Interface Characteristics of Neat Melts and Binary Mixtures of Polyethylenes from Atomistic Molecular Dynamics Simulations


Abstract
1. Introduction
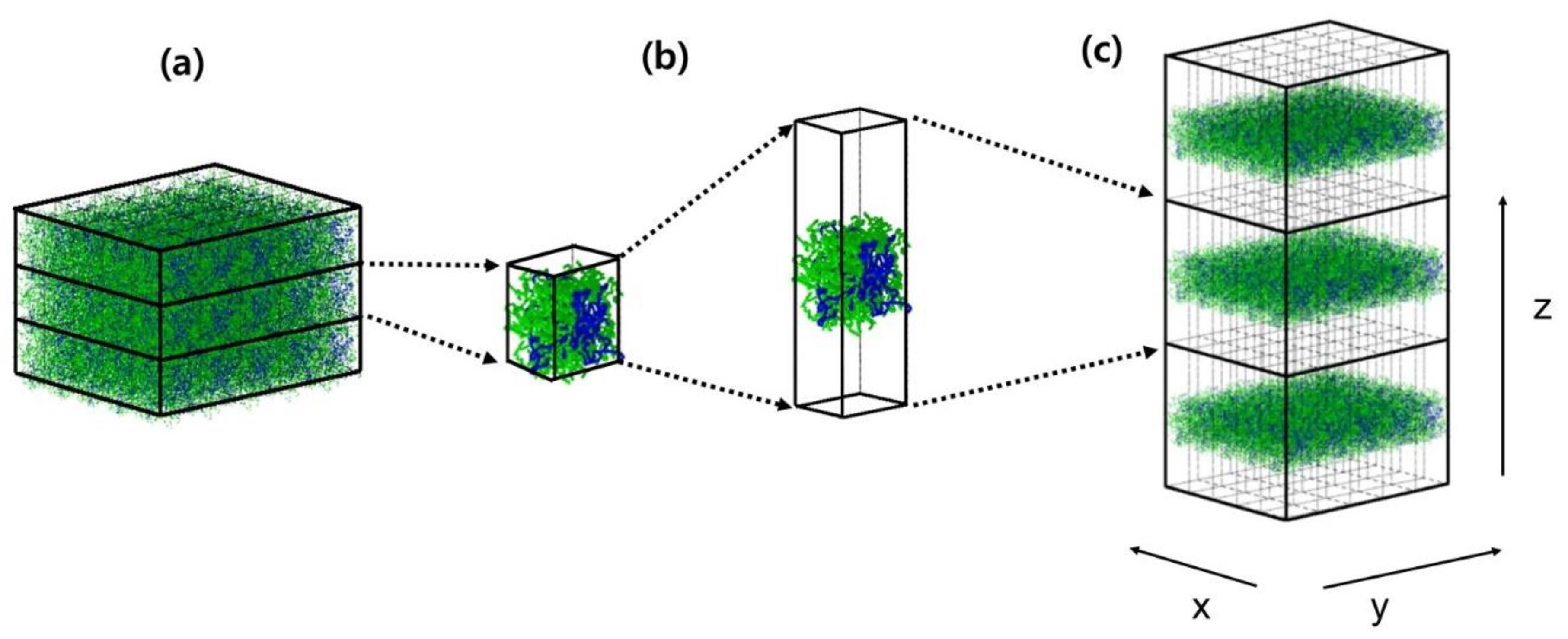
2. Simulation Methods and System Specifications
3. Results and Discussion
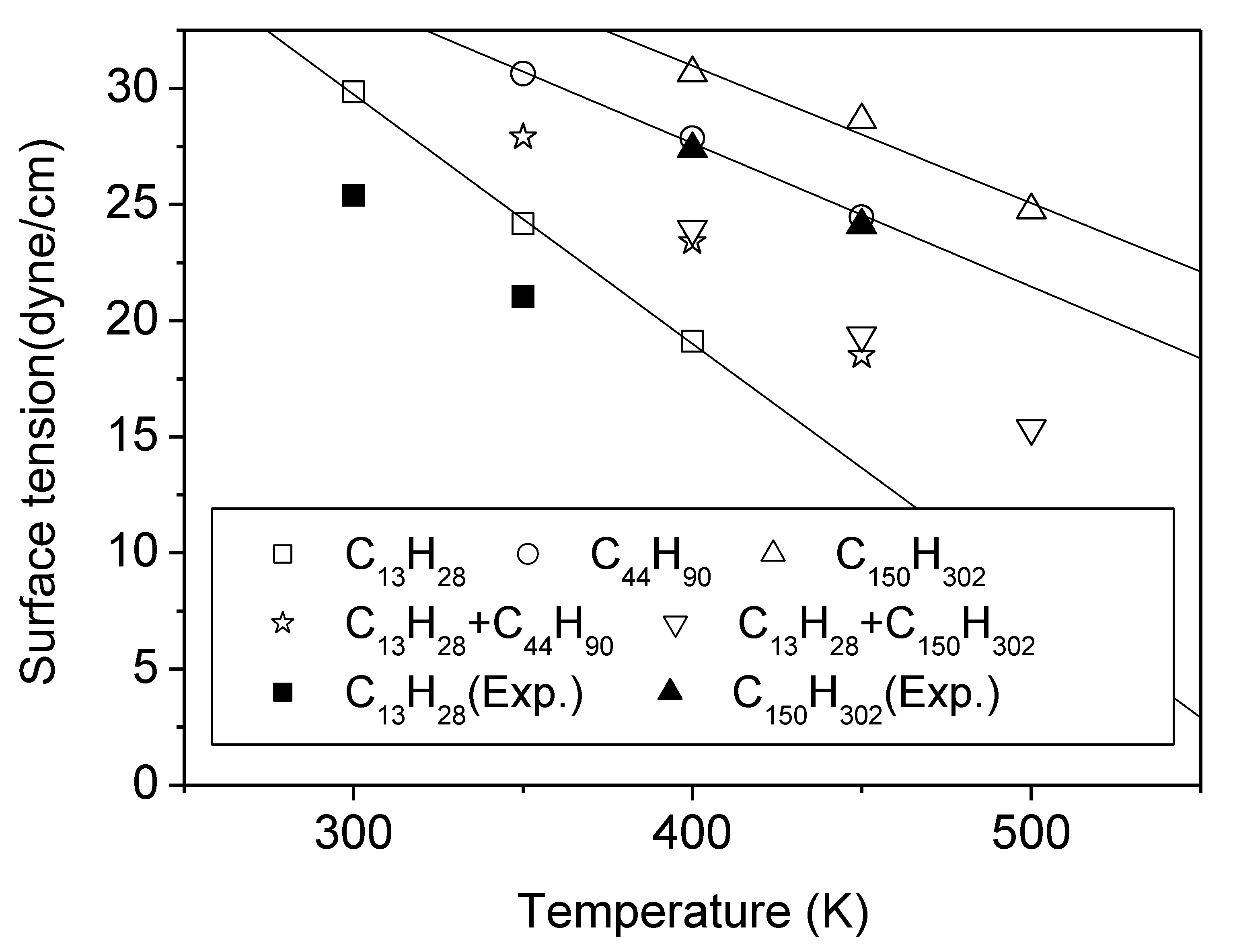
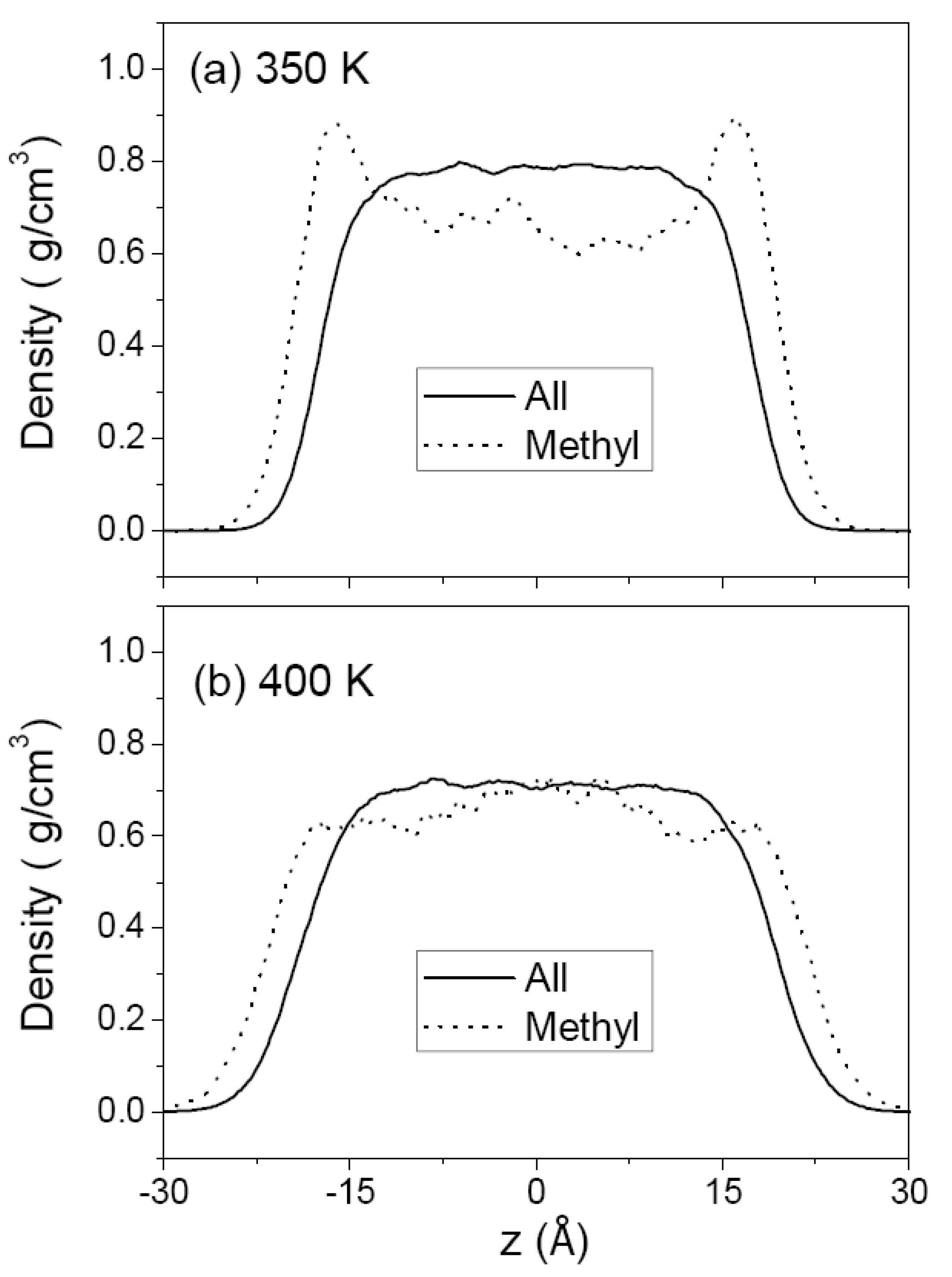
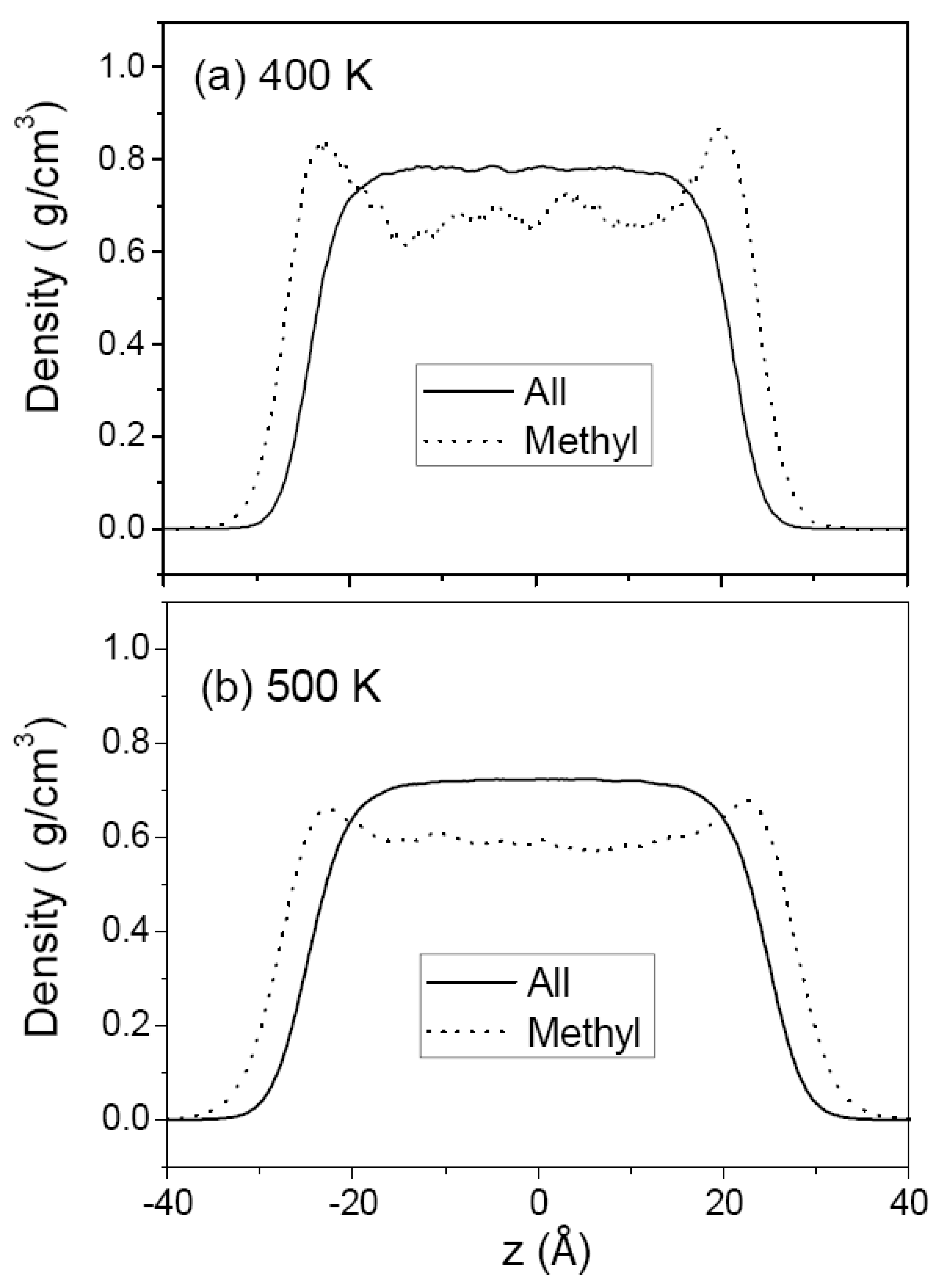
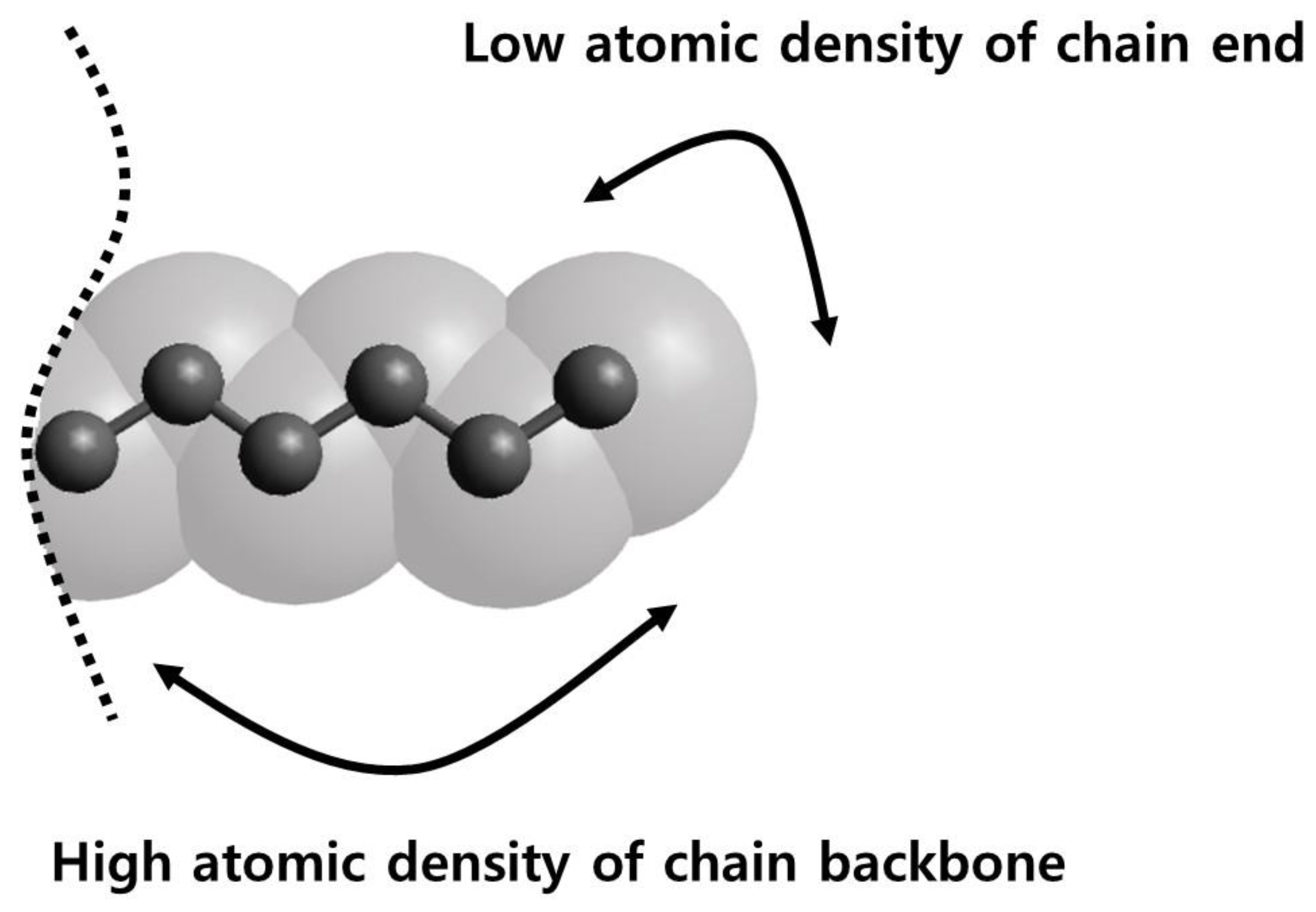
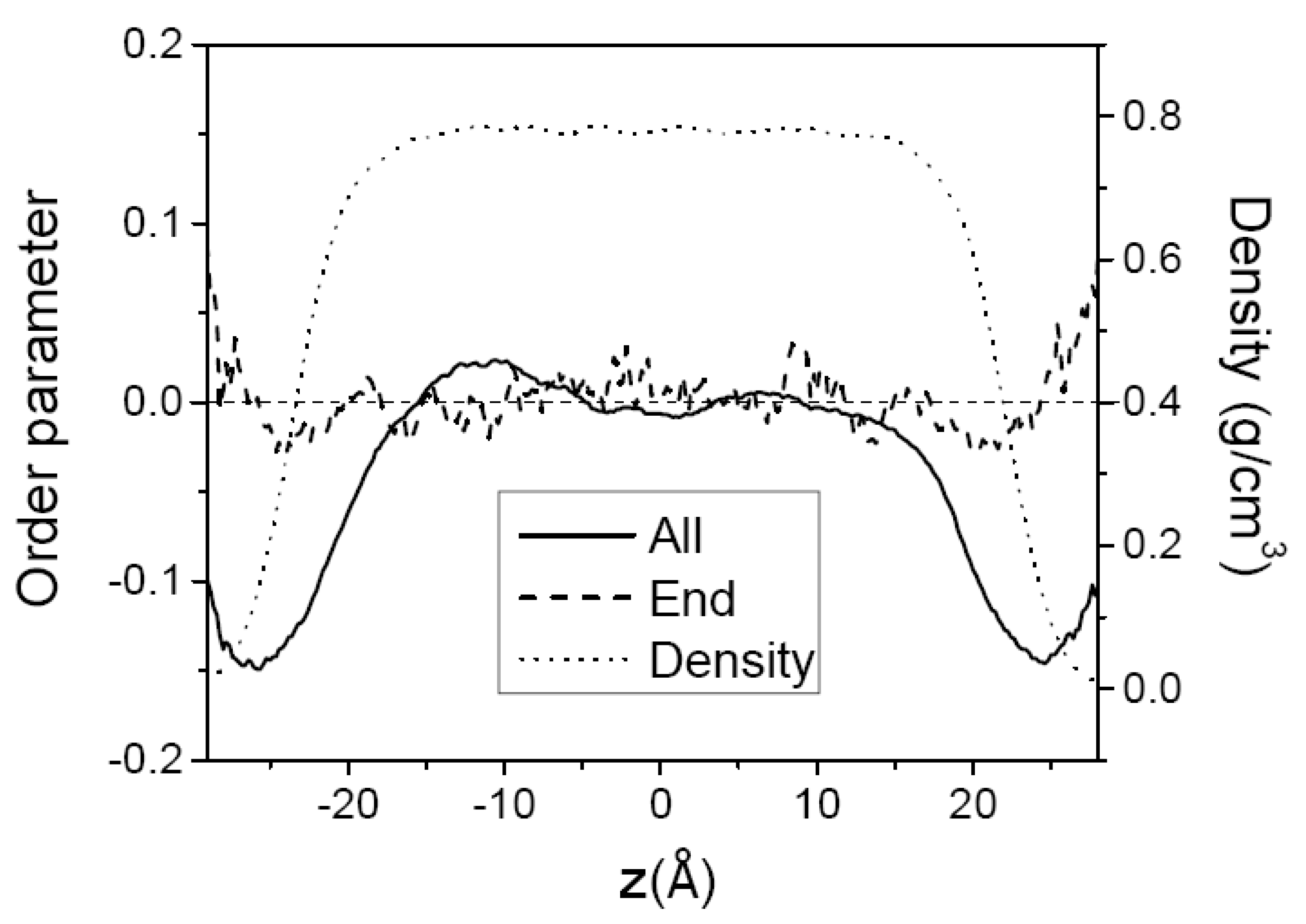
3.1. Pure Melts of C44H90 and C150H302
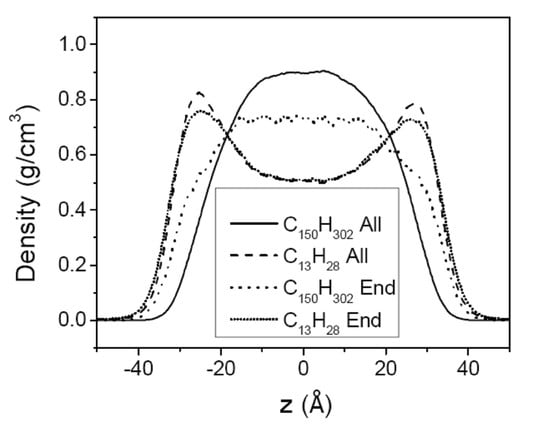
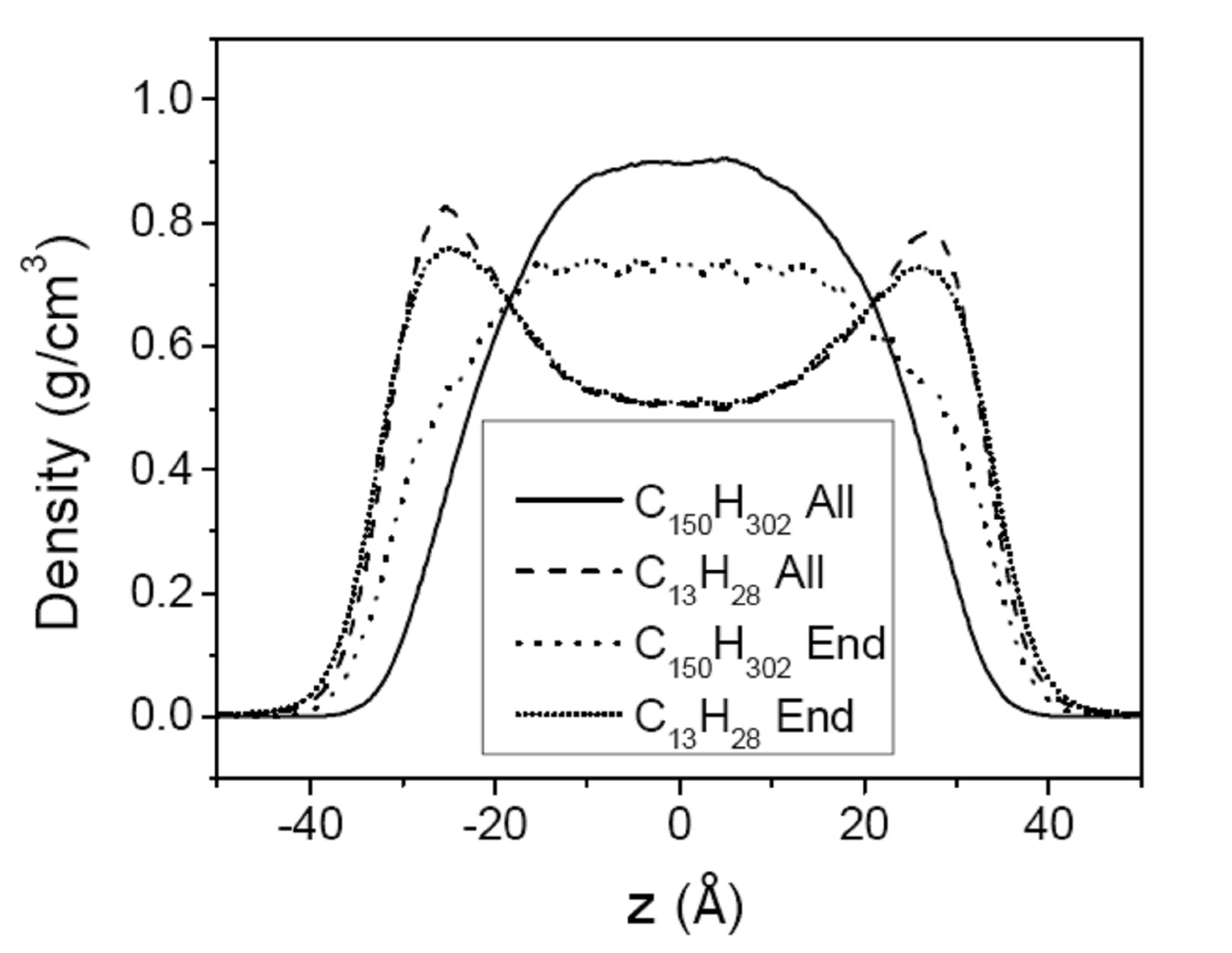
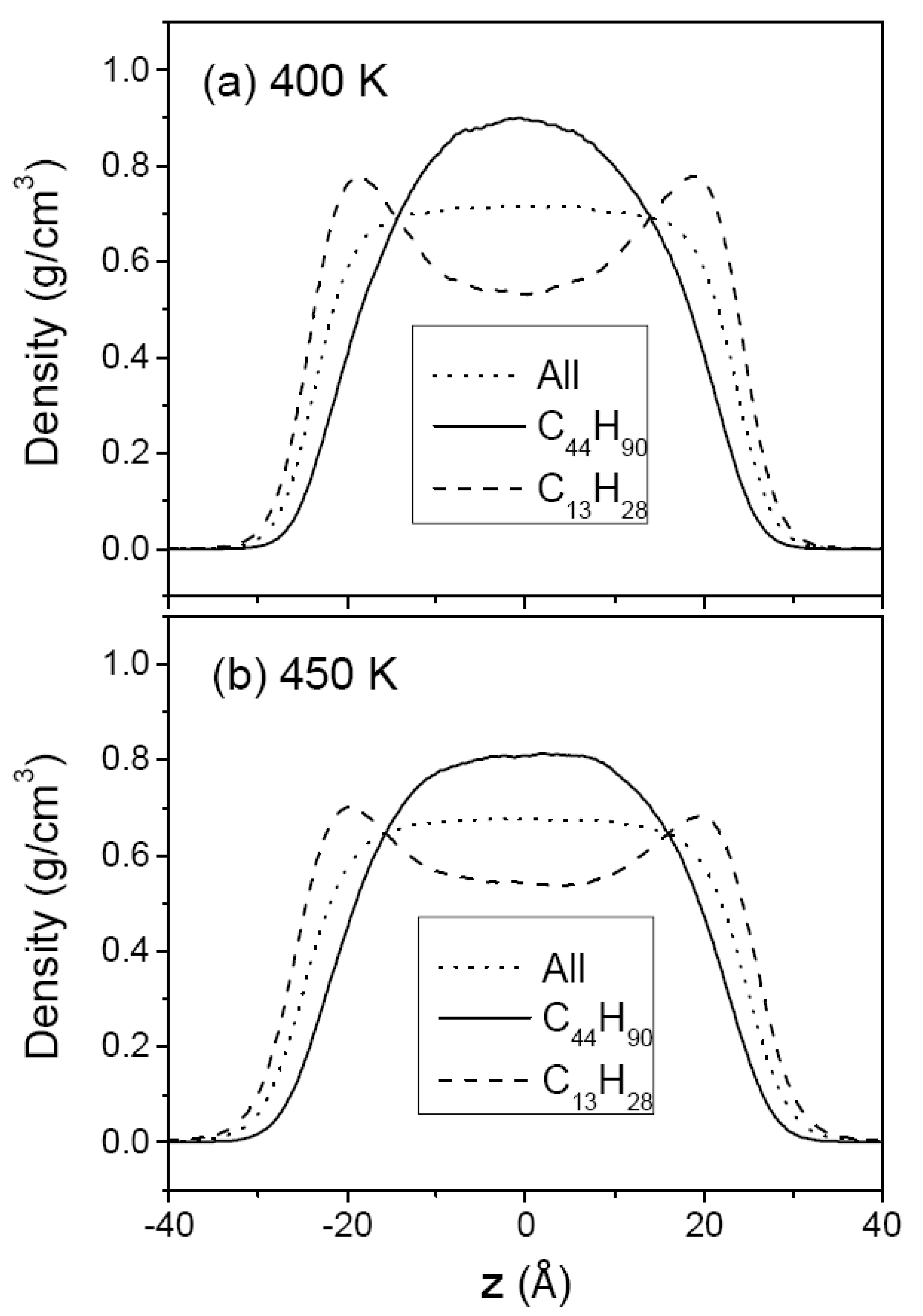
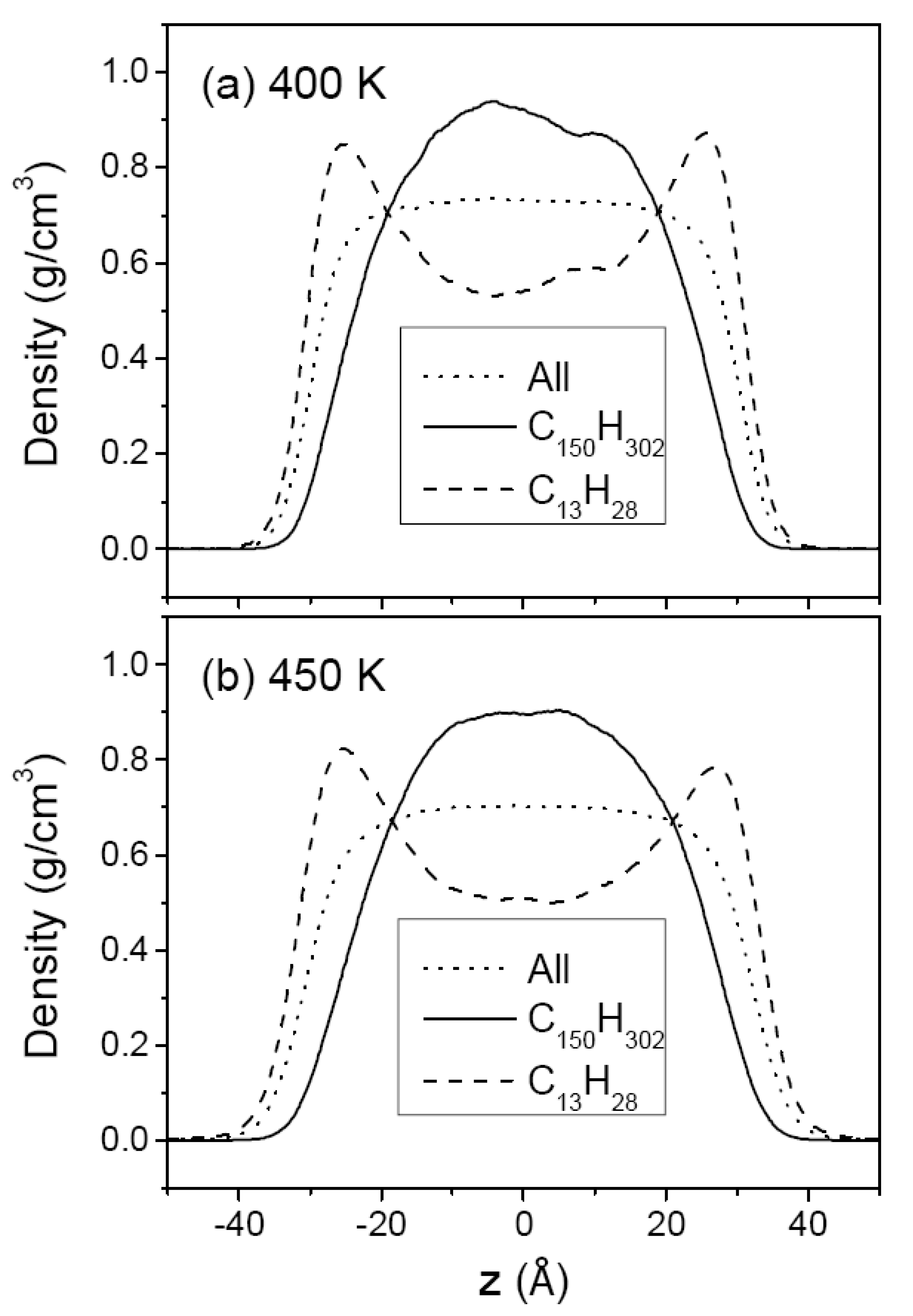
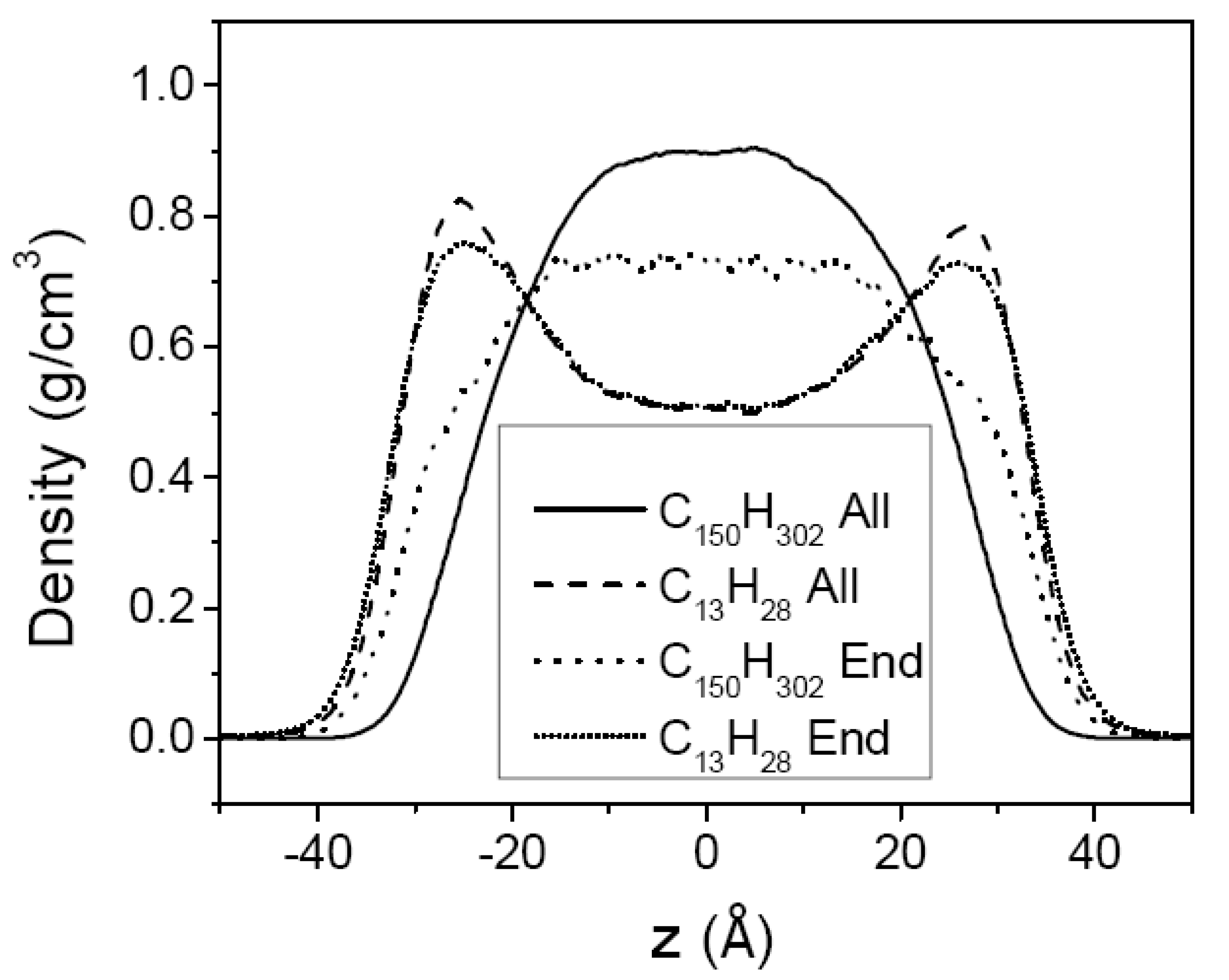
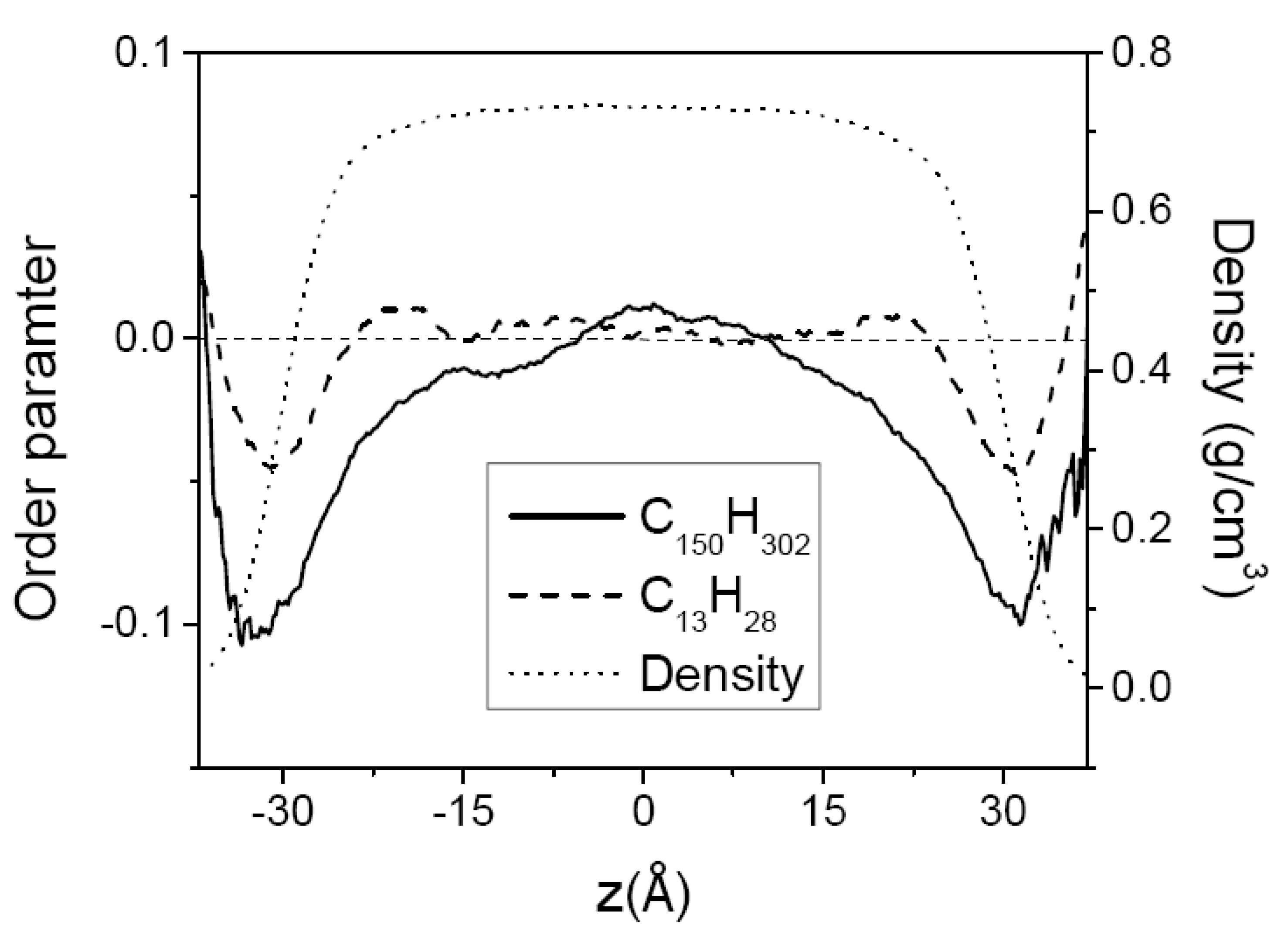
3.2. Mixtures of PE Chains with C13H28
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mansfield, K.F.; Theodorou, D.N. Molecular dynamics simulation of a glassy polymer surface. Macromolecules 1991, 24, 6283–6294. [Google Scholar] [CrossRef]
- Harris, J.G. Liquid-vapor interfaces of alkane oligomers: Structure and thermodynamics from molecular dynamics simulations of chemically realistic models. J. Phys. Chem. 1992, 96, 5077–5086. [Google Scholar] [CrossRef]
- Chang, J.; Han, J.; Yang, L.; Jaffe, R.L.; Yoon, D.Y. Structure and properties of polymethylene melt surfaces from molecular dynamics simulations. J. Chem. Phys. 2001, 115, 2831–2840. [Google Scholar] [CrossRef]
- Harmandaris, V.A.; Daoulas, K.C.; Mavrantzas, V.G. Molecular dynamics simulation of a polymer melts/solid interface: Local dynamics and chain mobility in a thin film of polyethylene melt adsorbed on graphite. Macromolecules 2005, 38, 5796–5809. [Google Scholar] [CrossRef]
- Peter, S.; Meyer, H.; Baschnagel, J. Molecular dynamics simulations of concentrated polymer solutions in thin film geometry. I. Equilibrium properties near the glass transition. J. Chem. Phys. 2009, 131, 014902. [Google Scholar] [CrossRef]
- Peter, S.; Meyer, H.; Baschnagel, J. Molecular dynamics simulations of concentrated polymer solutions in thin film geometry. II. Solvent evaporation near the glass transition. J. Chem. Phys. 2009, 131, 014903. [Google Scholar] [CrossRef]
- Torres, J.A.; Nealey, P.F.; de Pablo, J.J. Molecular simulation of ultrathin polymeric films near the glass transition. Phys. Rev. Lett. 2000, 85, 3221–3224. [Google Scholar] [CrossRef]
- Morita, H.; Tanaka, K.; Kajiyama, T.; Nishi, T.; Doi, M. Study of the glass transition temperature of polymer surface by coarse-grained molecular dynamics simulation. Macromolecules 2006, 39, 6233–6237. [Google Scholar] [CrossRef]
- Xia, W.; Hsu, D.D.; Keten, S. Molecular weight effects on the glass transition and confinement behavior of polymer thin films. Macromol. Rapid Commun. 2015, 36, 1422–1427. [Google Scholar] [CrossRef]
- Varnik, F.; Baschnagel, J.; Binder, K. Reduction of the glass transition temperature in polymer films: A molecular-dynamics study. Phys. Rev. E 2002, 65, 021507. [Google Scholar] [CrossRef]
- Varnik, F.; Baschnagel, J.; Binder, K. Molecular dynamics results on the pressure tensor of polymer films. J. Chem. Phys. 2000, 113, 4444–4453. [Google Scholar] [CrossRef]
- Kumar, N.; Manik, G. Molecular dynamics simulations of polyvinyl acetate-perfluorooctane based anti-stain coating. Polymer 2016, 100, 194–205. [Google Scholar] [CrossRef]
- Eslami, H.; Müller-Plathe, F. How thick is the interphase in an ultrathin polymer film? Coase-grained molecular dynamics simulations of polyamide-6,6 on graphene. J. Phys. Chem. C 2013, 117, 5249–5257. [Google Scholar] [CrossRef]
- Glynos, E.; Johnson, K.J.; Frieberg, B.; Chremos, A.; Narayanan, S.; Sakellariou, G.; Green, P.F. Free surface relaxations of star-shaped polymer films. Phys. Rev. Lett. 2017, 119, 227801. [Google Scholar] [CrossRef]
- Solar, M.; Binder, K.; Paul, W. Relaxation processes and glass transition of confined polymer melts: A molecular dynamics simulation of 1,4-polybutadiene between graphite walls. J. Chem. Phys. 2017, 146, 203308. [Google Scholar] [CrossRef]
- Jacobs, M.; Liang, H.; Pugnet, B.; Dobrynin, A.V. Molecular dynamics simulations of surface and interfacial of graft polymer melts. Langmuir 2018, 34, 12974–12981. [Google Scholar] [CrossRef]
- Lyulin, A.V.; Balabaev, N.K.; Baljon, A.R.C.; Mendoza, G.; Frank, C.W.; Yoon, D.Y. Interfacial and topological effects on the glass transition in free-standing polystyrene films. J. Chem. Phys. 2017, 146, 203314. [Google Scholar] [CrossRef]
- Lee, S.; Lyulin, A.V.; Frank, C.W.; Yoon, D.Y. Interface characteristics of polystyrene melts in free-standing thin films and on graphite surface from molecular dynamics simulations. Polymer 2017, 116, 540–548. [Google Scholar] [CrossRef]
- Li, S.; Ding, M.; Shi, T. Effect of bidispersity on structure and entanglement of confined polymer films. J. Phys. Chem. B 2017, 121, 7502–7507. [Google Scholar] [CrossRef]
- Li, S.; Chen, Q.; Ding, M.; Shi, T. Effect of bidispersity on dynamics of confined polymer films. Polymers 2017, 10, 1327. [Google Scholar] [CrossRef]
- Xia, W.; Lan, T. Interfacial dynamics governs the mechanical properties of glass polymer thin films. Macromolecules 2019, 52, 6547–6554. [Google Scholar] [CrossRef]
- Sorensen, R.A.; Liau, W.B.; Kesner, L.; Boyd, R.H. Prediction of polymer crystal structures and properties: Polyethylene and poly(oxymethylene). Macromolecules 1988, 21, 200–208. [Google Scholar] [CrossRef]
- Smith, G.D.; Yoon, D.Y. Equilibrium and dynamics properties of polymethylene melts from molecular dynamics simulations. I. n-Tridecane. J. Chem. Phys. 1994, 100, 649–658. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Chen, B.; Siepmann, J.I. Tranferable potential for phase equilibria. 3. Explicit-hydrogen description of normal alkane. J. Phys. Chem. B 1999, 103, 5370–5379. [Google Scholar] [CrossRef]
- Chang, J.; Sandler, S.I. Interatomic Lennard-Jones potentials of linear and branched alkanes calibrated by Gibbs ensemble simulations for vapor-liquid equilibria. J. Chem. Phys. 2004, 121, 7474–7483. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Madura, J.D.; Swenson, C.J. Optimized intermolecular potential functions for liquid hydrocarbons. J. Am. Chem. Soc. 1984, 106, 6638–6646. [Google Scholar] [CrossRef]
- Nath, S.K.; Escobedo, F.A.; de Pablo, J.J. On the simulation of vapor-liquid equilibria for alkanes. J. Chem. Phys. 1998, 108, 9905–9911. [Google Scholar] [CrossRef]
- Martin, M.G.; Siepmann, J.I. Transferable potentials for phase equilibria. 1. United-atom description of n-alkanes. J. Phys. Chem. B 1998, 102, 2569–2577. [Google Scholar] [CrossRef]
- Paul, W.; Yoon, D.Y.; Smith, G.D. An optimized united atom model for simulations of polymethylene melts. J. Chem. Phys. 1995, 103, 1702–1709. [Google Scholar] [CrossRef]
- Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef]
- Allen, M.P.; Tildesley, D.J. Computer Simulation of Liquids; Clarendon: Oxford, UK, 1990. [Google Scholar]
- Kirkwood, J.G.; Buff, F.P. The statistical mechanical theory of surface tension. J. Chem. Phys. 1949, 17, 338–343. [Google Scholar] [CrossRef]
- Rowlinson, J.S.; Widom, B. Molecular Theory of Capillarity; Clarendon: Oxford, UK, 1982. [Google Scholar]
- Mecke, M.; Winkelmann, J.; Fisher, J. Molecular dynamics simulation of the liquid-vapor interface: The Lennard-Jones fluid. J. Chem. Phys. 1997, 107, 9264–9270. [Google Scholar] [CrossRef]
- Thermodynamics Research Center. TRC Thermodynamic Tables, Hydrocarbons; The Texas A&M University System: College Station, TX, USA, 1990; Volume 3. [Google Scholar]
- Dee, G.; Sauer, B.B. The molecular weight and temperature dependence of polymer surface tension: Comparison of experiment with interface gradient theory. J. Colloid Interface Sci. 1992, 152, 85–103. [Google Scholar] [CrossRef]
- Doruker, P.; Mattice, W.L. Mobility of the surface and interior of thin films composed of amorphous polyethylene. Macromolecules 1999, 32, 194–198. [Google Scholar] [CrossRef]
- Yethiraj, A. Entropic and enthalpic surface segregation from blends of branched and linear polymers. Phys. Rev. Lett. 1995, 74, 2018–2021. [Google Scholar] [CrossRef]
- Scheutjens, J.M.H.M.; Fleer, G.J. Statistical theory of the adsorption of interacting chain molecules. 1. Partition function, segment density distribution, and adsorption isotherms. J. Phys. Chem. 1979, 83, 1619–1635. [Google Scholar] [CrossRef]
- Theodorou, D.N. Microscopic structure and thermodynamic properties of bulk copolymers and surface-active polymers at interfaces. 1. Theory. Macromolecules 1988, 21, 1411–1421. [Google Scholar] [CrossRef]
- Klein, J.; Kerle, T.; Zink, F.; Eiser, E. Segmental interaction parameters of binary polymer mixtures evaluated from binodals and from surface-segregation profiles: Comparison with small-angle neutron scattering. Macromolecules 2000, 33, 1298–1305. [Google Scholar] [CrossRef]
- Barton, A.F.M. Handbook of Solubility Parameters and Other Cohesion Parameters; CRC Press: Boca Raton, FL, USA, 1983. [Google Scholar]
- Hariharan, A.; Kumar, S.K.; Russell, T.P. A lattice model for the surface segregation of polymer chains due to molecular weight effects. Macromolecules 1990, 23, 3584–3592. [Google Scholar] [CrossRef]
- Van der Gucht, J.; Besseling, N.A.M.; Fleer, G.J. Surface segregation in polydisperse polymer melts. Macromolecules 2002, 35, 6732–6738. [Google Scholar] [CrossRef]
- Doruker, P. Simulation of polyethylene thin films composed of various chain lengths. Polymer 2002, 43, 425–430. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Temp. (K) | C13H28 | C44H90 | C150H302 | Mixture C13 + C44 C13 + C150 | ||||
|---|---|---|---|---|---|---|---|---|
| Sim. a | Exp. b | Sim. | Exp. c | Sim. | Exp. d | Sim. | Sim. | |
| 300 | 29.9 | 25.4 | … | … | … | … | … | … |
| 350 | 24.2 | 21.0 | 30.6 | 27.8 | … | … | 27.9 | … |
| 400 | 19.1 | … | 27.9 | … | 30.7 | 27.4 | 23.4 | 23.9 |
| 450 | … | … | 24.5 | … | 28.6 | 24.1 | 18.5 | 19.4 |
| 500 | … | … | … | … | 24.8 | … | … | … |
| Temp. (K) | Pure Melt | Mixture (50:50) | |||
|---|---|---|---|---|---|
| C13H28 a | C44H90 | C150H302 | C13 + C44 | C13 + C150 | |
| 350 | 1.383 | 1.232 | 1.300 | ||
| 400 | 1.443 | 1.269 | 1.233 | 1.357 | 1.335 |
| 450 | 1.520 | 1.274 | 1.398 | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.; Frank, C.W.; Yoon, D.Y. Interface Characteristics of Neat Melts and Binary Mixtures of Polyethylenes from Atomistic Molecular Dynamics Simulations. Polymers 2020, 12, 1059. https://doi.org/10.3390/polym12051059
Lee S, Frank CW, Yoon DY. Interface Characteristics of Neat Melts and Binary Mixtures of Polyethylenes from Atomistic Molecular Dynamics Simulations. Polymers. 2020; 12(5):1059. https://doi.org/10.3390/polym12051059
Chicago/Turabian StyleLee, Sanghun, Curtis W. Frank, and Do Y. Yoon. 2020. "Interface Characteristics of Neat Melts and Binary Mixtures of Polyethylenes from Atomistic Molecular Dynamics Simulations" Polymers 12, no. 5: 1059. https://doi.org/10.3390/polym12051059
APA StyleLee, S., Frank, C. W., & Yoon, D. Y. (2020). Interface Characteristics of Neat Melts and Binary Mixtures of Polyethylenes from Atomistic Molecular Dynamics Simulations. Polymers, 12(5), 1059. https://doi.org/10.3390/polym12051059