH: Effect of chemical composition and depolymerisation. Carbohydr. Polym. 1994, 25, 65–70. [Google Scholar] [CrossRef]
- Yuan, Y.; Chesnutt, B.M.; Haggard, W.O.; Bumgardner, J.D. Deacetylation of chitosan: Material characterization and in vitro evaluation via albumin adsorption and pre-osteoblastic cell cultures. Materials 2011, 4, 1399–1416. [Google Scholar] [CrossRef]
- Shrotri, A.; Kobayashi, H.; Fukuoka, A. Catalytic conversion of structural carbohydrates and lignin to chemicals. In Advances in Catalysis; Elsevier: Amsterdam, The Netherlands, 2017; Volume 60, pp. 59–123. [Google Scholar]
- Pa, J.-H.; Yu, T.L. Light scattering study of chitosan in acetic acid aqueous solutions. Macromol. Chem. Phys. 2001, 202, 985–991. [Google Scholar] [CrossRef]
- Wu, C.; Zhou, S.; Wang, W. A dynamic laser light-scattering study of chitosan in aqueous solution. Biopolymers 1995, 35, 385–392. [Google Scholar] [CrossRef]
- Dzherayan, T.G.; Vanifatova, N.G.; Burmistrov, A.A.; Lazareva, E.V.; Rudnev, A.V. Detection and quantification of chitosan aggregates by pressure-assisted capillary zone electrophoresis. J. Anal. Chem. 2017, 72, 309–315. [Google Scholar] [CrossRef]
- Pillai, C.; Paul, W.; Sharma, C.P. Chitin and chitosan polymers: Chemistry, solubility and fiber formation. Prog. Polym. Sci. 2009, 34, 641–678. [Google Scholar] [CrossRef]
- Anthonsen, M.W.; Vårum, K.M.; Hermansson, A.M.; Smidsrød, O.; Brant, D.A. Aggregates in acidic solutions of chitosans detected by static laser light scattering. Carbohydr. Polym. 1994, 25, 13–23. [Google Scholar] [CrossRef]
- Foster, L.J.R.; Ho, S.; Hook, J.M.; Basuki, M.; Marçal, H. Chitosan as a biomaterial: Influence of degree of deacetylation on its physiochemical, material and biological properties. PLoS ONE 2015, 10, e0135153. [Google Scholar] [CrossRef] [PubMed]
- Tikhomirov, S.G.; Semenov, M.E.; Khaustov, I.A.; Bitukov, V.K.; Khvostov, A.A.; Karmanova, O.V.; Popov, A.P. Polymer degradation in a solution: Monitoring and predicting quality parameters. Theor. Found. Chem. Eng. 2018, 52, 568–573. [Google Scholar] [CrossRef]
- Couillaud, B.M.; Espeau, P.; Mignet, N.; Corvis, Y. Cover feature: State of the art of pharmaceutical solid forms: From crystal property issues to nanocrystals formulation (ChemMedChem 1/2019). ChemMedChem 2019, 14, 2. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Entry | Sample | Mp | Mn | Mw | PD | Heating Conditions | Yield | % DD | % Mn Decrease |
|---|---|---|---|---|---|---|---|---|---|
| 1 | CS-A | 191,500 | 90,000 | 253,500 | 2.82 | - | - | 85 | - |
| 2 | CS-B | 282,500 | 174,500 | 392,000 | 2.25 | - | - | 99 | - |
| 3 c | dCS-A | n.d. | n.d. | n.d. | n.d. | (80°, 48 h) a | 50% | 100 | |
| 4 c | dCS-B | n.d. | n.d. | n.d. | n.d. | (80°, 48 h) a | 49% | 100 | |
| 5 d | dCS-B | n/a | n/a | n/a | n/a | (100°, 19’) b | <1% | 99 | 95 |
| 6 | dCS-A | 13,600 | 12,600 | 17,700 | 1.41 | (100°, 19’) b | 15% | 85 | 86 |
| Entry | Sample | Starting pH a | Final pH b |
|---|---|---|---|
| 1 | CS-A | 6.40 | 2.19 |
| 2 | CS-B | 7.48 | 1.93 |
| 3 | dCS-A | 6.86 | 5.97 |
| Sample | Mp | Mn | Mw | PD | Yield |
|---|---|---|---|---|---|
| dCS (14) | 16,100 | 15,100 | 19,700 | 1.31 | 10% |
| dCS (7) | 16,600 | 15,000 | 20,500 | 1.37 | 14% |
| dCS (3.5) | 14,200 | 13,400 | 18,100 | 1.36 | 15% |
| Entry | Sample a | Mp | Mn | Mw | PD | Yield | % DD |
|---|---|---|---|---|---|---|---|
| 1 | dCS (prec pH 6.7) | 41,900 | 32,800 | 54,800 | 1.67 | 4% | 86 |
| 2 | dCS (prec pH 6.8) | 36,300 | 29,300 | 50,900 | 1.74 | 12% | 83 |
| 3 | dCS (prec pH 6.9) | 35,500 | 28,400 | 48,200 | 1.70 | 18% | 84 |
| 4 | dCS (prec pH 7.0) | 33,700 | 28,700 | 46,900 | 1.64 | 12% | 82 |
| 5 | dCS (prec pH 7.4) | 28,200 | 24,500 | 38,500 | 1.57 | 9% | 86 |
| 6 b | dCS (sol pH 7.4) | 15,100 | 12,900 | 18,800 | 1.46 | 19% | 85 |
| Sample | mg/mL |
|---|---|
| dCS-A (sol I) a | 0.22 ± 0.02 |
| dCS-A (cycle 0) b | 0.19 ± 0.02 |
| dCS-A (cycle I) b | 0.16 ± 0.02 |
| dCS-A (cycle II) b | 0.16 ± 0.02 |
| dCS-A (cycle III) b | 0.15 ± 0.02 |
| dCS-A (cycle IV–VIII) b | 0.15 ± 0.01 |
| dCS-A (sol II) c | 0.15 ± 0.01 |
| Sample | Mp | Mn | Mw | PD | DD |
|---|---|---|---|---|---|
| dial-dCS-B | 7600 | 8900 | 12,100 | 1.36 | 99% |
| prec-dCS-B | 8500 | 11,000 | 15,100 | 1.38 | 99% |
| liq-dCS-B | 6800 | 7200 | 8700 | 1.20 | 99% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Journot, C.M.A.; Nicolle, L.; Lavanchy, Y.; Gerber-Lemaire, S. Selection of Water-Soluble Chitosan by Microwave-Assisted Degradation and pH-Controlled Precipitation. Polymers 2020, 12, 1274. https://doi.org/10.3390/polym12061274
Journot CMA, Nicolle L, Lavanchy Y, Gerber-Lemaire S. Selection of Water-Soluble Chitosan by Microwave-Assisted Degradation and pH-Controlled Precipitation. Polymers. 2020; 12(6):1274. https://doi.org/10.3390/polym12061274
Chicago/Turabian StyleJournot, Céline M. A., Laura Nicolle, Yann Lavanchy, and Sandrine Gerber-Lemaire. 2020. "Selection of Water-Soluble Chitosan by Microwave-Assisted Degradation and pH-Controlled Precipitation" Polymers 12, no. 6: 1274. https://doi.org/10.3390/polym12061274
APA StyleJournot, C. M. A., Nicolle, L., Lavanchy, Y., & Gerber-Lemaire, S. (2020). Selection of Water-Soluble Chitosan by Microwave-Assisted Degradation and pH-Controlled Precipitation. Polymers, 12(6), 1274. https://doi.org/10.3390/polym12061274