Controlling Molecule Aggregation and Electronic Spatial Coherence in the H-Aggregate and J-Aggregate Regime at Room Temperature



Abstract
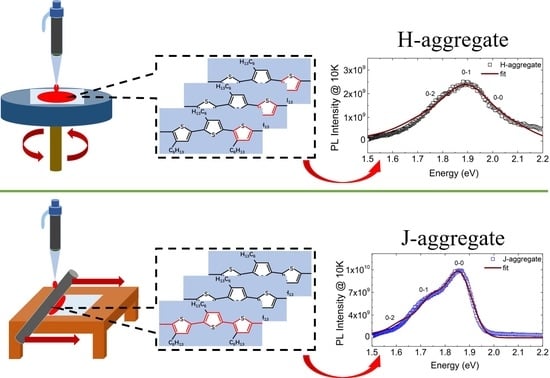
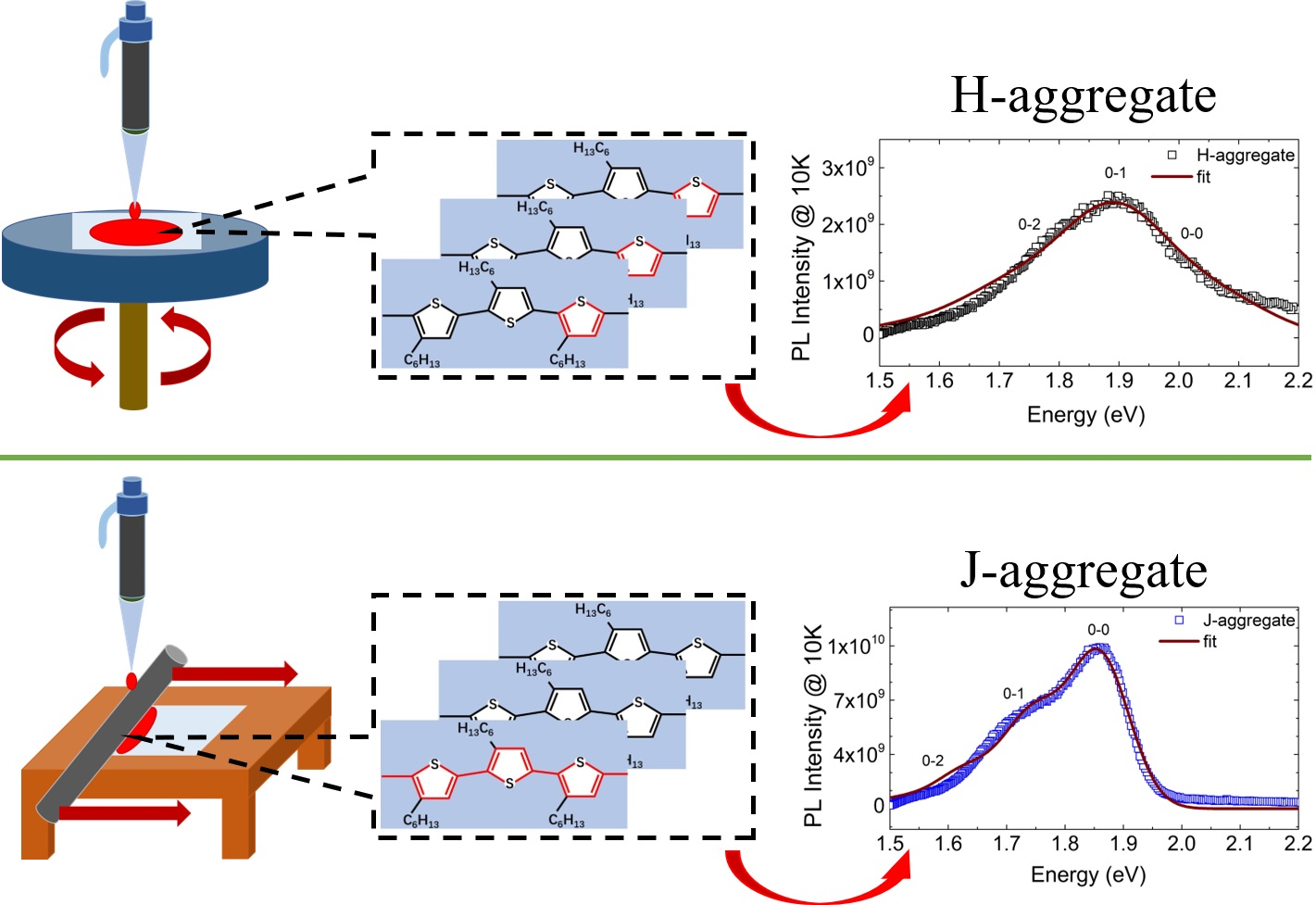
1. Introduction
2. Experimental Methods
3. Results and Discussion
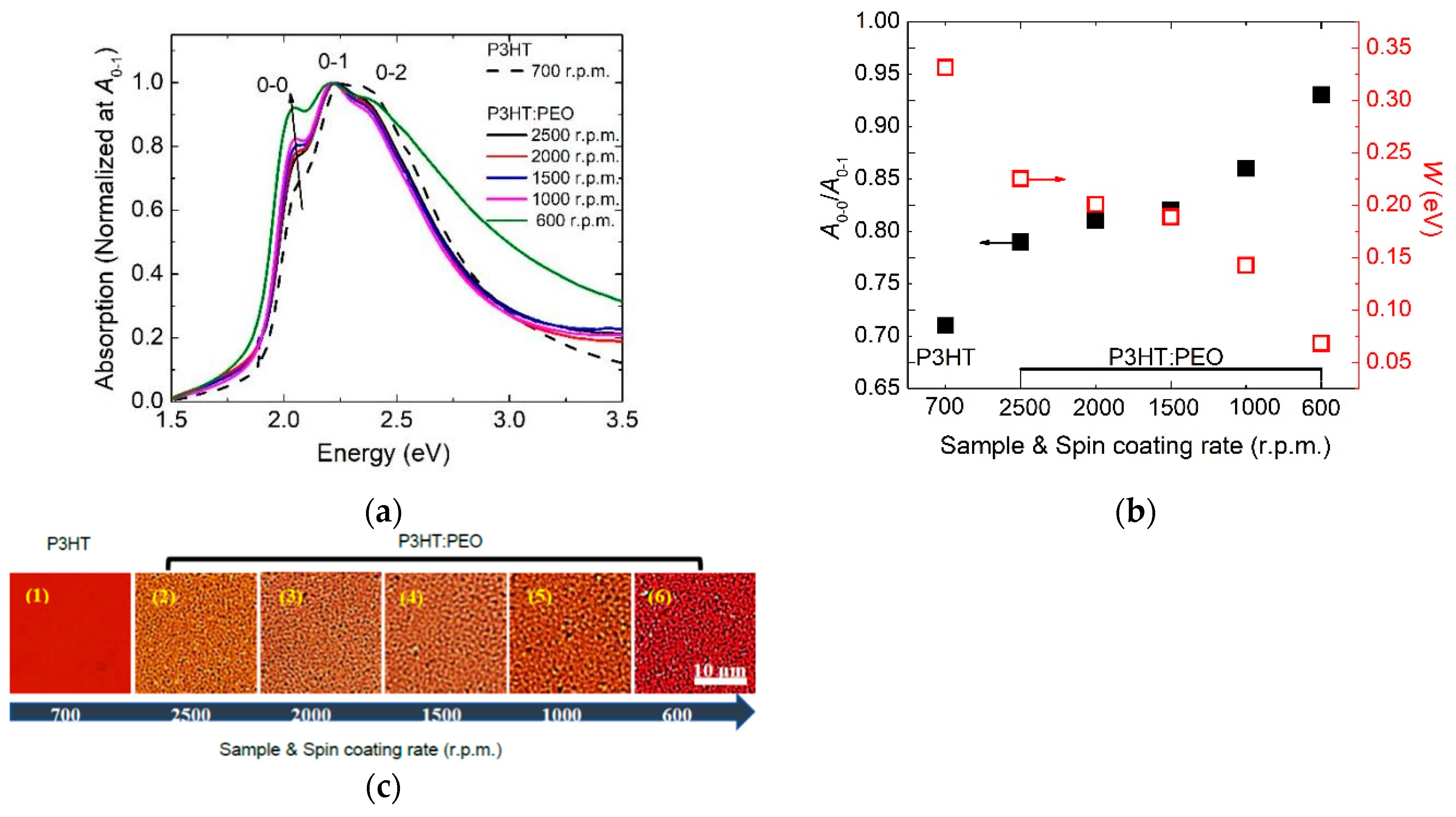
3.1. Controlling of the Molecule Intrachain Ordering in the H-aggregate Regime
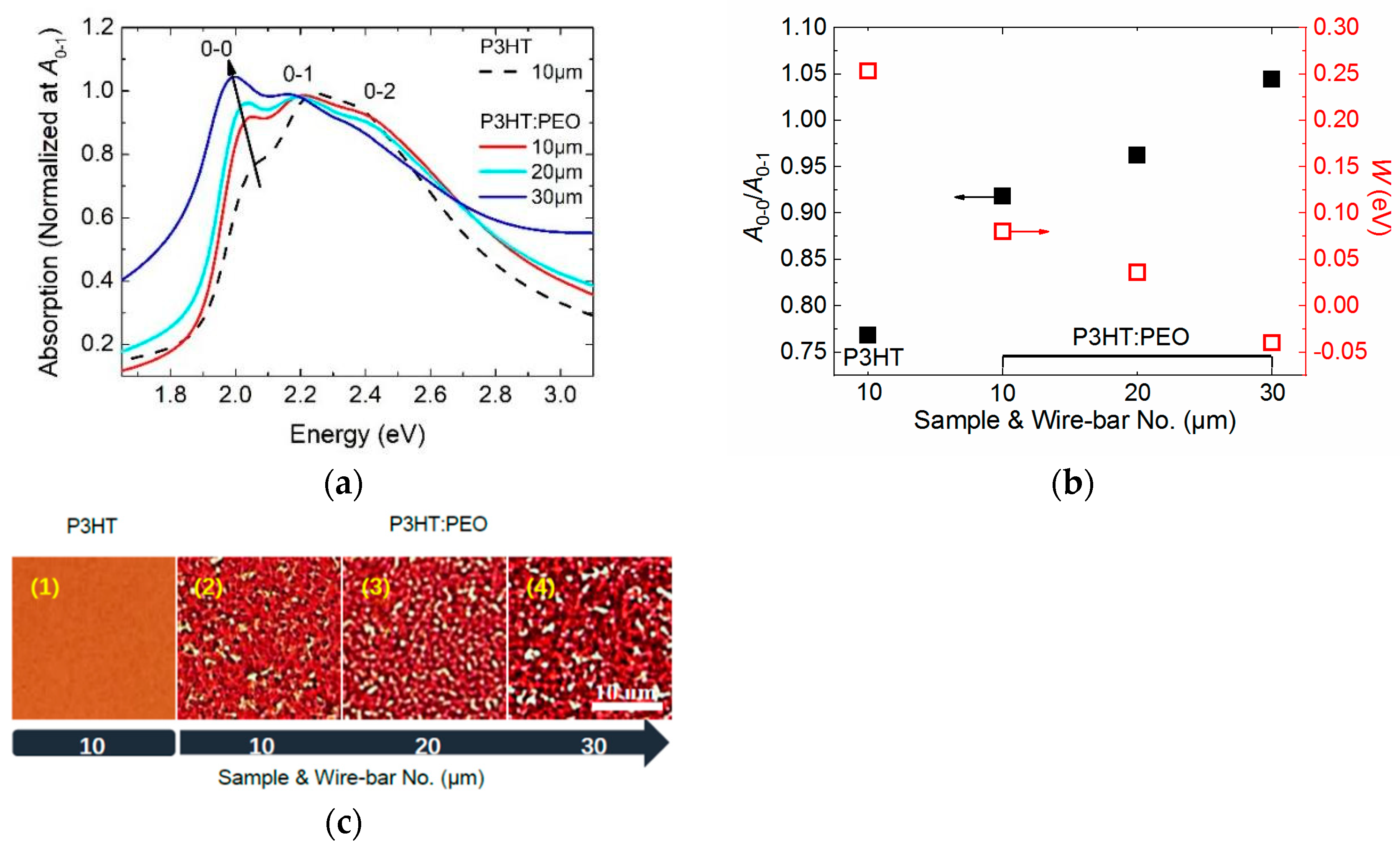
3.2. Controlling of the Molecule Intrachain Ordering in the J-Aggregate Regime
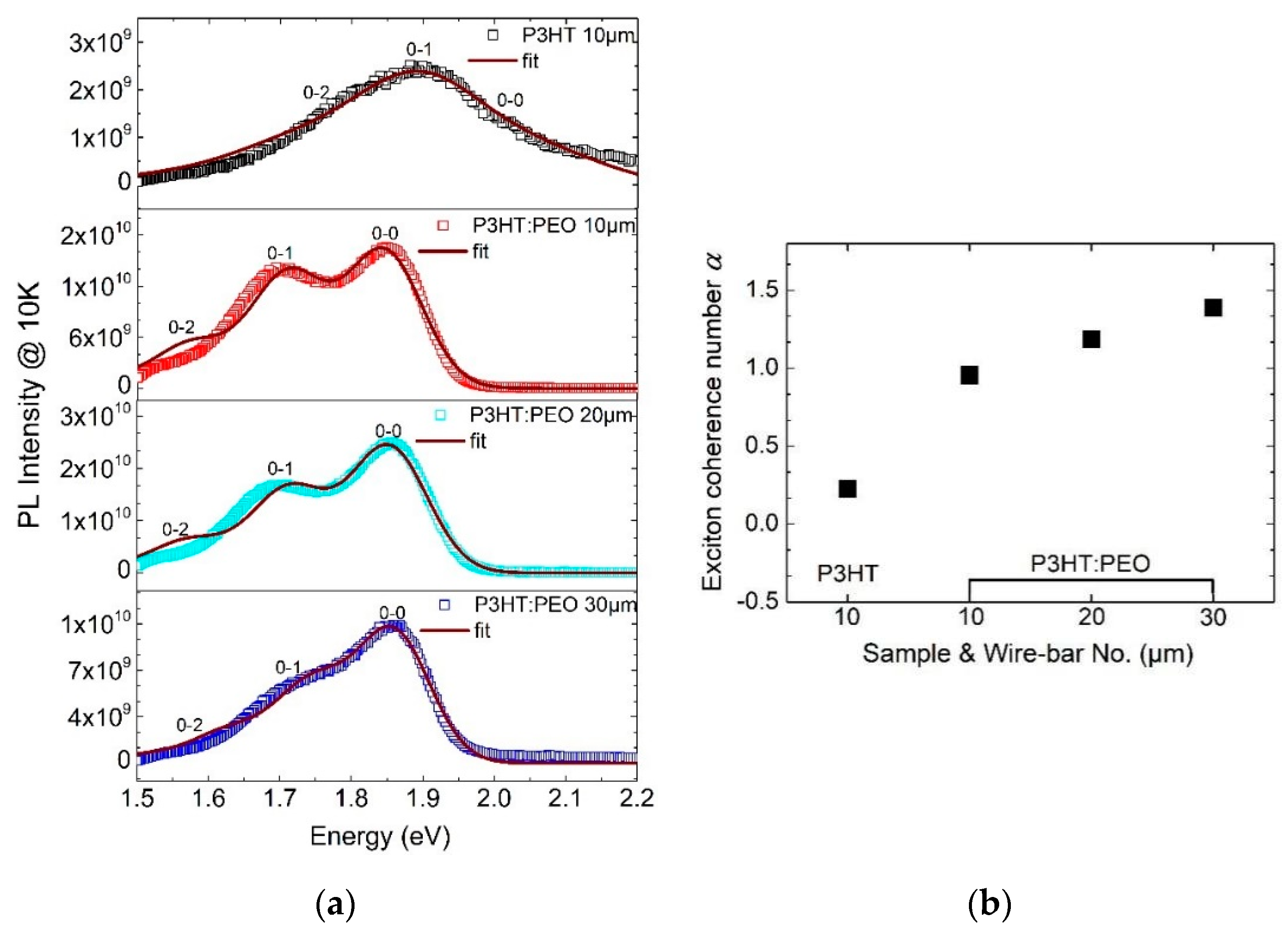
3.3. Exciton Generations in H- and J-Aggregate Regimes
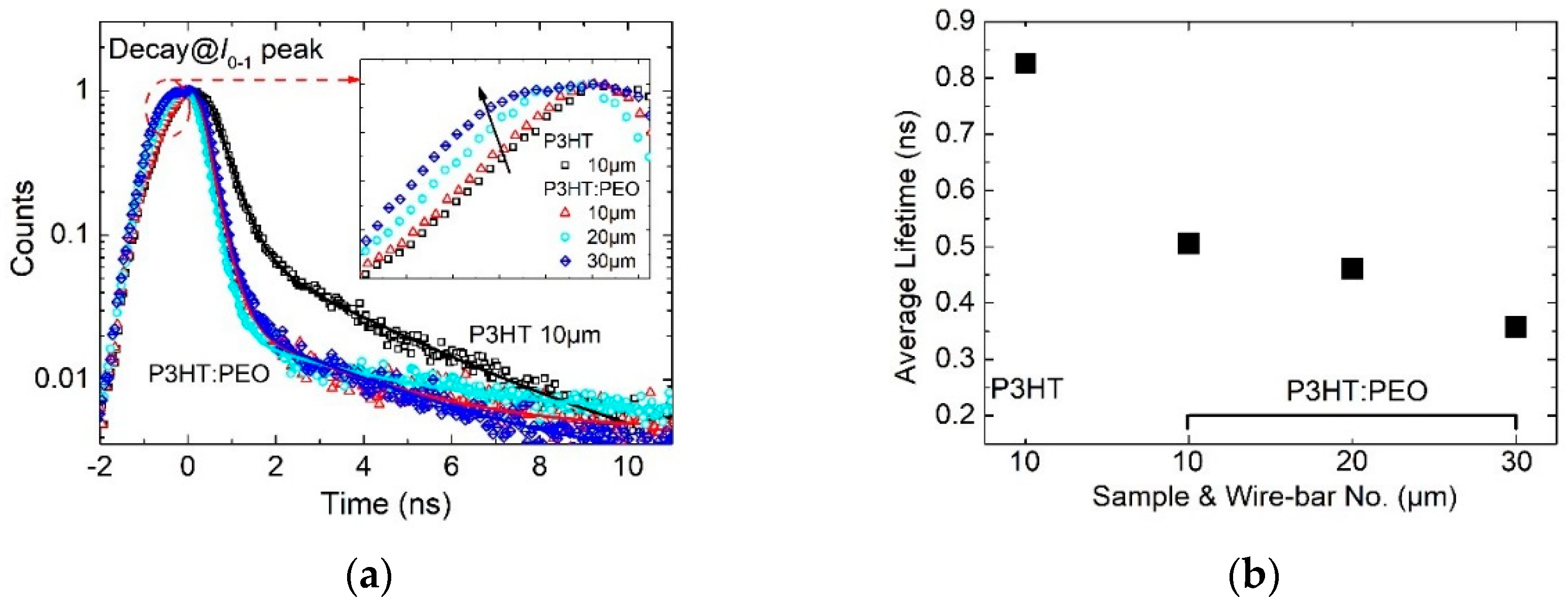
3.4. Exciton Dynamics in H- and J-Aggregate Regimes
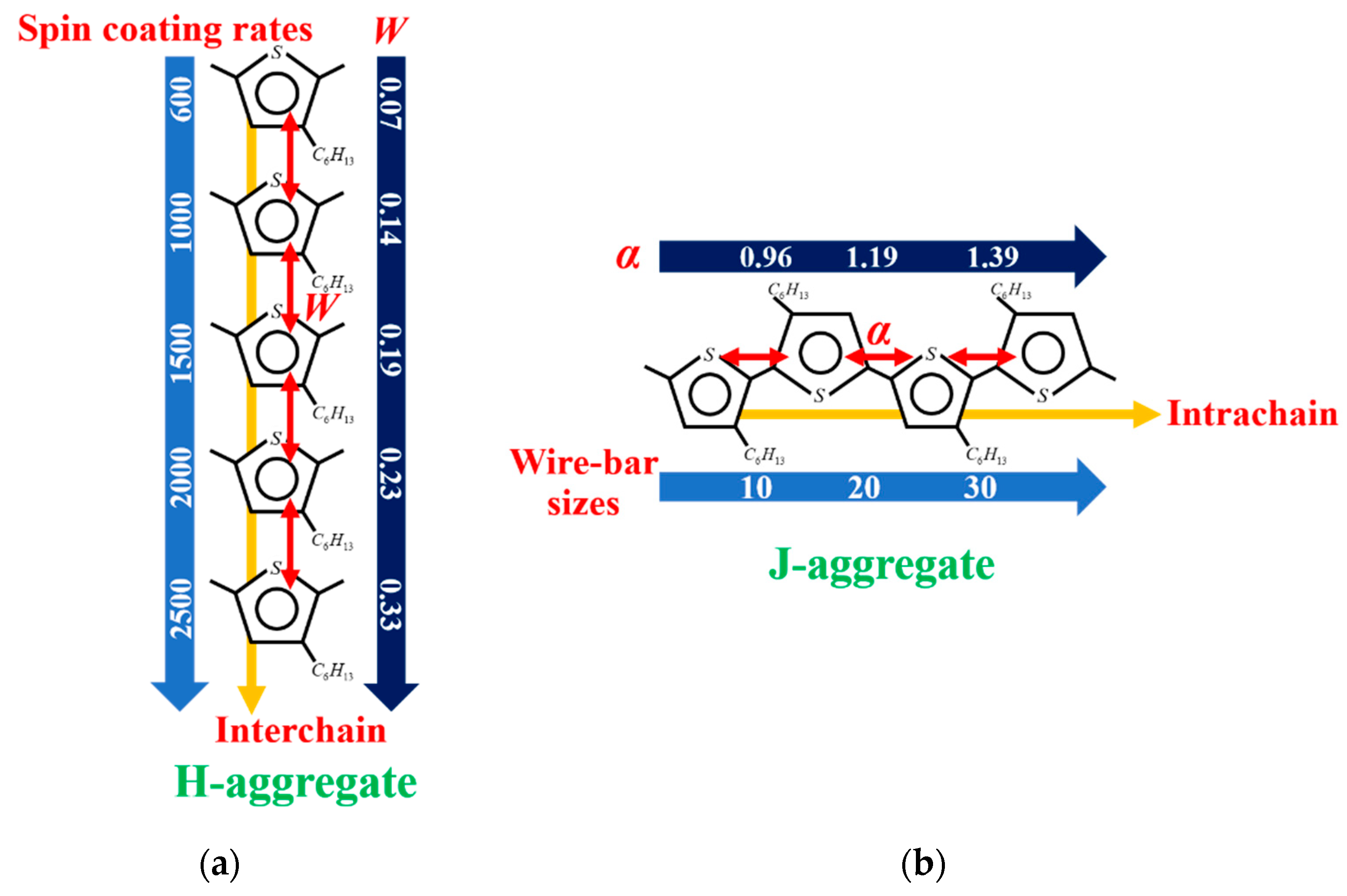
3.5. The Relationship between Processing Methods, Molecule Aggregation, Spatial Electronic Coherence, and Exciton Dynamics in H- and J-Aggregate Regimes
- (1)
- The formation of the H-aggregation of P3HT molecules requires a relatively fast solidification rate during film processing in order to prevent phase separation, such as spin-coated film in our investigation.
- (2)
- Excitons in H-aggregated films denote a larger interchain free exciton bandwidth with W > 0 eV.
- (3)
- The average lifetime of interchain-coherence-dominated excitons in H-aggregated films lasts longer, which is longer than 0.8 ns.
- (1)
- The formation of the J-aggregation of P3HT molecules requires a relatively slow solidification rate during the film processing in order to promote phase separation, such as wire-bar-coated film in our investigation.
- (2)
- The excitons in J-aggregated film show a larger intrachain exciton coherence number with α > 1.
- (3)
- The average lifetime of intrachain coherence dominated excitons in J-aggregated film is shorter (shorter than 0.5 ns).
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yao, H.; Li, Y.; Hu, H.; Chow, P.C.Y.; Chen, S.; Zhao, J.; Li, Z.; Carpenter, J.H.; Lai, J.Y.L.; Yang, G.; et al. A Facile Method to Fine-Tune Polymer Aggregation Properties and Blend Morphology of Polymer Solar Cells Using Donor Polymers with Randomly Distributed Alkyl Chains. Adv. Energy Mater. 2018, 8, 1701895. [Google Scholar] [CrossRef]
- Zhu, L.; Zhong, W.; Qiu, C.; Lyu, B.; Zhou, Z.; Zhang, M.; Song, J.; Xu, J.; Wang, J.; Ali, J.; et al. Aggregation-Induced Multilength Scaled Morphology Enabling 11.76% Efficiency in All-Polymer Solar Cells Using Printing Fabrication. Adv. Mater. 2019, 31, 1902899. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhao, J.; Li, Z.; Mu, C.; Ma, W.; Hu, H.; Jiang, K.; Lin, H.; Ade, H.; Yan, H. Aggregation and morphology control enables multiple cases of high-efficiency polymer solar cells. Nat. Commun. 2014, 5, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Pan, X.; Meng, X.; Liu, Y.; Wei, D.; Ma, W.; Huo, L.; Sun, X.; Lee, T.H.; Huang, M.; et al. Alkyl Side-Chain Engineering in Wide-Bandgap Copolymers Leading to Power Conversion Efficiencies over 10%. Adv. Mater. 2017, 29, 1604251. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.-G.; Li, Y. Side-chain engineering of high-efficiency conjugated polymer photovoltaic materials. Sci. China Chem. 2015, 58, 192–209. [Google Scholar] [CrossRef]
- Lee, C.; Kang, H.; Lee, W.; Kim, T.; Kim, K.-H.; Woo, H.Y.; Wang, C.; Kim, B.J. High-Performance All-Polymer Solar Cells Via Side-Chain Engineering of the Polymer Acceptor: The Importance of the Polymer Packing Structure and the Nanoscale Blend Morphology. Adv. Mater. 2015, 27, 2466–2471. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Ye, L.; Zhao, W.; Yan, H.; Yang, B.; Liu, D.; Li, W.; Ade, H.; Hou, J. A Wide Band Gap Polymer with a Deep Highest Occupied Molecular Orbital Level Enables 14.2% Efficiency in Polymer Solar Cells. J. Am. Chem. Soc. 2018, 140, 7159–7167. [Google Scholar] [CrossRef] [PubMed]
- Hellmann, C.; Paquin, F.; Treat, N.D.; Bruno, A.; Reynolds, L.X.; Haque, S.A.; Stavrinou, P.N.; Silva, C.; Stingelin, N. Controlling the interaction of light with polymer semiconductors. Adv. Mater. 2013, 25, 4906–4911. [Google Scholar] [CrossRef] [PubMed]
- Hellmann, C.; Treat, N.D.; Scaccabarozzi, A.D.; Hollis, J.R.; Fleischli, F.D.; Bannock, J.H.; Mello, J.D.; Michels, J.J.; Kim, J.-S.; Stingelin, N. Solution Processing of Polymer Semiconductors: Insulator Blends-Tailored Optical Properties through Liquid–Liquid Phase Separation Control. J. Polym. Sci. Polym. Phys. 2015, 53, 304–310. [Google Scholar] [CrossRef]
- Pan, S.; Zhu, M.; He, L.; Zhang, H.; Qiu, F.; Lin, Z.; Peng, J. Transformation from Nanofibers to Nanoribbons in Poly(3-hexylthiophene) Solution by Adding Alkylthiols. Macromol. Rapid Commun. 2018, 39, 1800048. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.; Lee, J.; Kleinhenz, N.; Fu, B.; Reichmanis, E. Photoinduced Anisotropic Supramolecular Assembly and Enhanced Charge Transport of Poly(3-hexylthiophene) Thin Films. Adv. Funct. Mater. 2014, 24, 4457–4465. [Google Scholar] [CrossRef]
- McDearmon, B.; Lim, E.; Lee, I.-H.; Kozycz, L.M.; O’Hara, K.; Robledo, P.I.; Venkatesan, N.R.; Chabinyc, M.L.; Hawker, C.J. Effects of Side-Chain Topology on Aggregation of Conjugated Polymers. Macromolecules 2018, 51, 2580–2590. [Google Scholar] [CrossRef]
- Reichenberger, M.; Kroh, D.; Matrone, G.M.M.; Schötz, K.; Pröller, S.; Filonik, O.; Thordardottir, M.E.; Herzig, E.M.; Bässler, H.; Stingelin, N.; et al. Controlling aggregate formation in conjugated polymers by spin-coating below the critical temperature of the disorder–order transition. J. Polym. Sci. Polym. Phys. 2017, 56, 532–542. [Google Scholar] [CrossRef]
- Panzer, F.; Bässler, H.; Lohwasser, R.; Thelakkat, M.; Köhler, A. The Impact of Polydispersity and Molecular Weight on the Order–Disorder Transition in Poly (3-hexylthiophene). J. Phys. Chem. Lett. 2014, 5, 2742–2747. [Google Scholar] [CrossRef] [PubMed]
- Spano, F.C.; Silva, C. H-and J-Aggregate Behavior in Polymeric Semiconductors. Annu. Rev. Phys. Chem. 2014, 65, 477–500. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.; Silva, C.; Friend, R.H.; Spano, F.C. Role of Intermolecular Coupling in the Photophysics of Disordered Organic Semiconductors: Aggregate Emission in Regioregular Polythiophene. Phys. Rev. Lett. 2007, 98, 206406. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, H.-Y.; Gao, B.-R.; Wang, L.; Yang, Z.-Y.; Du, X.-B.; Chen, Q.-D.; Song, J.-F.; Sun, H.-B. Exciton diffusion and charge transfer dynamics in nano phase-separated P3HT/PCBM blend films. Nanoscale 2011, 3, 2280–2285. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples & Bar-Size | B1 | τ1 (ns) | B2 | τ2 (ns) | τaverage (ns) |
|---|---|---|---|---|---|
| P3HT (10 μm) | 3.582 | 0.364 | 0.098 | 2.926 | 0.826 |
| P3HT:PEO (10 μm) | 2.375 | 0.261 | 0.027 | 2.640 | 0.506 |
| P3HT:PEO (20 μm) | 2.385 | 0.242 | 0.019 | 2.820 | 0.461 |
| P3HT:PEO (30 μm) | 4.690 | 0.254 | 0.046 | 1.820 | 0.357 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dou, F.; Li, J.; Men, H.; Zhang, X. Controlling Molecule Aggregation and Electronic Spatial Coherence in the H-Aggregate and J-Aggregate Regime at Room Temperature. Polymers 2020, 12, 786. https://doi.org/10.3390/polym12040786
Dou F, Li J, Men H, Zhang X. Controlling Molecule Aggregation and Electronic Spatial Coherence in the H-Aggregate and J-Aggregate Regime at Room Temperature. Polymers. 2020; 12(4):786. https://doi.org/10.3390/polym12040786
Chicago/Turabian StyleDou, Fei, Jiawei Li, Huijie Men, and Xinping Zhang. 2020. "Controlling Molecule Aggregation and Electronic Spatial Coherence in the H-Aggregate and J-Aggregate Regime at Room Temperature" Polymers 12, no. 4: 786. https://doi.org/10.3390/polym12040786
APA StyleDou, F., Li, J., Men, H., & Zhang, X. (2020). Controlling Molecule Aggregation and Electronic Spatial Coherence in the H-Aggregate and J-Aggregate Regime at Room Temperature. Polymers, 12(4), 786. https://doi.org/10.3390/polym12040786