Unusual Structures of Interpolyelectrolyte Complexes: Vesicles and Perforated Vesicles



Abstract
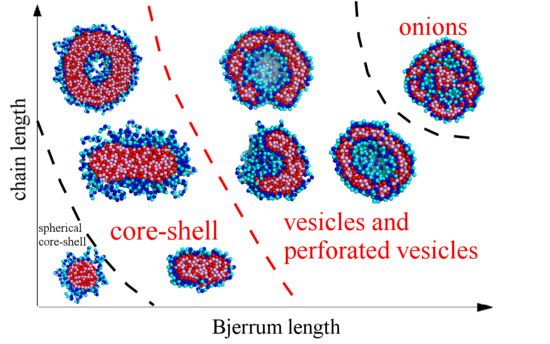
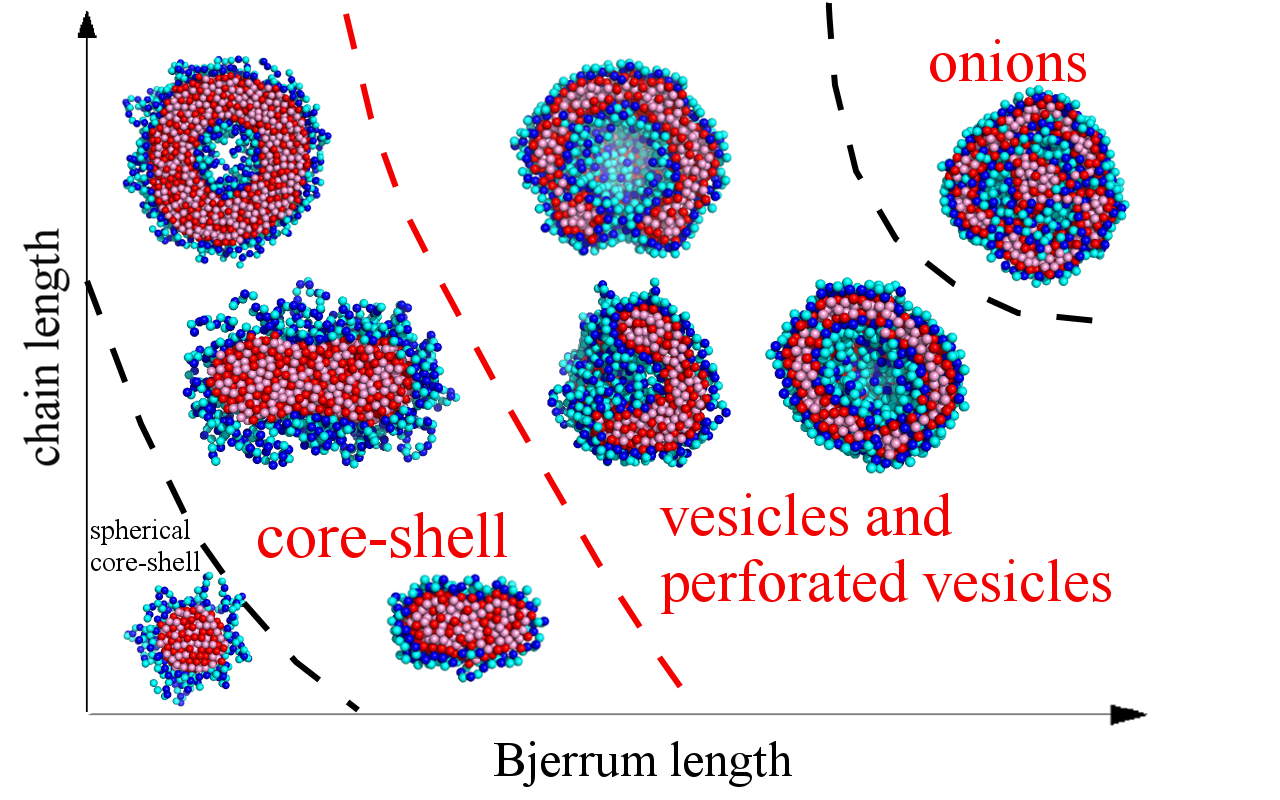{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Computer Simulation
2.1. Model and Simulation Technique
2.2. Results and Discussion
3. Analytical Theory
3.1. Model
3.1.1. The “Core–Shell” Structure
3.1.2. Vesicle
3.2. Analytical Results
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
Appendix B
References
- Dorigo, B.; Schalch, T.; Kulangara, A.; Duda, A.; Schroeder, R.R.; Richmond, T.J. Nucleosome arrays reveal the two-start organization of the chromatin fiber. Science 2004, 306, 1571–1573. [Google Scholar] [CrossRef]
- Kabanov, V.A. Basic Properties of Soluble Interpolyelectrolyte Complexes Applied to Bioengineering and Cell Transformations. In Macromolecular Complexes in Chemistry and Biology; Dubin, P., Bock, J., Davies, R.M., Schulz, D.N., Thies, C., Eds.; Springer: Berlin, Germany, 1994; pp. 151–174. [Google Scholar]
- Liao, I.-C.; Wan, A.C.A.; Yim, E.K.F.; Leong, K.W. Controlled release from fibers of polyelectrolyte complexes. J. Control. Release 2005, 104, 347–358. [Google Scholar] [CrossRef]
- Müller, M. Sizing, shaping and pharmaceutical applications of polyelectrolyte complex nanoparticles. Adv. Polym. Sci. 2014, 256, 197–260. [Google Scholar]
- Thomas, M.; Klibanov, A.M. Non-viral gene therapy: Polycation-mediated DNA delivery. Appl. Microbiol. Biotechnol. 2003, 62, 27–34. [Google Scholar] [CrossRef]
- Safinya, C. Structures of lipid-DNA complexes: Supramolecular assembly and gene delivery. Curr. Opin. Struct. Biol. 2001, 11, 440–448. [Google Scholar] [CrossRef]
- Boustta, M.; Leclercq, L.; Vert, M.; Vasilevskaya, V.V. Experimental and Theoretical Studies of Polyanion-Polycation Complexation in Salted Media in the Context of Nonviral Gene Transfection. Macromolecules 2014, 47, 3574–3581. [Google Scholar] [CrossRef]
- Bertin, A. Polyelectrolyte complexes of DNA and polycations as gene delivery vectors. Adv. Polym. Sci. 2014, 256, 103–196. [Google Scholar]
- Giron-Gonzalez, M.D.; Salto-Gonzalez, R.; Lopez-Jaramillo, F.J.; Salinas-Castillo, A.; Jodar-Reyes, A.B.; Ortega-Muñoz, M.; Hernandez-Mateo, F.; Santoyo-Gonzalez, F. Polyelectrolyte Complexes of Low Molecular Weight PEI and Citric Acid as Efficient and Nontoxic Vectors for in Vitro and in Vivo Gene Delivery. Bioconj. Chem. 2016, 27, 549–561. [Google Scholar] [CrossRef]
- Yu, L.; Liu, X.K.; Yuan, W.C.; Brown, L.J.; Wang, D.Y. Confined flocculation of ionic pollutants by poly(L-dopa)-based polyelectrolyte complexes in hydrogel beads for three-dimensional, quantitative efficient water decontamination. Langmuir 2015, 31, 6351–6366. [Google Scholar] [CrossRef]
- Panova, I.G.; Sybachin, A.V.; Spiridonov, V.V.; Kydralieva, K.; Jorobekova, S.; Zezin, A.B.; Yaroslavov, A.A. Non-stoichiometric interpolyelectrolyte complexes: Promising candidates for protection of soils. Geoderma 2017, 307, 91–97. [Google Scholar] [CrossRef]
- Ou, Z.; Muthukumar, M. Entropy and enthalpy of polyelectrolyte complexation: Langevin dynamics simulations. J. Chem. Phys. 2006, 124, 154902. [Google Scholar] [CrossRef]
- Grosberg, A.Y.; Khokhlov, A.R. Statistical Physics of Macromolecules; AIP Press: New York, NY, USA, 1994. [Google Scholar]
- Priftis, D.; Laugel, N.; Tirrell, M. Thermodynamic characterization of polypeptide complex coacervation. Langmuir 2012, 28, 15947–15957. [Google Scholar] [CrossRef]
- Winkler, R.G. Universal properties of complexes formed by two oppositely charged flexible polyelectrolytes. New J. Phys. 2004, 6, 11. [Google Scholar] [CrossRef]
- Tsuchida, E. Formation of polyelectrolyte complexes and their structures. J. Macromol. Sci. Pure Appl. Chem. 1994, 31, 1–15. [Google Scholar] [CrossRef]
- Shovsky, A.; Varga, I.; Makuška, R.; Claesson, P.M. Formation and Stability of Water-Soluble, Molecular Polyelectrolyte Complexes: Effects of Charge Density, Mixing Ratio, and Polyelectrolyte Concentration. Langmuir 2009, 25, 6113–6121. [Google Scholar] [CrossRef]
- Machinskaya, A.E.; Leclercq, L.; Boustta, M.; Vert, M.; Vasilevskaya, V.V. Salt Effects on Macrophase Separations in Non-Stoichiometric Mixtures of Oppositely Charged Macromolecules: Theory and Experiment. J. Pol. Sci. B Pol. Phys. 2016, 54, 1717–1730. [Google Scholar] [CrossRef]
- Overbeek, J.T.G.; Voorn, M.J. Phase separation in polyelectrolyte solutions. Theory of complex coacervation. J. Cell. Compar. Physiol. 1957, 49, 7–26. [Google Scholar] [CrossRef]
- Rumyantsev, A.M.; Potemkin, I.I. Explicit description of complexation between oppositely charged polyelectrolytes as an advantage of the random phase approximation over the scaling approach. Phys. Chem. Chem. Phys. 2017, 19, 27580–27592. [Google Scholar] [CrossRef]
- Ali, S.; Bleuel, M.; Prabhu, V.M. Lower critical temperature in polyelectrolyte complex coacervates. ACS Macro Lett. 2019, 8, 289–293. [Google Scholar] [CrossRef]
- Harada, A.; Kataoka, K. Formation of Polyion Complex Micelles in an Aqueous Milieu from a Pair of Oppositely-Charged Block Copolymers with Poly (ethylene glycol) Segments. Macromolecules 1995, 28, 5294–5299. [Google Scholar] [CrossRef]
- Kabanov, A.V.; Bronich, V.K.; Kabanov, V.A.; Yu, K.; Eisenberg, A. Soluble Stoichiometric Complexes from Poly(N-ethyl-4-vinylpyridinium) Cations and Poly(ethylene oxide)-block-polymethacrylate Anions. Macromolecules 1996, 29, 6797–6802. [Google Scholar] [CrossRef]
- Etrych, T.; Leclercq, L.; Boustta, M.; Vert, M. Polyelectrolyte complex formation and stability when mixing polyanions and polycations in salted media: A model study related to the case of body fluids. Eur. J. Pharm. Sci. 2005, 25, 281–288. [Google Scholar] [CrossRef]
- Vasilevskaya, V.V.; Leclercq, L.; Boustta, M.; Vert, M.; Khokhlov, A.R. Study of Interpolymer Complexes of Oppositely Charged Macromolecules with Different Affinity to Solvent. Macromolecules 2007, 40, 5934–5940. [Google Scholar] [CrossRef]
- Glagoleva, A.A.; Vasilevskaya, V.V. On Conditions of Formation of Hollow Particles by an Interpolylectrolyte Complex. Polym. Sci. Ser. A 2019, 61, 780–788. [Google Scholar] [CrossRef]
- Vasilevskaya, V.V.; Klochkov, A.A.; Lazutin, A.A.; Khalatur, P.G.; Khokhlov, A.R. HA (Hydrophobic/Amphiphilic) Copolymer Model: Coil−Globule Transition versus Aggregation. Macromolecules 2004, 37, 5444–5460. [Google Scholar] [CrossRef]
- Kale, T.S.; Klaikherd, A.; Popere, B.; Thayumanavan, S. Supramolecular Assemblies of Amphiphilic Homopolymers. Langmuir 2009, 25, 9660–9670. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, K.; Mullen, K.; Yin, M. Self-assemblies of amphiphilic homopolymers: Synthesis, morphology studies and biomedical applications. Chem. Commun. 2015, 51, 11541–11555. [Google Scholar] [CrossRef]
- Glagoleva, A.A.; Vasilevskaya, V.V.; Khokhlov, A.R. Vesicle-Like Globules of Amphiphilic Macromolecules. Macromol. Theory Simul. 2015, 24, 393–398. [Google Scholar] [CrossRef]
- Glagoleva, A.A.; Vasilevskaya, V.V. Formation of a Vesicle-Like Globule under Steric Restrictions. Polym. Sci. Ser. A 2016, 58, 292–301. [Google Scholar] [CrossRef]
- Larin, D.E.; Glagoleva, A.A.; Govorun, E.N.; Vasilevskaya, V.V. Morphological diagram of amphiphilic H-graft-P macromolecules: Theory and computer experiment. Polymer 2018, 146, 230–241. [Google Scholar] [CrossRef]
- Vasilevskaya, V.V.; Govorun, E.N. Hollow and Vesicle Particles from Macromolecules with Amphiphilic Monomer Units. Polym. Rev. 2019, 59, 625–650. [Google Scholar] [CrossRef]
- Vasilevskaya, V.V.; Ermilov, V.A. Computer simulation of macromolecular systems with amphiphilic monomer units: Biomimetic models. Polym. Sci. Ser. A 2011, 53, 846–866. [Google Scholar] [CrossRef]
- Kharel, S.; Gautam, A.; Dickescheid, A.; Loo, S.C.J. Hollow Microparticles as a Superior Delivery System over Solid Microparticles for the Encapsulation of Peptides. Pharm. Res. 2018, 35, 185. [Google Scholar] [CrossRef]
- Antonietti, M.; Forster, S. Vesicles and liposomes: A self-assembly principle beyond lipids. Adv. Mater. 2003, 15, 1323–1333. [Google Scholar] [CrossRef]
- Muller, L.K.; Landfester, K. Natural liposomes and synthetic polymeric structures for biomedical applications. Biochem. Bioph. Res. Commun. 2015, 468, 411–418. [Google Scholar] [CrossRef]
- Yaroslavov, A.A.; Zaborova, O.V.; Sybachin, A.V.; Kalashnikova, I.V.; Kesselman, E.; Schmidt, J.; Talmon, Y.; Rodriguez, A.R.; Deming, T.J. Biodegradable containers composed of anionic liposomes and cationic polypeptide vesicles. RSC Adv. 2015, 5, 98687–98691. [Google Scholar] [CrossRef][Green Version]
- Discher, B.M.; Won, Y.-Y.; Ege, D.S.; Lee, J.C.-M.; Bates, F.S.; Discher, D.E.; Hammer, D.A. Polymersomes: Tough vesicles made from diblock copolymers. Science 1999, 284, 1143–1146. [Google Scholar] [CrossRef]
- Kishimura, A. Development of polyion complex vesicles (PICsomes) from block copolymers for biomedical applications. Polym. J. 2013, 45, 892–897. [Google Scholar] [CrossRef]
- Anraku, Y.; Kishimura, A.; Oba, M.; Yamasaki, Y.; Kataoka, K. Spontaneous formation of nanosized unilamellar polyion complex vesicles with tunable size and properties. J. Am. Chem. Soc. 2010, 132, 1631–1636. [Google Scholar] [CrossRef]
- Cha, J.N.; Birkedal, H.; Euliss, L.E.; Bartl, M.H.; Wong, M.S.; Deming, T.J.; Stucky, G.D. Spontaneous Formation of Nanoparticle Vesicles from Homopolymer Polyelectrolytes. J. Am. Chem. Soc. 2003, 125, 8285–8289. [Google Scholar] [CrossRef]
- Donath, E.; Sukhorukov, G.B.; Caruso, F.; Davies, S.A.; Möhwald, H. Novel Hollow Polymer Shells by Colloid-Templated Assembly of Polyelectrolytes. Angew. Chem. Int. Ed. 1998, 37, 2202–2205. [Google Scholar] [CrossRef]
- Decher, G. Fuzzy Nanoassemblies: Toward Layered Polymeric Multicomposites. Science 1997, 277, 1232–1237. [Google Scholar] [CrossRef]
- Driver, K.; Baco, S.; Khutoryanskiy, V.V. Hollow capsules formed in a single stage via interfacial hydrogen-bonded complexation of methylcellulose with poly(acrylic acid) and tannic acid. Eur. Pol. J. 2013, 49, 4249–4256. [Google Scholar] [CrossRef]
- Wang, Q.; Schlenoff, J.B. Single-and Multicompartment Hollow Polyelectrolyte Complex Microcapsules by One-Step Spraying. Adv. Mater. 2015, 27, 2077–2082. [Google Scholar] [CrossRef]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics; Sandia National Labs.: Albuquerque, NM, USA, 1993.
- Pollock, E.L.; Glosli, J. Comments on p3M, FMM, and the Ewald method for large periodic Coulombic systems. Comput. Phys. Commun. 1996, 95, 93–110. [Google Scholar] [CrossRef]
- Almgren, M. Vesicle Transformations Resulting from Curvature Tuning in Systems with Micellar, Lamellar, and Bicontinuous Cubic Phases. J. Dispers. Sci. Technol. 2007, 28, 43–54. [Google Scholar] [CrossRef]
- Alakoskela, J.-M.; Parry, M.J.; Kinnunen, P.K.J. The Intermediate State of DMPG Is Stabilized by Enhanced Positive Spontaneous Curvature. Langmuir 2010, 26, 4892–4900. [Google Scholar] [CrossRef]
- Berlepsch, H.V.; Thota, B.N.S.; Wyszogrodzka, M.; de Carlo, S.; Haag, R.; Böttcher, C. Controlled self-assembly of stomatosomes by use of single-component fluorinated dendritic amphiphiles. Soft Matter 2018, 14, 5256–5269. [Google Scholar] [CrossRef]
- He, X.; Schmid, F. Spontaneous Formation of Complex Micelles from a Homogeneous Solution. Phys. Rev. Lett. 2008, 100, 137802. [Google Scholar] [CrossRef]
- Kong, W.; Li, B.; Jin, Q.; Ding, D.; Shi, A.C. Complex micelles from self-assembly of ABA triblock copolymers in B-selective solvents. Langmuir 2010, 26, 4226–4232. [Google Scholar] [CrossRef]
- Arfken, G.B.; Weber, H. Mathematical Methods for Physics; Academic Press: London, UK, 1995. [Google Scholar]
- Steinhardt, P.J.; Nelson, D.R.; Ronchetti, M. Bond-orientational order in liquids and glasses. Phys. Rev. B 1983, 28, 784–805. [Google Scholar] [CrossRef]
- Terrones, H.; Mackay, A.L. The characterisation of coordination polyhedral by invariants. J. Math. Chem. 1994, 15, 157–181. [Google Scholar] [CrossRef]
- Whitford, P.C.; Phillies, G.D.J. Enhanced septahedral ordering in cold Lennard-Jones fluids. Phys. Rev. E 2005, 72, 021203. [Google Scholar] [CrossRef]
- Scott, D.; Mountjoy, G. Rotational invariants of network former and modifier cations in silicate glasses. J. Non Cryst. Solids 2014, 401, 54–59. [Google Scholar] [CrossRef]
- Wang, L.; Fujimoto, K.; Yoshii, N.; Okazaki, S. A molecular dynamics study of the breathing and deforming modes of the spherical ionic SDS and nonionic C12E8 micelles. J. Chem. Phys. 2016, 144, 034903. [Google Scholar] [CrossRef]
- Yoshii, N.; Nimura, Y.; Fujimoto, K.; Okazaki, S. Spherical harmonics analysis of surface density fluctuations of spherical ionic SDS and nonionic C12E8 micelles: A molecular dynamics study. J. Chem. Phys. 2017, 147, 034906. [Google Scholar] [CrossRef]
- Erukhimovich, I. Weak Segregation Theory and Multicompartment Spherical Micelles. Polym. Sci. Ser. C 2018, 60, S49–S55. [Google Scholar] [CrossRef]
- Borukhov, I.; Andelman, D.; Orland, H. Polyelectrolyte Solutions between Charged Surfaces. Europhys. Lett. 1995, 32, 499–504. [Google Scholar] [CrossRef]
- Lifshitz, I.M. Some problems of the statistical theory of biopolymers. Sov. Phys. JETP-USSR 1969, 28, 1280–1286. [Google Scholar]
- Lifshitz, I.M.; Grosberg, A.Y.; Khokhlov, A.R. Some problems of the statistical physics of polymer chains with volume interactions. Rev. Mod. Phys. 1978, 50, 683–713. [Google Scholar] [CrossRef]
- Sadovnichy, V.; Tikhonravov, A.; Voevodin, V.; Opanasenko, V. “Lomonosov”: Supercomputing at Moscow State University. In Contemporary High Performance Computing: From Petascale toward Exascale (Chapman & Hall/CRC Computational Science); CRC Press: Boca Raton, FL, USA, 2013; pp. 283–307. [Google Scholar]










© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Glagoleva, A.A.; Larin, D.E.; Vasilevskaya, V.V. Unusual Structures of Interpolyelectrolyte Complexes: Vesicles and Perforated Vesicles. Polymers 2020, 12, 871. https://doi.org/10.3390/polym12040871
Glagoleva AA, Larin DE, Vasilevskaya VV. Unusual Structures of Interpolyelectrolyte Complexes: Vesicles and Perforated Vesicles. Polymers. 2020; 12(4):871. https://doi.org/10.3390/polym12040871
Chicago/Turabian StyleGlagoleva, A. A., D. E. Larin, and V. V. Vasilevskaya. 2020. "Unusual Structures of Interpolyelectrolyte Complexes: Vesicles and Perforated Vesicles" Polymers 12, no. 4: 871. https://doi.org/10.3390/polym12040871
APA StyleGlagoleva, A. A., Larin, D. E., & Vasilevskaya, V. V. (2020). Unusual Structures of Interpolyelectrolyte Complexes: Vesicles and Perforated Vesicles. Polymers, 12(4), 871. https://doi.org/10.3390/polym12040871