Solubility Improvement of Progesterone from Solid Dispersions Prepared by Solvent Evaporation and Co-milling





Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Preparation of PG Solid Dispersions Using Solvent Evaporation
2.2.2. Preparation of PG Solid Dispersions Using Mechano-chemical Activation
2.2.3. Preparation of Physical Mixtures
2.2.4. Differential Scanning Calorimetry (DSC)
2.2.5. Fourier-Transform Infrared Spectroscopy (FTIR) for Solid Dispersions
2.2.6. FTIR Studies of Solid Dispersions Dissolved in D2O and Methanol
2.2.7. Solubility Studies
2.2.8. Drug–Polymer Miscibility
Solubility Parameters
Flory–Huggins Thermodynamic Analysis
3. Results and Discussion
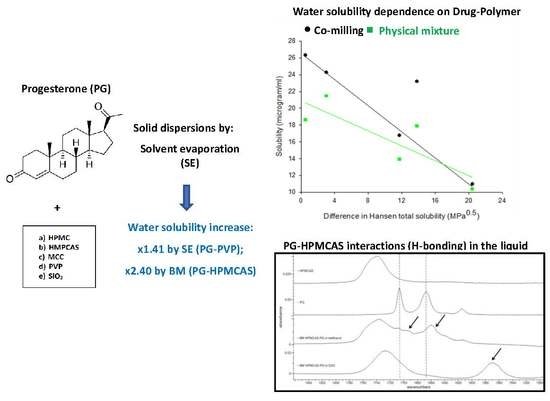
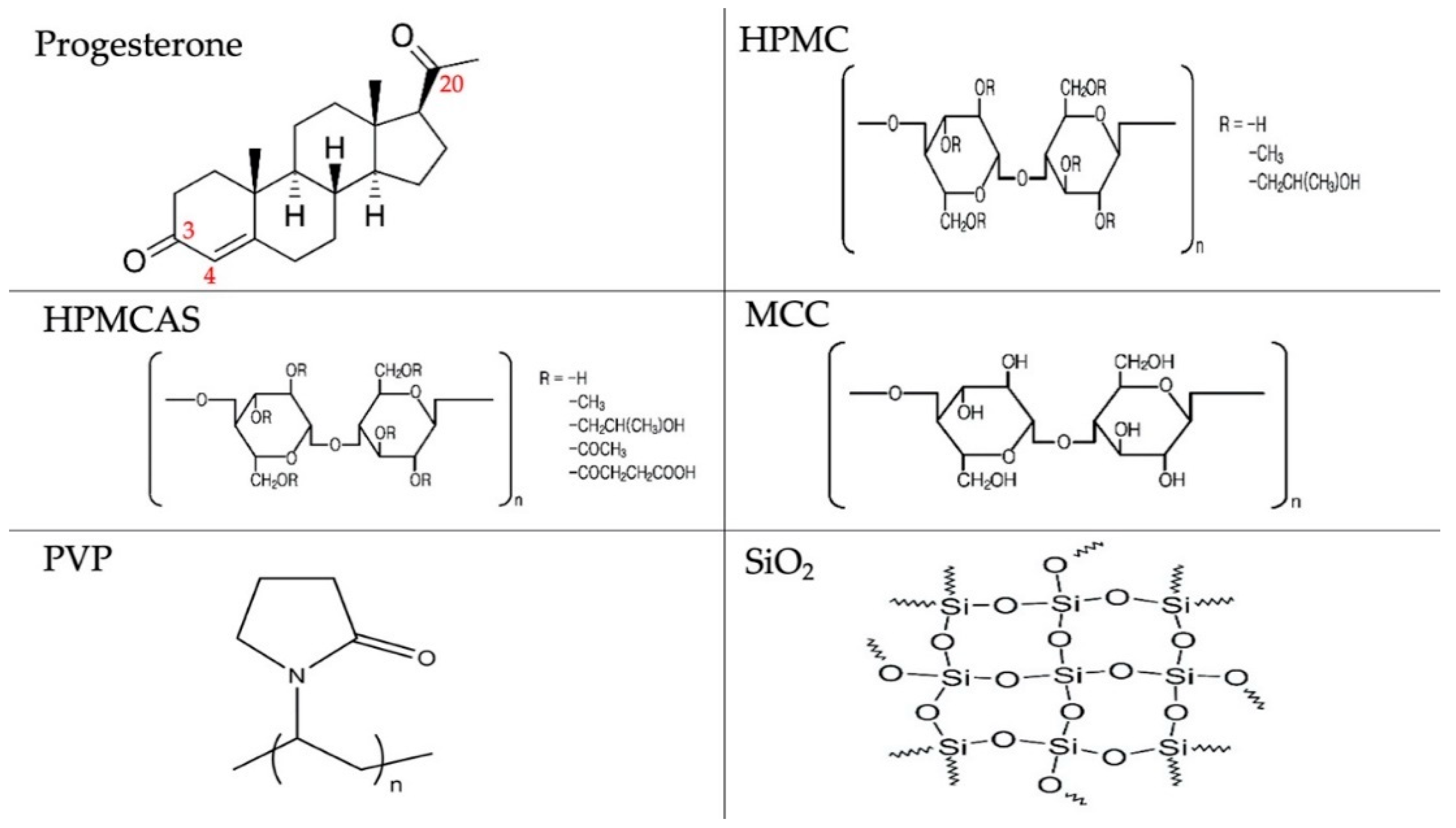
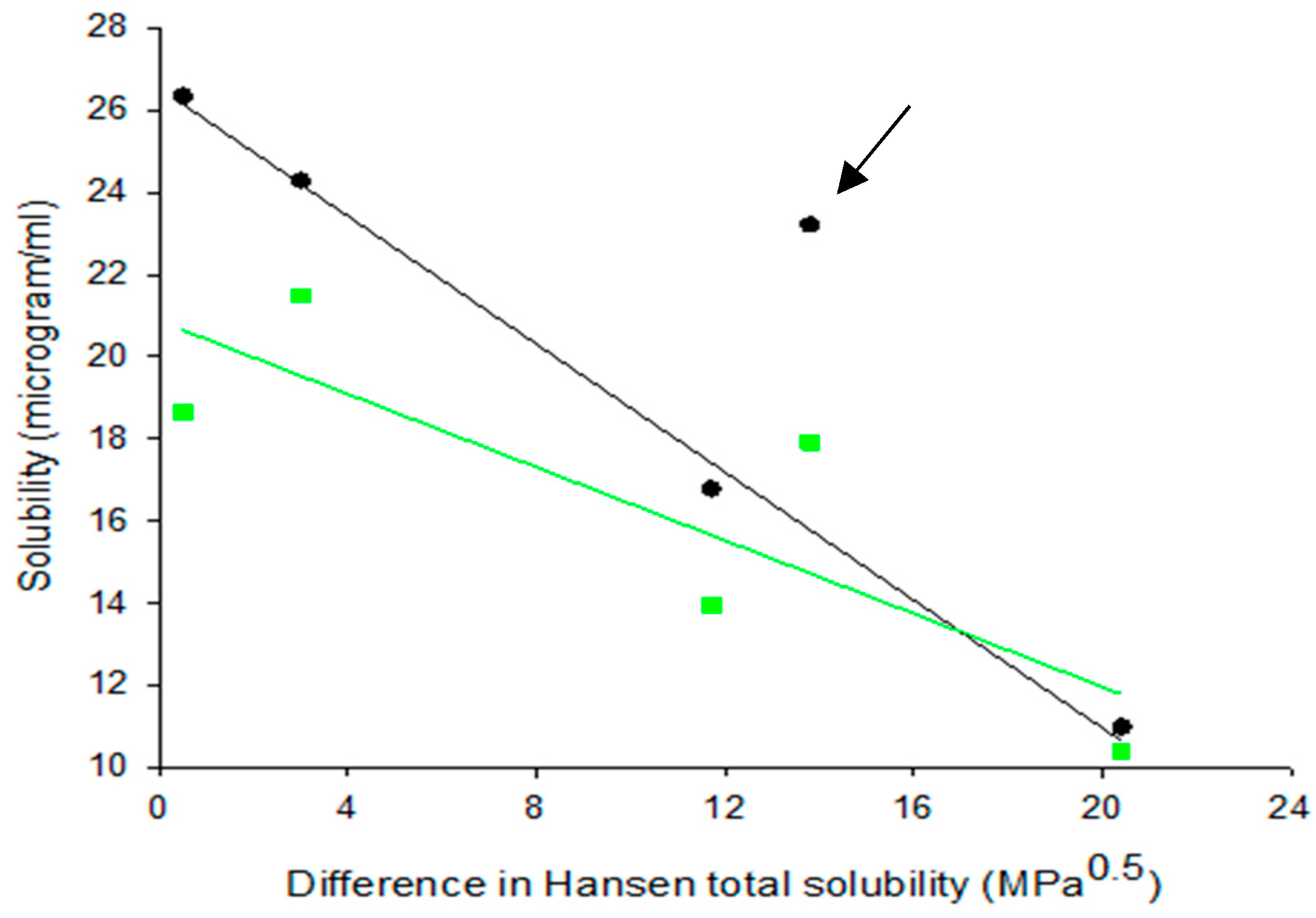
3.1. Hansen Solubility Parameters
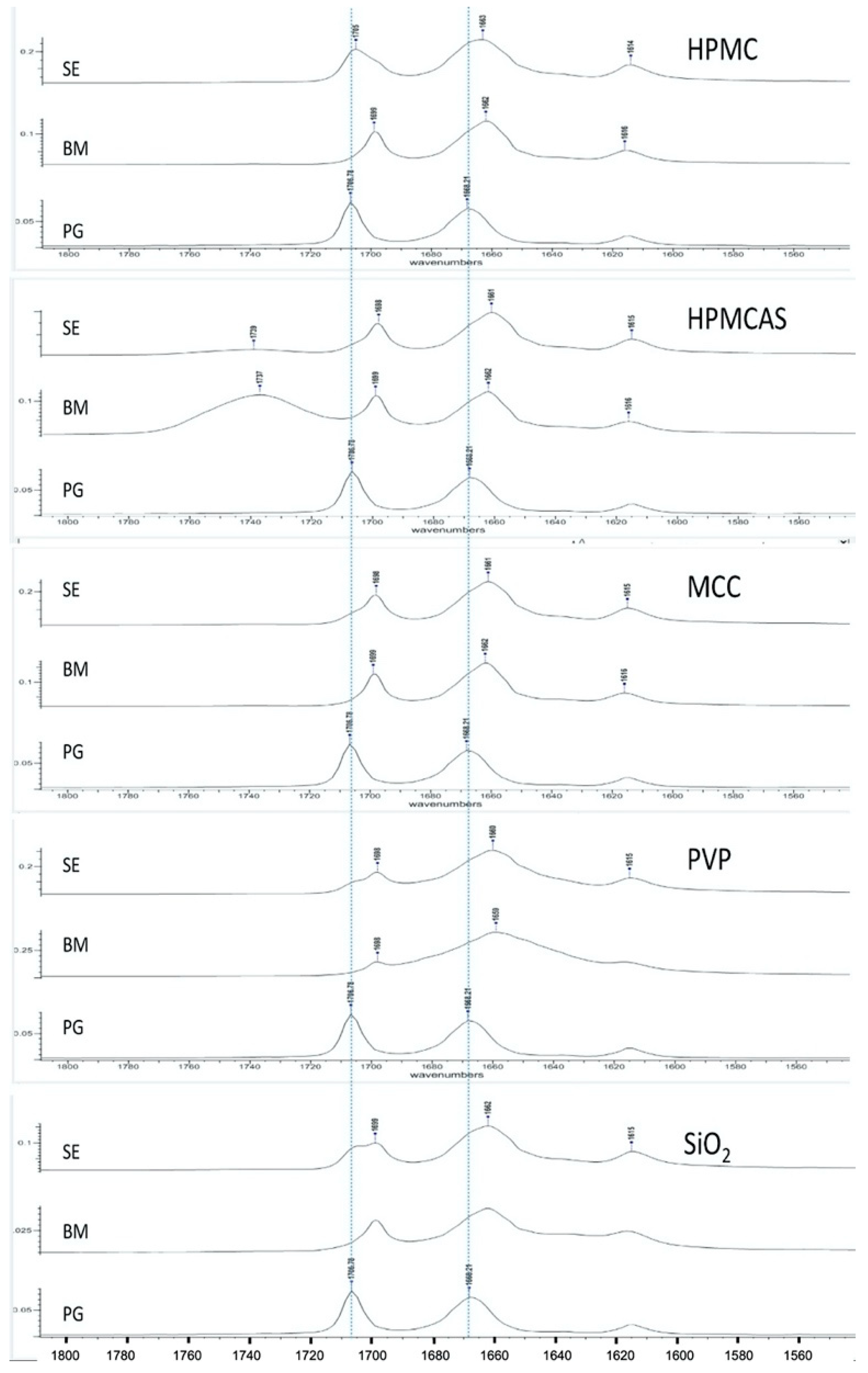
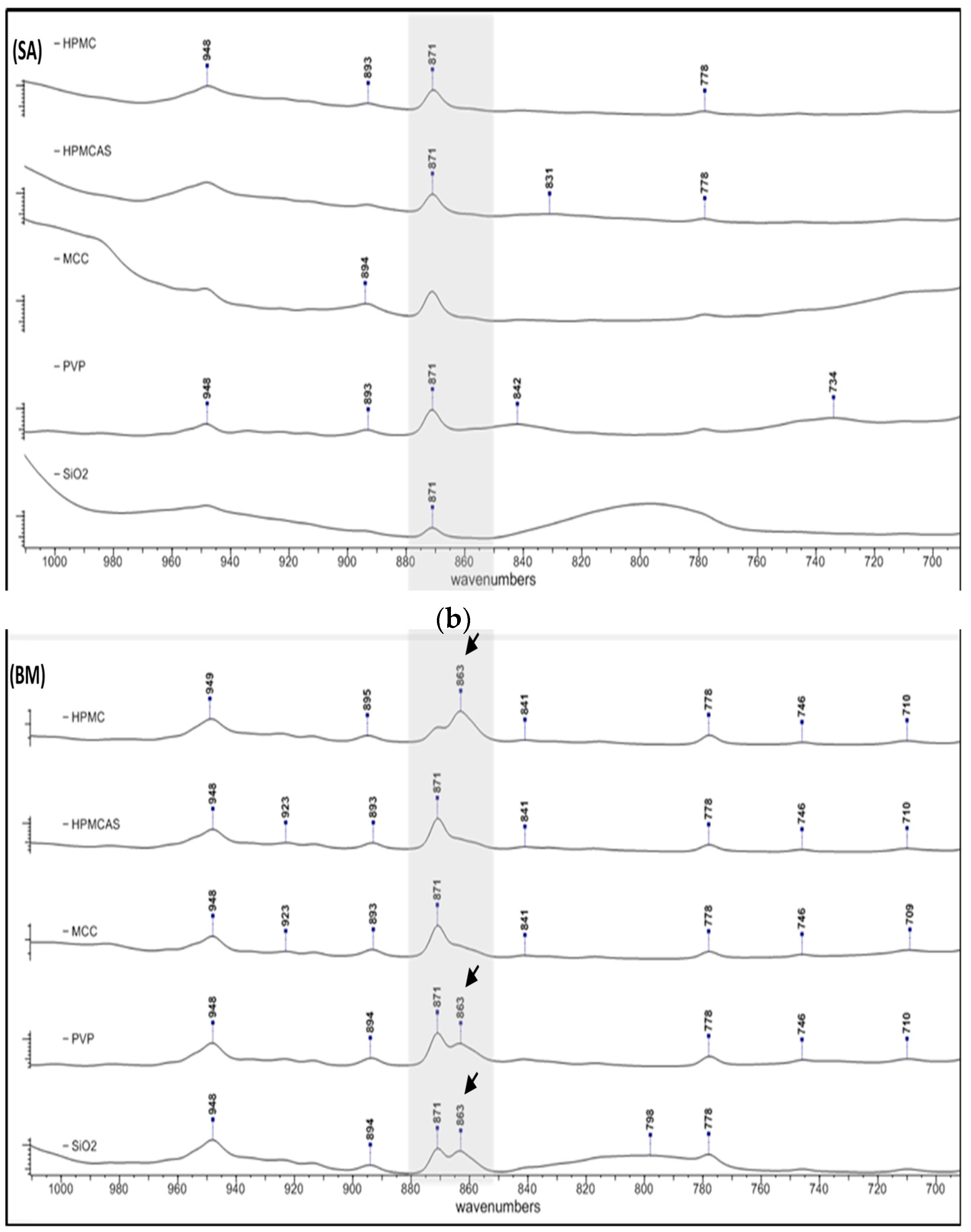
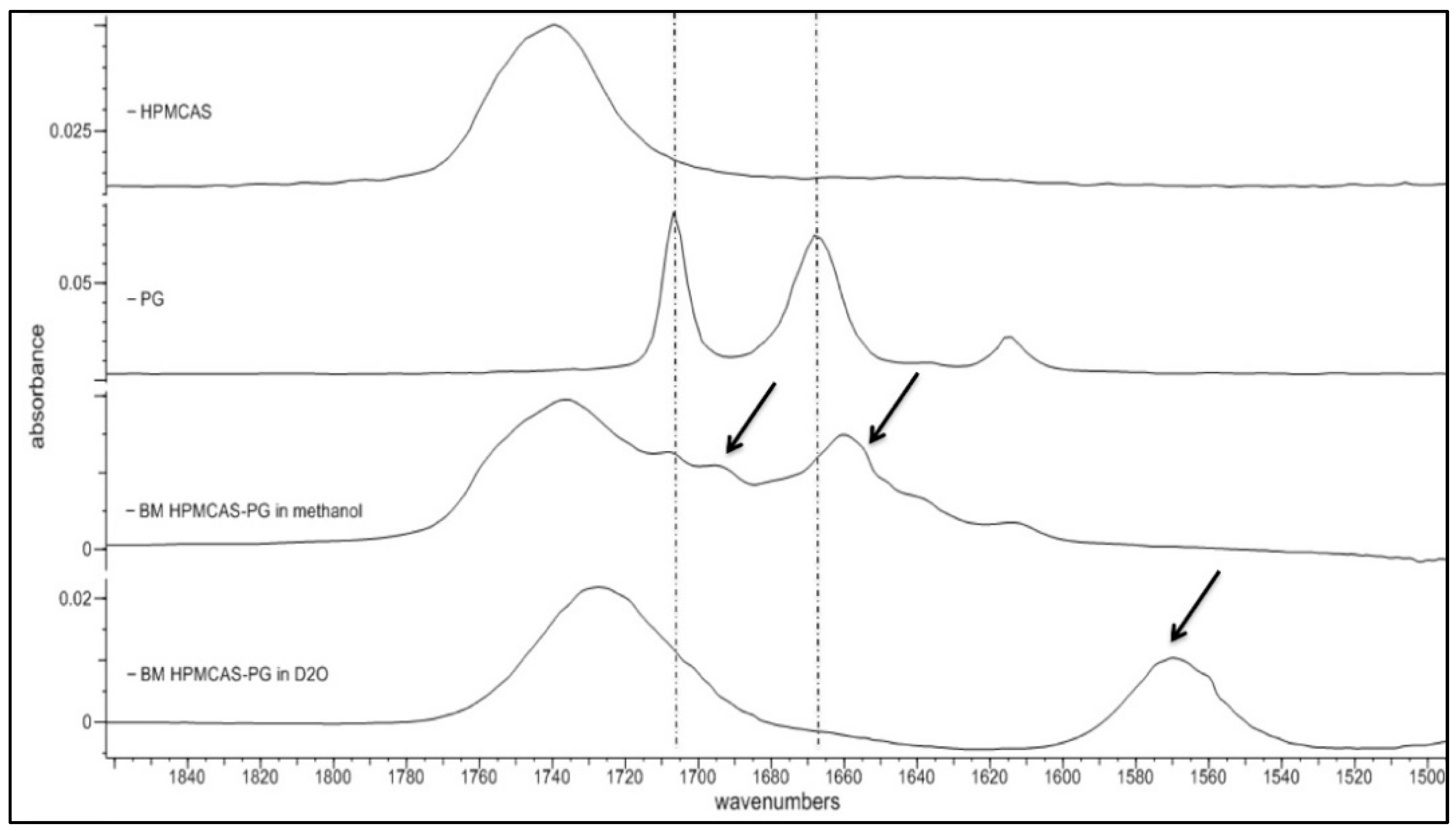
3.2. Spectroscopic Analysis of PG Solid Dispersions
3.2.1. Drug–Polymer Interactions
3.2.2. Polymorphism
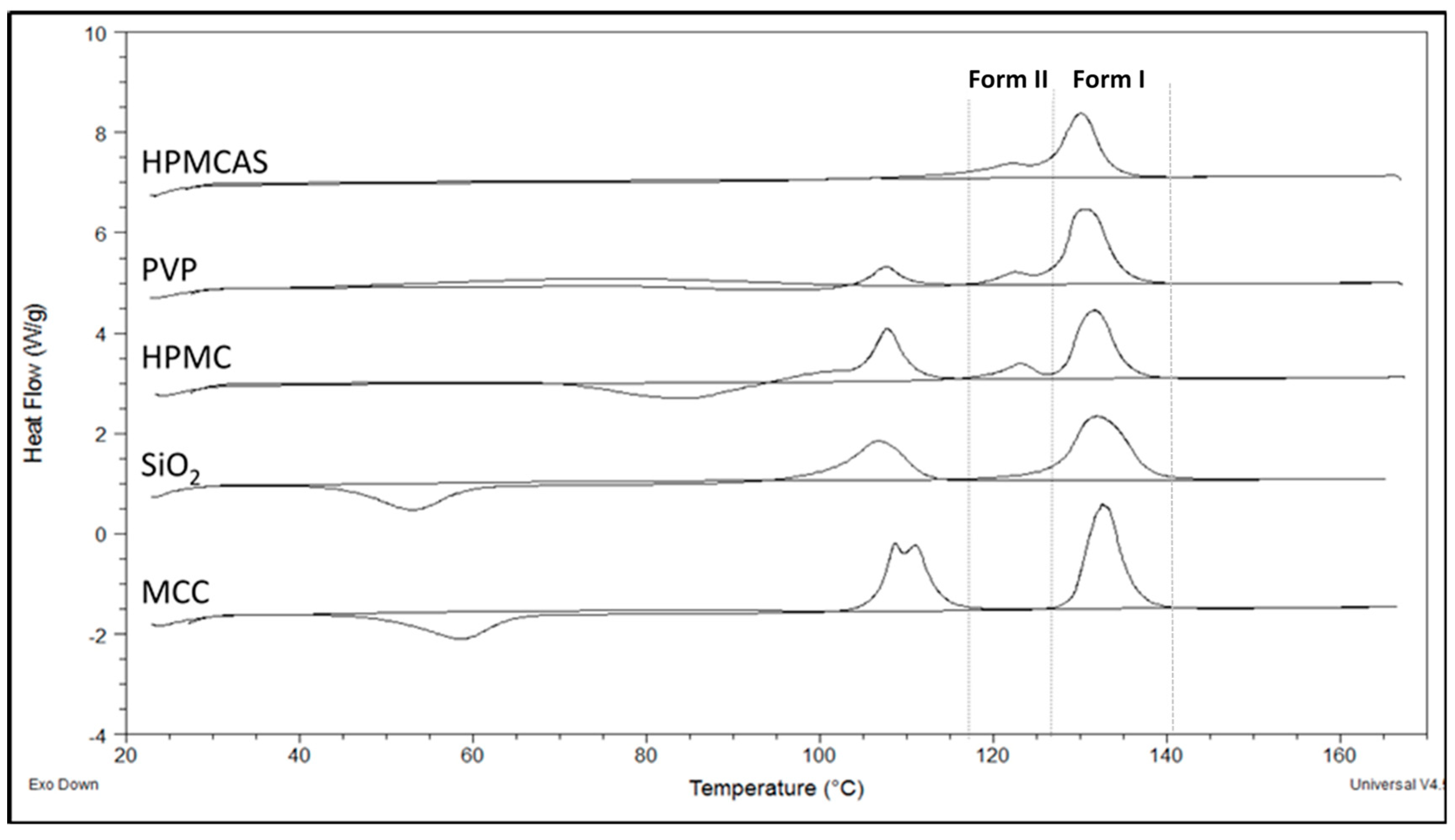
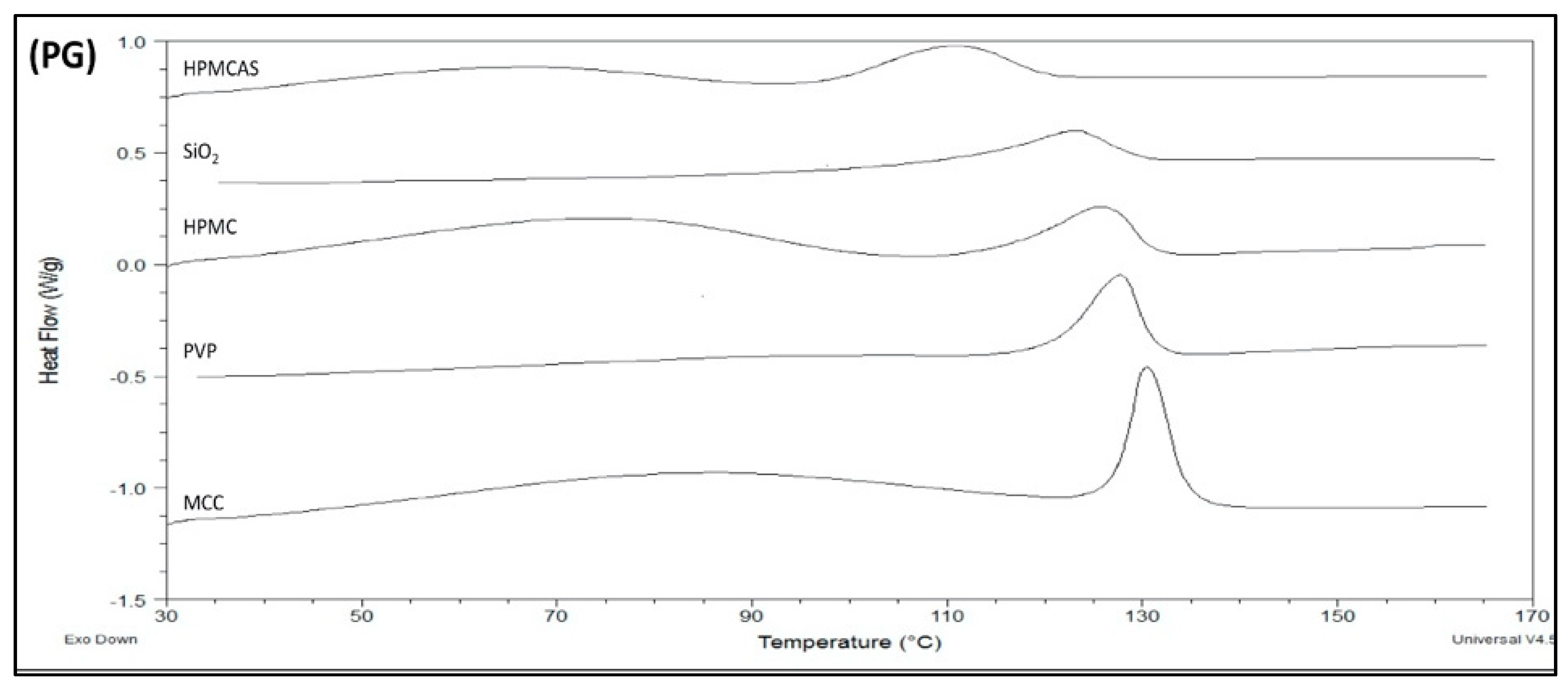
3.3. Thermal Analysis of PG Solid Dispersions
3.3.1. DSC of PG Solid Dispersions Prepared by Solvent Evaporation
3.3.2. DSC of PG Solid Dispersions Prepared by Co-milling
3.4. Solubility Studies
3.4.1. Drug Solubility from Solid Dispersions Prepared by Solvent Evaporation (SE)
3.4.2. Drug Solubility from Solid Dispersions Prepared by Co-milling (BM)
3.5. Spectroscopic Analysis of PG–HPMCAS Interactions in the Liquid State
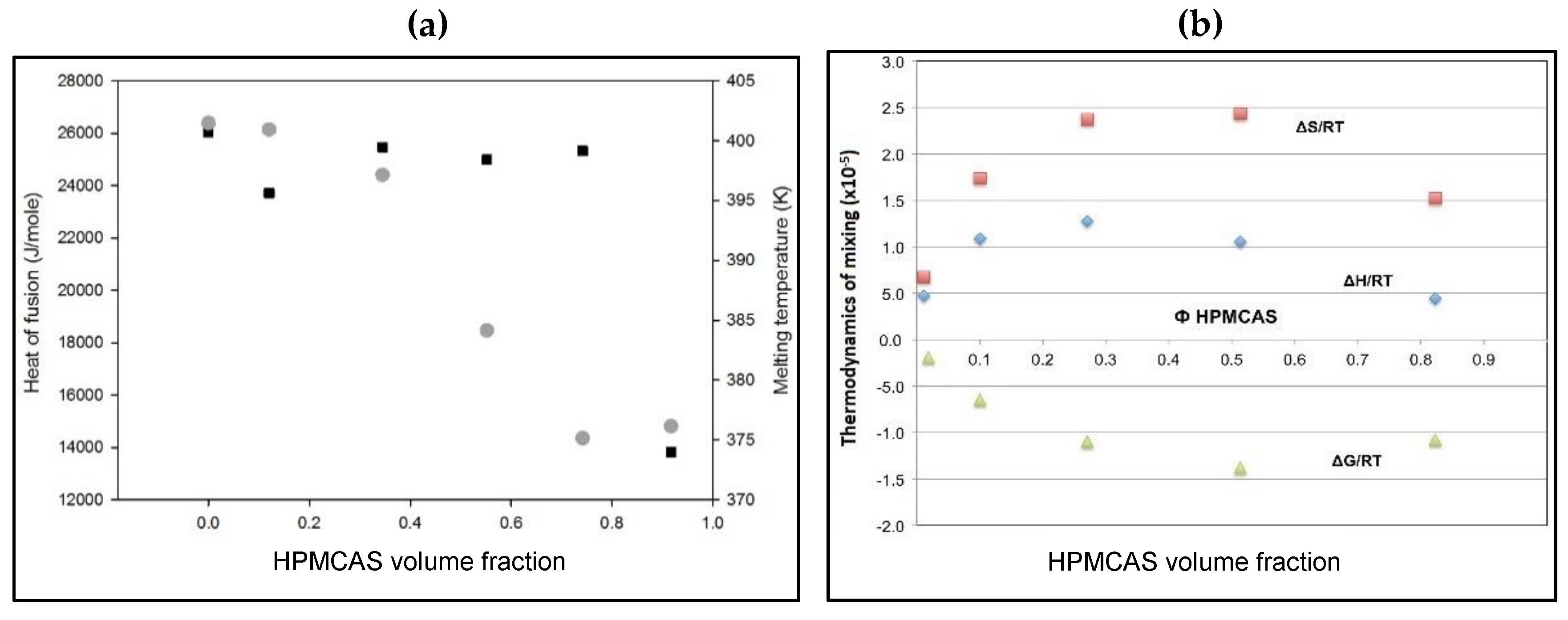
3.6. Flory–Huggins Thermodynamic Analysis of PG–HPMCAS Interactions
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Al-Obaidi, H.; Buckton, G. Evaluation of griseofulvin binary and ternary solid dispersions with HPMCAS. AAPS PharmSciTech 2009, 10, 1172–1177. [Google Scholar] [CrossRef] [PubMed]
- Al-Obaidi, H.; Brocchini, S.; Buckton, G. Anomalous properties of spray dried solid dispersions. J. Pharm. Sci. 2009, 98, 4724–4737. [Google Scholar] [CrossRef]
- Al-Obaidi, H.; Lawrence, M.J.; Shah, S.; Moghul, H.; Al-Saden, N.; Bari, F. Effect of drug-polymer interactions on the aqueous solubility of milled solid dispersions. Int. J. Pharm. 2013, 446, 100–105. [Google Scholar] [CrossRef]
- Barzegar-Jalali, M.; Valizadeh, H.; Shadbad, M.R.S.; Adibkia, K.; Mohammadi, G.; Farahani, A.; Arash, Z.; Nokhodchi, A. Cogrinding as an approach to enhance dissolution rate of a poorly water-soluble drug (gliclazide). Powder. Technol. 2010, 197, 150–158. [Google Scholar] [CrossRef]
- Koutsamanis, I.; Paudel, A.; Nickisch, K.; Eggenreich, K.; Roblegg, E.; Eder, S. Controlled-Release from High-Loaded Reservoir-Type Systems-A Case Study of Ethylene-Vinyl Acetate and Progesterone. Pharmaceutics 2020, 12, 103. [Google Scholar] [CrossRef] [PubMed]
- Hussain, A.; Smith, G.; Khan, K.A.; Bukhari, N.I.; Pedge, N.I.; Ermolina, I. Solubility and dissolution rate enhancement of ibuprofen by co-milling with polymeric excipients. Eur. J. Pharm. Sci. 2018, 123, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Stolle, A.; Schmidt, R.; Jacob, K. Scale-up of organic reactions in ball mills: Process intensification with regard to energy efficiency and economy of scale. Faraday Discuss. 2014, 170, 267–286. [Google Scholar] [CrossRef] [PubMed]
- Metta, N.; Verstraeten, M.; Ghijs, M.; Kumar, A.; Schafer, E.; Singh, R.; De Beer, T.; Nopens, I.; Cappuyns, P.; Van Assche, I.; et al. Model development and prediction of particle size distribution, density and friability of a comilling operation in a continuous pharmaceutical manufacturing process. Int. J. Pharm. 2018, 549, 271–282. [Google Scholar] [CrossRef]
- Hassan, A.S.; Soliman, G.M.; El-Mahdy, M.M.; El-Gindy, G.E.A. Solubilization and Enhancement of Ex Vivo Vaginal Delivery of Progesterone Using Solid Dispersions, Inclusion Complexes and Micellar Solubilization. Curr. Drug. Deliv. 2018, 15, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.S.; Soliman, G.M.; Ali, M.F.; El-Mahdy, M.M.; El-Gindy, G.E.A. Mucoadhesive tablets for the vaginal delivery of progesterone: In vitro evaluation and pharmacokinetics/pharmacodynamics in female rabbits. Drug. Dev. Ind. Pharm. 2018, 44, 224–232. [Google Scholar] [CrossRef]
- Vegeto, E.; Shahbaz, M.M.; Wen, D.X.; Goldman, M.E.; O’Malley, B.W.; McDonnell, D.P. Human progesterone receptor A form is a cell-and promoter-specific repressor of human progesterone receptor B function. Mol. Endocrinol. 1993, 7, 1244–1255. [Google Scholar] [PubMed]
- de Lignieres, B.; Dennerstein, L.; Backstrom, T. Influence of route of administration on progesterone metabolism. Maturitas 1995, 21, 251–257. [Google Scholar] [CrossRef]
- Nandi, I.; Bateson, M.; Bari, M.; Joshi, H.N. Synergistic effect of PEG-400 and cyclodextrin to enhance solubility of progesterone. AAPS PharmSciTech 2003, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Kincl, F.A.; Ciaccio, L.A.; Benagiano, G. Increasing oral bioavailability of progesterone by formulation. J. Steroid Biochem. 1978, 9, 83–84. [Google Scholar] [CrossRef]
- Morville, R.; Dray, F.; Reynier, J.; Barrat, J. The bioavailability of natural progesterone given by mouth. Measurement of steroid concentrations in plasma, endometrium and breast tissue. J. Gynecol. Obstet. Biol. Reprod. 1982, 11, 355–363. [Google Scholar]
- Maxson, W.S.; Hargrove, J.T. Bioavailability of oral micronized progesterone**Supported in part by a grant from PMS (Premenstrual Syndrome) Action, Madison, Wisconsin. Fertil. Steril. 1985, 44, 622–626. [Google Scholar] [CrossRef]
- Hargrove, J.T.; Maxson, W.S.; Colston Wentz, A. Absorption of oral progesterone is influenced by vehicle and particle size. Am. J. Obstet. Gynecol. 1989, 161, 948–951. [Google Scholar] [CrossRef]
- Simon, J.A.; Robinson, D.E.; Andrews, M.C.; Hildebrand, J.R.; Rocci, M.L.; Blake, R.E.; Hodgen, G.D. The absorption of oral micronized progesterone: The effect of food, dose proportionality, and comparison with intramuscular progesterone*†*Supported in part by a grant from Besins-Iscovesco, Paris, France.†Presented in part at the 35th Annual Meeting of the Society for Gynecologic Investigation, Baltimore, Maryland, March 17 to 20, 1988. Fertil. Steril. 1993, 60, 26–33. [Google Scholar] [CrossRef]
- Al-Obaidi, H.; Kowalczyk, R.M.; Kalgudi, R.; Zariwala, M.G. Griseofulvin solvate solid dispersions with synergistic effect against fungal biofilms. Colloids. Surf. B Biointerfaces 2019, 184, 110540. [Google Scholar] [CrossRef]
- Al-Obaidi, H.; Ke, P.; Brocchini, S.; Buckton, G. Characterization and stability of ternary solid dispersions with PVP and PHPMA. Int. J. Pharm. 2011, 419, 20–27. [Google Scholar] [CrossRef]
- Hansen, C. Hansen Solubility Parameters: A User’s Handbook, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2012; p. 546. [Google Scholar]
- Stefanis, E.; Panayiotou, C. Prediction of Hansen Solubility Parameters with a New Group-Contribution Method. Int. J. Thermophys. 2008, 29, 568–585. [Google Scholar] [CrossRef]
- Tsutsumi, S.; Kondo, K.; Kato, Y.; Fujiwara, N.; Yamamoto, H. Determination of Hansen solubility parameters of particles using a capillary penetration method. Chem. Phys. 2019, 521, 115–122. [Google Scholar] [CrossRef]
- Archer, W.L. Hansen solubility parameters for selected cellulose ether derivatives and their use in the pharmaceutical industry. Drug Dev. Ind. Pharm. 1992, 18, 599–616. [Google Scholar] [CrossRef]
- Marsac, P.J.; Konno, H.; Rumondor, A.C.; Taylor, L.S. Recrystallization of nifedipine and felodipine from amorphous molecular level solid dispersions containing poly(vinylpyrrolidone) and sorbed water. Pharm. Res. 2008, 25, 647–656. [Google Scholar] [CrossRef]
- Djuris, J.; Nikolakakis, I.; Ibric, S.; Djuric, Z.; Kachrimanis, K. Preparation of carbamazepine-Soluplus solid dispersions by hot-melt extrusion, and prediction of drug-polymer miscibility by thermodynamic model fitting. Eur. J. Pharm. Biopharm. 2013, 84, 228–237. [Google Scholar] [CrossRef]
- Varghese, S.; Ghoroi, C. Improving the wetting and dissolution of ibuprofen using solventless co-milling. Int. J. Pharm. 2017, 533, 145–155. [Google Scholar] [CrossRef]
- Gupta, P.; Thilagavathi, R.; Chakraborti, A.K.; Bansal, A.K. Role of molecular interaction in stability of celecoxib-PVP amorphous systems. Mol. Pharm. 2005, 2, 384–391. [Google Scholar] [CrossRef]
- Yang, K.Y.; Glemza, R.; Jarowski, C.I. Effects of amorphous silicon dioxides on drug dissolution. J. Pharm. Sci. 1979, 68, 560–565. [Google Scholar] [CrossRef]
- Bounartzi, M.; Panagopoulou, A.; Kantiranis, N.; Malamataris, S.; Nikolakakis, I. Effect of plasticiser type on the hot melt extrusion of venlafaxine hydrochloride. J. Pharm. Pharmacol. 2014, 66, 297–308. [Google Scholar] [CrossRef]
- Kolter, K.; Karl, M.; Gryczke, A.; Ludwigshafen am Rhein, B. Hot-Melt Extrusion with BASF Pharma Polymers: Extrusion Compendium; BASF SE: Ludwigshafen, Germany, 2012. [Google Scholar]
- Lehmkemper, K.; Kyeremateng, S.O.; Bartels, M.; Degenhardt, M.; Sadowski, G. Physical stability of API/polymer-blend amorphous solid dispersions. Eur. J. Pharm. Biopharm. 2018, 124, 147–157. [Google Scholar] [CrossRef]
- Szcześniak, L.; Rachocki, A.; Tritt-Goc, J. Glass transition temperature and thermal decomposition of cellulose powder. Cellulose 2008, 15, 445–451. [Google Scholar] [CrossRef]
- Le Questel, J.Y.; Boquet, G.; Berthelot, M.; Laurence, C. Hydrogen bonding of progesterone: A combined theoretical, spectroscopic, thermodynamic, and crystallographic database study. J. Phys. Chem. B 2000, 104, 11816–11823. [Google Scholar] [CrossRef]
- Sheskey, P.J.; Cook, W.G.; Cable, C.G.; American Pharmacists, A. Handbook of Pharmaceutical Excipients; Pharmaceutical Press: London, UK, 2017. [Google Scholar]
- Al-Oweini, R.; El-Rassy, H. Synthesis and characterization by FTIR spectroscopy of silica aerogels prepared using several Si(OR)(4) and R″ Si(OR′)(3) precursors. J. Mol. Struct. 2009, 919, 140–145. [Google Scholar] [CrossRef]
- Tripathi, R.; Biradar, S.V.; Mishra, B.; Paradkar, A.R. Study of polymorphs of progesterone by novel melt sonocrystallization technique: A technical note. AAPS PharmSciTech 2010, 11, 1493–1498. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, A.; Ragab, D.; Rohani, S. Polymorphism of Progesterone: A New Approach for the Formation of Form II and the Relative Stabilities of Form I and Form II. Cryst. Growth. Des 2014, 14, 4574–4582. [Google Scholar] [CrossRef]
- Bernabei, M.T.; Gamberini, G.; Cameroni, R. [Polymorphism of progesterone. 3. Solubility and thermodynamic studies of 2 crystalline forms]. Farm. Prat 1974, 29, 184–191. [Google Scholar]
- Cameroni, R.; Gamberini, G.; Bernabei, M.T.; Facchini, M. Polymorphism of progesterone. I. Preparation and characterization of polymorphic forms. Farm. Prat 1973, 28, 621–635. [Google Scholar]
- Cameroni, R.; Gamberini, G.; Bernabei, M.T. Polymorphism of progesterone. II. Use of differential calorimetry in the study of crystalline forms. Farm. Prat 1973, 28, 636–641. [Google Scholar]
- Lancaster, R.W.; Karamertzanis, P.G.; Hulme, A.T.; Tocher, D.A.; Lewis, T.C.; Price, S.L. The polymorphism of progesterone: Stabilization of a ’disappearing’ polymorph by co-crystallization. J. Pharm. Sci. 2007, 96, 3419–3431. [Google Scholar] [CrossRef]
- Legendre, B.; Feutelais, Y.; Defossemont, G. Importance of heat capacity determination in homogeneous nucleation: Application to progesterone. Thermochim. Acta 2003, 400, 213–219. [Google Scholar] [CrossRef]
- Drebushchak, V.A.; Shakhtshneider, T.P.; Apenina, S.A.; Medvedeva, A.S.; Safronova, L.P.; Boldyrev, V.V. Thermoanalyticalinvestigation of drug–excipient interaction. J. Therm. Anal. Calorim. 2006, 86, 303–309. [Google Scholar] [CrossRef]
- Najib, N.M.; Suleiman, M.; Malakh, A. Characteristics of the in vitro release of ibuprofen from polyvinylpyrrolidone solid dispersions. Int. J. Pharm. 1986, 32, 229–236. [Google Scholar] [CrossRef]
- Nikolakakis, I.; Tsarvouli, K.; Malamataris, S. Water retention and drainage in different brands of microcrystalline cellulose: Effect of measuring conditions. Eur. J. Pharm. Biopharm. 2006, 63, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Kachrimanis, K.; Nikolakakis, I.; Malamataris, S. Tensile strength and disintegration of tableted silicified microcrystalline cellulose: Influences of interparticle bonding. J. Pharm. Sci. 2003, 92, 1489–1501. [Google Scholar] [CrossRef] [PubMed]
- Stiopkin, I.V.; Weeraman, C.; Pieniazek, P.A.; Shalhout, F.Y.; Skinner, J.L.; Benderskii, A.V. Hydrogen bonding at the water surface revealed by isotopic dilution spectroscopy. Nature 2011, 474, 192–195. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Progesterone | Group Contributions (Ci’s) | Occurrences | NiCi | ||||
| First-Order Groups | δd’ | δp’ | δhb’ | Ni | δd’ | δp’ | δhb’ |
| CH3– | −123.01 | −1.6444 | −0.7458 | 3 | −369.03 | −4.9332 | −2.2374 |
| –CH2- | 1.82 | −0.3141 | −0.3877 | 9 | 16.38 | −2.8269 | −3.4893 |
| –CH< | 82.94 | 0.6051 | −0.2064 | 3 | 248.82 | 1.8153 | −0.6192 |
| >C< | 182.13 | 2.0249 | −0.0113 | 2 | 364.26 | 4.0498 | −0.0226 |
| >C=O | −127.16 | 0.7691 | 1.7033 | 1 | −127.16 | 0.7691 | 1.7033 |
| –CH=C< | 62.48 | −1.1018 | −1.7171 | 1 | 62.48 | −1.1018 | −1.7171 |
| ΣNiCi: | 195.75 | −2.23 | −6.38 | ||||
| Second-Order Groups | δd’’ | δp’’ | δhb’’ | Μj | δd’’ | δp’’ | δhb’’ |
| Ccyclic=O | −46.57 | 0.1972 | −0.4496 | 1 | −46.57 | 0.1972 | −0.4496 |
| ΣMjDj: | −46.57 | 0.1972 | −0.4496 | ||||
| HPMCAS | Group Contributions (Ci’s) | Occurrences | NiCi | ||||
| First-Order Groups | δd’ | δp’ | δhb’ | (Ni) | δd’ | δp’ | δhb’ |
| –CHO- | 111.46 | 1.6001 | 0.4873 | 2 | 222.92 | 3.2002 | 0.9746 |
| –CH< | 82.94 | 0.6051 | −0.2064 | 3 | 248.82 | 1.8153 | −0.6192 |
| –CH2– | 1.82 | −0.3141 | −0.3877 | 1 | 1.82 | −0.3141 | −0.3877 |
| –OH | −29.97 | 1.0587 | 7.3609 | 1.5 | −44.96 | 1.5881 | 11.0414 |
| CH3COO– | −53.86 | −0.6075 | 1.7051 | 0.36 | −19.39 | −0.2187 | 0.6138 |
| –CH2COO– | 89.11 | 3.4942 | 1.3893 | 0.18 | 16.04 | 0.6290 | 0.2501 |
| –CH2– | 1.82 | −0.3141 | −0.3877 | 0.18 | 0.33 | −0.0565 | −0.0698 |
| –COOH | −38.16 | 0.7153 | 3.8422 | 0.18 | −6.87 | 0.1288 | 0.6916 |
| –OCH3 | −68.07 | 0.0089 | 0.2676 | 0.72 | −49.01 | 0.0064 | 0.1927 |
| –CH3 | −123.01 | −1.6444 | −0.7458 | 0.24 | −29.52 | −0.3947 | −0.1790 |
| –CH< | 82.94 | 0.6051 | −0.2064 | 0.24 | 19.91 | 0.1452 | −0.0495 |
| –OH | −29.97 | 1.0587 | 7.3609 | 0.24 | −7.19 | 0.2541 | 1.7666 |
| –CH2O– | 13.4 | 0.8132 | −0.1196 | 0.24 | 3.22 | 0.1952 | −0.0287 |
| ΣNiCi: | 356.11 | 6.98 | 14.20 | ||||
| Material | δd | δp | δhb | δt | Δδt (MPa)1/2 | Tg (°C) |
|---|---|---|---|---|---|---|
| Progesterone | 18.0 | 5.6 | 0.9 | 18.9 | – | 10 |
| HPMC | 18.0 | 15.3 | 19.4 | 30.6 | 11.7 | 162 |
| HPMCAS | 19.4 | 14.6 | 21.9 | 32.7 | 13.8 | 120 |
| MCC | 19.4 | 12.7 | 31.3 | 39.3 | 20.4 | 143 |
| PVP | 17.4 | 0.6 | 8.6 | 19.4 | 0.5 | 168 |
| SiO2 | 19.4 | 7.5 | 6.7 | 21.9 | 3.0 | 250 |
| Polymorph | HPMC | HPMCAS | MCC | PVP | SiO2 | ||
|---|---|---|---|---|---|---|---|
| First heating cycle | Melting temp. (onset, °C) | I | 128.3 ± 0.2 | 126.1 ± 1.1 | 129.1 ± 1.2 | 127.3 ± 0.7 | 127.3 ± 0.3 |
| II | 120 ± 0.4 | 118.8 ± 0.3 | N/A | 120.1 ± 0.4 | N/A | ||
| Heat of fusion (J/g) | I | 73 ± 1.4 | 78.8 ± 1.3 | 83.3 ± 0.6 | 81.9 ± 0.5 | 83.1 ± 0.7 | |
| II | 10.4 ± 0.9 | 2.1 ± 0.2 | N/A | 2.8 ± 0.2 | N/A | ||
| Second heating cycle | Melting temp. (onset, °C) | III | 107.8 ± 0.3 | 104.6 ± 0.8 | 106.4 ± 0.2 | 107.6 ± 0.4 | 100.4 ± 0.5 |
| Crystallization temp (onset, °C) | – | 69.9 ± 0.7 | N/A | 49.5 ± 0.4 | N/A | 53 ± 0.3 | |
| Heat of fusion/crystallization (J/g) | III | 26.1 ± 0.8/59.1 ± 1.2 | 0.7 ± 0.1/NA | 63.1 ± 1.1/37.9 ± 1.3 | 18.4 ± 1.1/NA | 57.6 ± 0.1/35.9 ± 0.3 | |
| HPMC | HPMCAS | MCC | PVP | SiO2 | ||
|---|---|---|---|---|---|---|
| First Heating Cycle | Melting temp. (onset (°C)) | 115.4 ± 0.2 | 98.2 ± 0.2 | 127.4 ± 0.2 | 120.8 ± 0.3 | 110.8 ± 0.3 |
| Heat of fusion (J/g) | 52.4 ± 1.4 | 51.2 ± 1.3 | 73.6 ± 0.6 | 58.8 ± 0.5 | 46.4 ± 0.7 | |
| Crystallinity (%) | 62% | 61% | 88% | 70% | 55% | |
| Polymer | HPMC | HPMCAS | MCC | PVP | SiO2 |
|---|---|---|---|---|---|
| SE | 16.79 ± 0.25 | 23.21 ± 0.92 | 10.99 ± 0.52 | 26.34 ± 1.64 | 24.29 ± 5.26 |
| PM | 13.95 ± 0.59 | 17.92 ± 0.58 | 10.37 ± 0.87 | 18.64 ± 2.55 | 21.48 ± 0.57 |
| PG | 11.49 ± 0.46 | ||||
| Polymer | HPMC | HPMCAS | MCC | PVP | SiO2 |
|---|---|---|---|---|---|
| BM | 56.68 ± 2.35 | 82.13 ± 4.32 | 14.67 ± 1.31 | 54.42 ± 5.05 | 25.18 ± 0.85 |
| PM | 42.52 ± 1.53 | 34.21 ± 1.66 | 15.67 ± 0.80 | 50.90 ± 2.45 | 32.94 ± 0.96 |
| PG | 11.49 ± 0.46 | ||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, X.; Partheniadis, I.; Nikolakakis, I.; Al-Obaidi, H. Solubility Improvement of Progesterone from Solid Dispersions Prepared by Solvent Evaporation and Co-milling. Polymers 2020, 12, 854. https://doi.org/10.3390/polym12040854
Chen X, Partheniadis I, Nikolakakis I, Al-Obaidi H. Solubility Improvement of Progesterone from Solid Dispersions Prepared by Solvent Evaporation and Co-milling. Polymers. 2020; 12(4):854. https://doi.org/10.3390/polym12040854
Chicago/Turabian StyleChen, Xing, Ioannis Partheniadis, Ioannis Nikolakakis, and Hisham Al-Obaidi. 2020. "Solubility Improvement of Progesterone from Solid Dispersions Prepared by Solvent Evaporation and Co-milling" Polymers 12, no. 4: 854. https://doi.org/10.3390/polym12040854
APA StyleChen, X., Partheniadis, I., Nikolakakis, I., & Al-Obaidi, H. (2020). Solubility Improvement of Progesterone from Solid Dispersions Prepared by Solvent Evaporation and Co-milling. Polymers, 12(4), 854. https://doi.org/10.3390/polym12040854