Hybrid Complex Coacervate

 , , , , and
, , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
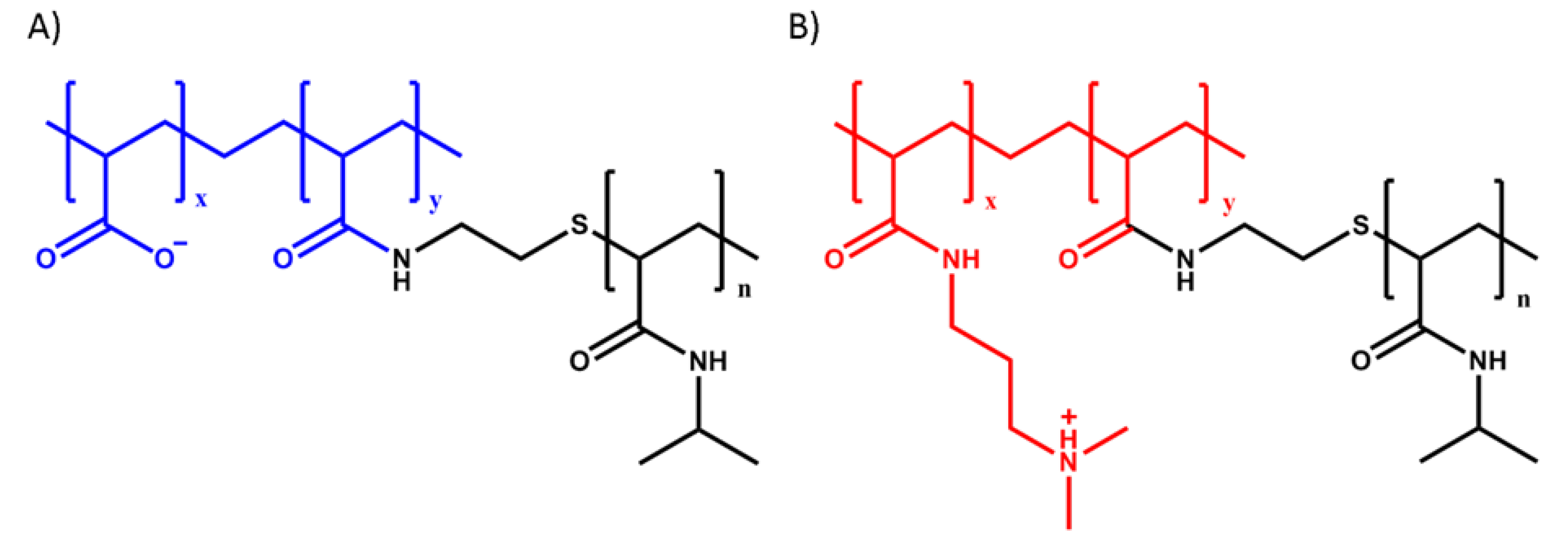
2.2. Polymer Synthesis
2.3. Complex Coacervation
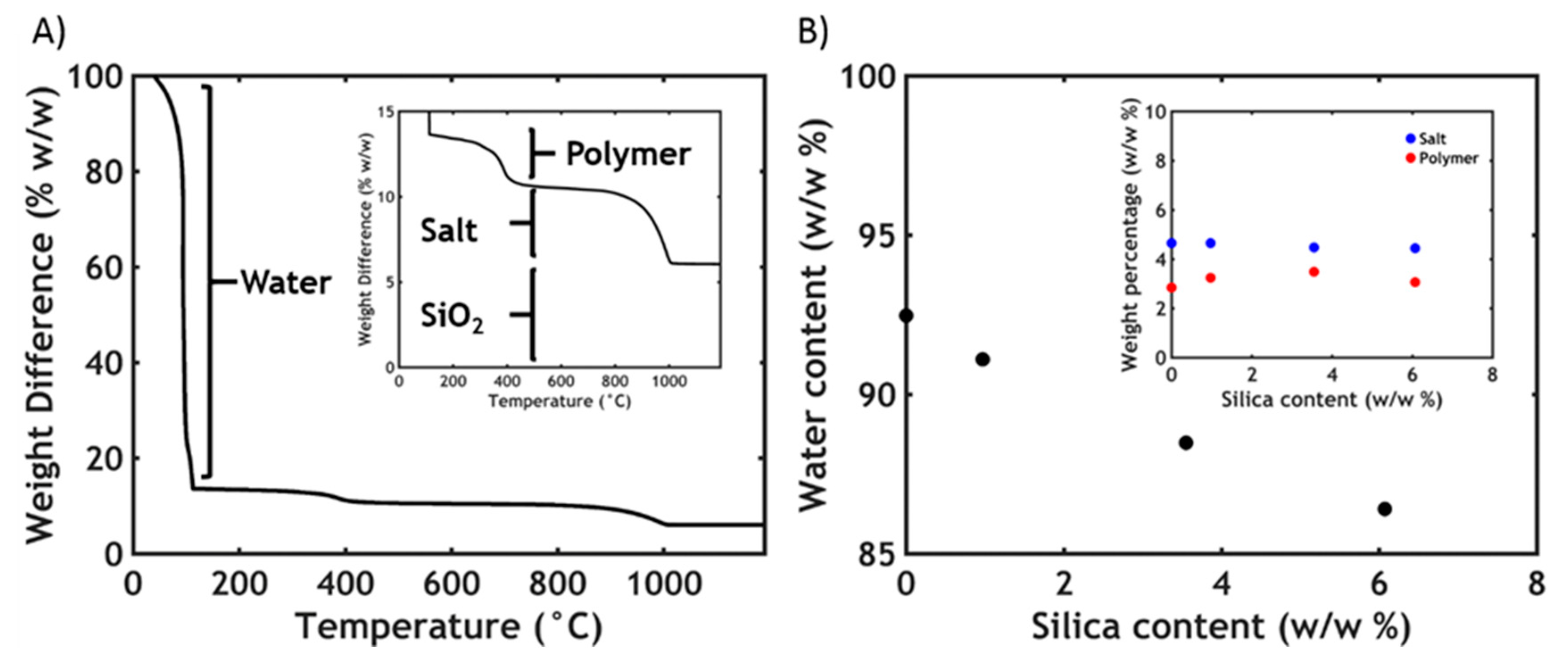
2.4. Thermogravimetric Analysis (TGA)
2.5. Rheology
2.5.1. Linear Rheology
2.5.2. Non-Linear Rheology
2.6. Differential Scanning Calorimetry (DSC)
2.7. Underwater Adhesion
2.8. PAA Hydrogel Thin Film Synthesis
3. Results and Discussion
3.1. Complex Coacervation
3.2. Thermogravimetric Analysis
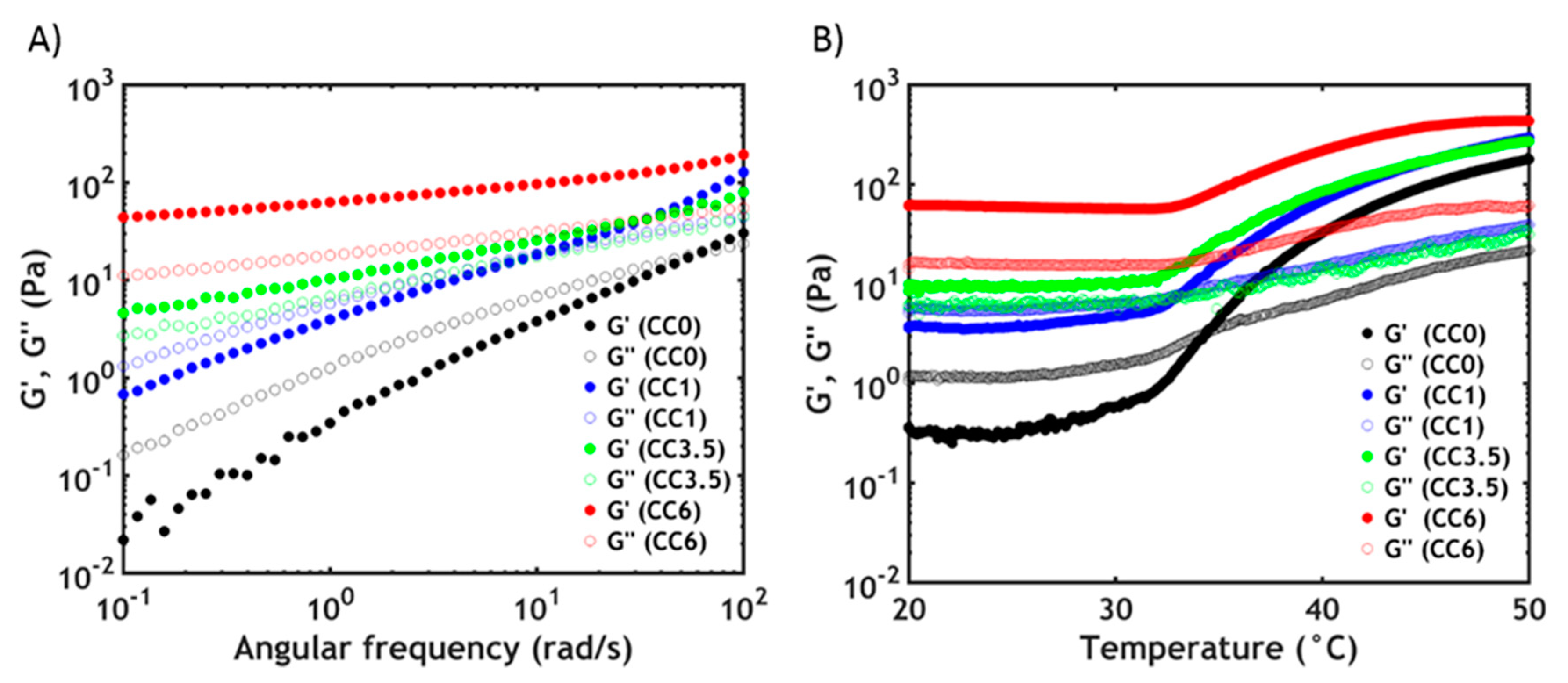
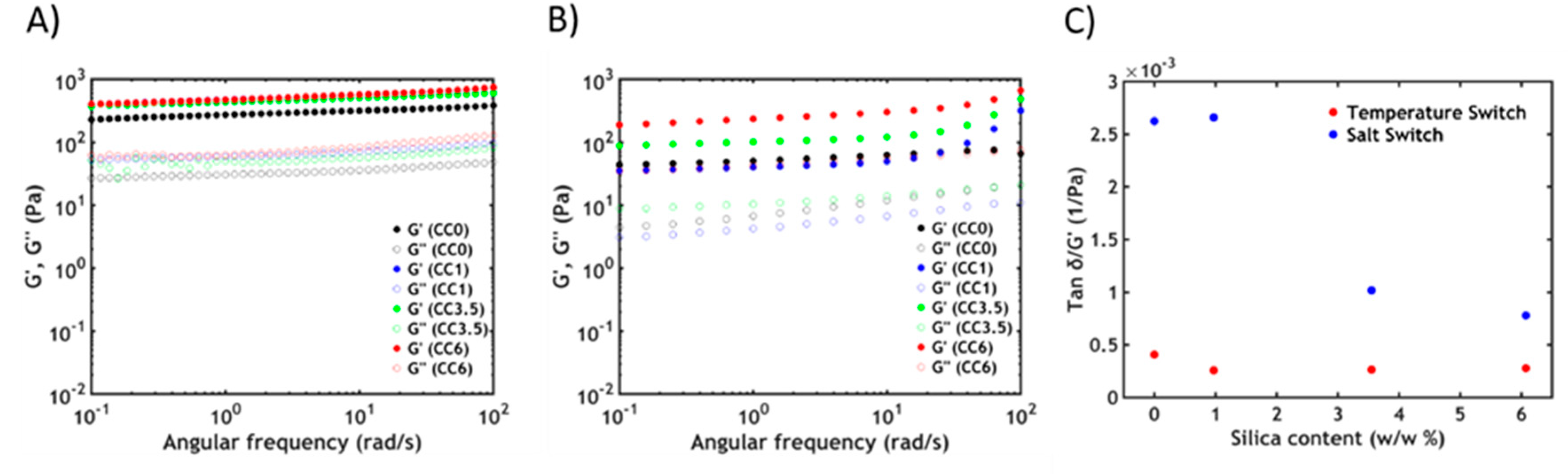
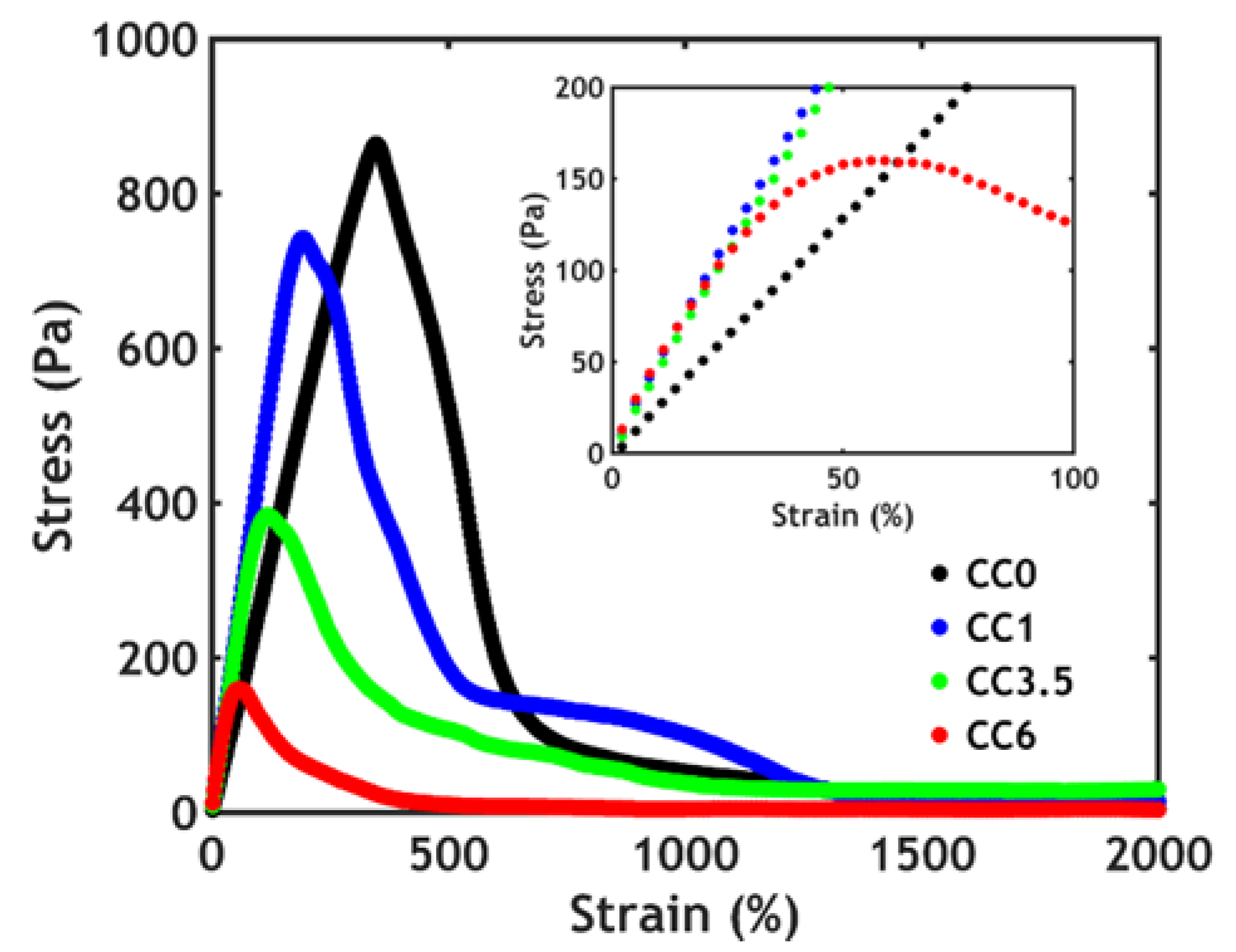
3.3. Rheology
3.3.1. Linear Rheology
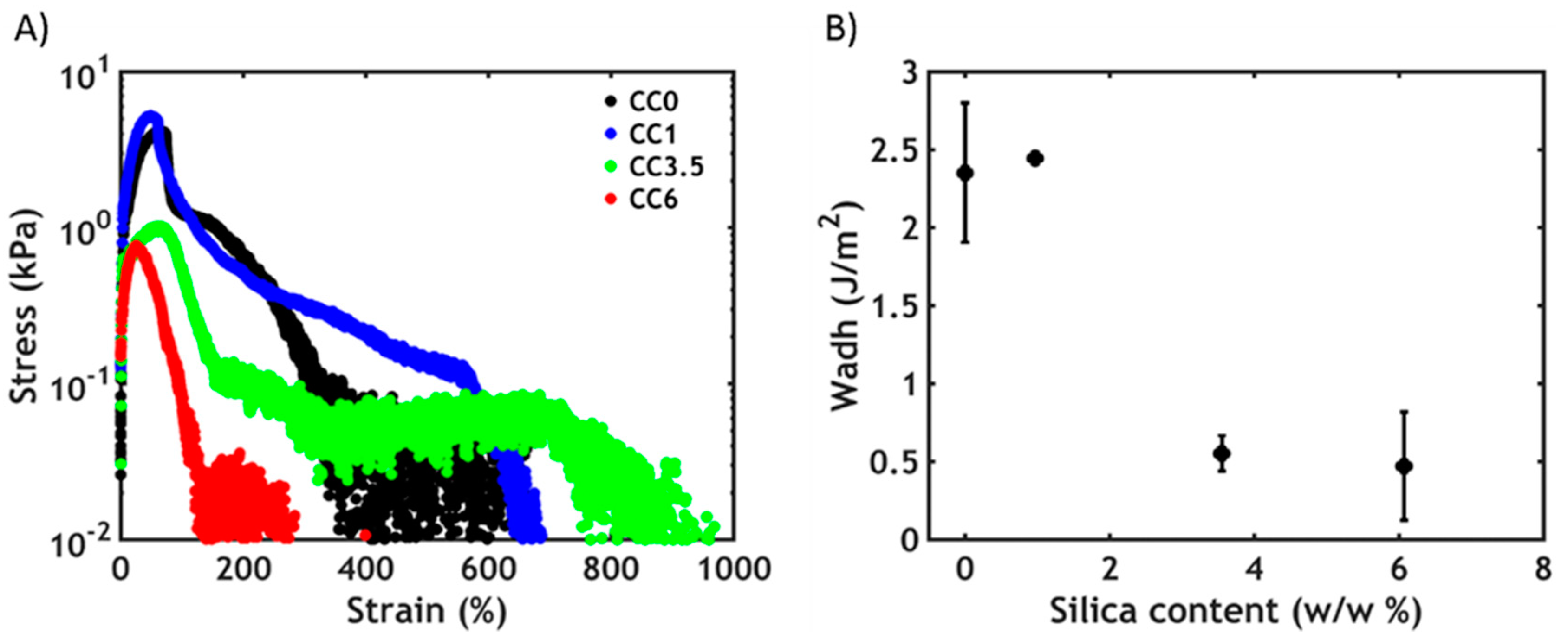
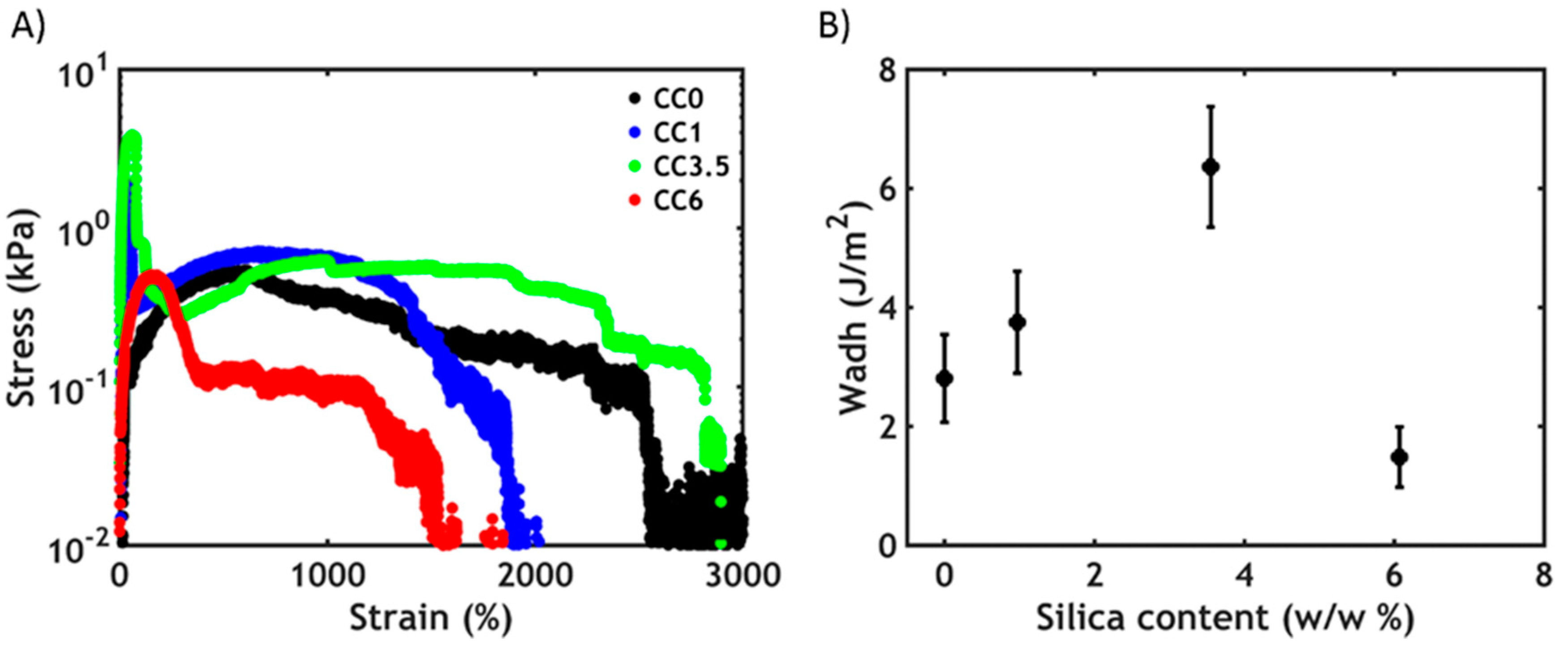
3.3.2. Non-Linear Rheology
3.4. Underwater Adhesion
4. Conclusions
5. Patents
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- da Silva, L.F.M.; Öchsner, A.; Adams, R.D. Introduction to Adhesive Bonding Technology. In Handbook of Adhesion Technology; da Silva, L.F.M., Öchsner, A., Adams, R.D., Eds.; Springer: Cham, Switzerland, 2018; Volume 2, pp. 1–7. [Google Scholar]
- Waite, J.H. Nature’s underwater adhesive specialist. Int. J. Adhes. Adhes. 1987, 7, 9–14. [Google Scholar] [CrossRef]
- Bré, L.P.; Zheng, Y.; Pêgo, A.P.; Wang, W. Taking tissue adhesives to the future: From traditional synthetic to new biomimetic approaches. Biomater. Sci. 2013, 1, 239–253. [Google Scholar] [CrossRef]
- Bhagat, V.; Becker, M.L. Degradable Adhesives for Surgery and Tissue Engineering. Biomacromolecules 2017, 18, 3009–3039. [Google Scholar] [CrossRef]
- Bouten, P.J.M.; Zonjee, M.; Bender, J.; Yauw, S.T.K.; van Goor, H.; van Hest, J.C.M.; Hoogenboom, R. The chemistry of tissue adhesive materials. Prog. Polym. Sci. 2014, 39, 1375–1405. [Google Scholar] [CrossRef]
- Dragu, A.; Unglaub, F.; Schwarz, S.; Beier, J.P.; Kneser, U.; Bach, A.D.; Horch, R.E. Foreign body reaction after usage of tissue adhesives for skin closure: A case report and review of the literature. Arch. Orthop. Trauma Surg. 2008, 129, 167–169. [Google Scholar] [CrossRef]
- Montanaro, L.; Arciola, C.R.; Cenni, E.; Ciapetti, G.; Savioli, F.; Filippini, F.; Barsanti, L.A. Cytotoxicity, blood compatibility and antimicrobial activity of two cyanoacrylate glues for surgical use. Biomaterials 2000, 22, 59–66. [Google Scholar] [CrossRef]
- Patel, S.; Rodriguez-Merchan, E.C.; Haddad, F.S. The use of fibrin glue in surgery of the knee. J. Bone Jt. Surg. Br. 2010, 92, 1325–1331. [Google Scholar] [CrossRef][Green Version]
- Spotnitz, W.D. Fibrin Sealant: Past, Present, and Future: A Brief Review. World J. Surg. 2010, 34, 632–634. [Google Scholar] [CrossRef]
- Sawhney, A.S.; Pathak, C.P.; Hubbell, J.A. Bioerodible hydrogels based on photopolymerized poly(ethylene glycol)-co-poly(.alpha.-hydroxy acid) diacrylate macromers. Macromolecules 1993, 26, 581–587. [Google Scholar] [CrossRef]
- Wallace, D.G.; Cruise, G.M.; Rhee, W.M.; Schroeder, J.A.; Prior, J.J.; Ju, J.; Maroney, M.; Duronio, J.; Ngo, M.H.; Estridge, T.; et al. A tissue sealant based on reactive multifunctional polyethylene glycol. J. Biomed. Mater. Res. 2001, 58, 545–555. [Google Scholar] [CrossRef]
- Bochyńska, A.I.; Hannink, G.; Grijpma, D.W.; Buma, P. Tissue adhesives for meniscus tear repair: An overview of current advances and prospects for future clinical solutions. J. Mater. Sci. Mater. Med. 2016, 27, 85. [Google Scholar] [CrossRef]
- Stewart, R.J.; Ransom, T.C.; Hlady, V. Natural underwater adhesives. J. Polym. Sci. Part B Polym. Phys. 2011, 49, 757–771. [Google Scholar] [CrossRef]
- Waite, J.H. Mussel adhesion—Essential footwork. J. Exp. Biol. 2017, 220, 517–530. [Google Scholar] [CrossRef]
- Stewart, R.J.; Weaver, J.C.; Morse, D.E.; Waite, J.H. The tube cement of Phragmatopoma californica: A solid foam. J. Exp. Biol. 2004, 207, 4727–4734. [Google Scholar] [CrossRef]
- Gucht, J.; Spruijt, E.; Lemmers, M.; Cohen Stuart, M.A. Polyelectrolyte complexes: Bulk phases and colloidal systems. J. Colloid Interface Sci. 2011, 361, 407–422. [Google Scholar] [CrossRef]
- Spruijt, E.; Westphal, A.H.; Borst, J.W.; Cohen Stuart, M.A.; van der Gucht, J. Binodal Compositions of Polyelectrolyte Complexes. Macromolecules 2010, 43, 6476–6484. [Google Scholar] [CrossRef]
- Stewart, R.J.; Wang, C.S.; Shao, H. Complex coacervates as a foundation for synthetic underwater adhesives. Adv. Colloid Interface Sci. 2011, 167, 85–93. [Google Scholar] [CrossRef]
- Stewart, R.J.; Wang, C.S.; Song, I.T.; Jones, J.P. The role of coacervation and phase transitions in the sandcastle worm adhesive system. Adv. Colloid Interface Sci. 2017, 239, 88–96. [Google Scholar] [CrossRef]
- Spruijt, E.; Sprakel, J.; Cohen Stuart, M.A.; van der Gucht, J. Interfacial tension between a complex coacervate phase and its coexisting aqueous phase. Soft Matter 2010, 6, 172–178. [Google Scholar] [CrossRef]
- Jones, J.P.; Sima, M.; O’Hara, R.G.; Stewart, R.J. Water-Borne Endovascular Embolics Inspired by the Undersea Adhesive of Marine Sandcastle Worms. Adv. Healthc. Mater. 2016, 5, 795–801. [Google Scholar] [CrossRef]
- Ahn, B.K.; Das, S.; Linstadt, R.; Kaufman, Y.; Martinez-Rodriguez, N.R.; Mirshafian, R.; Kesselman, E.; Talmon, Y.; Lipshutz, B.H.; Israelachvili, J.N.; et al. High-performance mussel-inspired adhesives of reduced complexity. Nat. Commun. 2015, 6, 8663. [Google Scholar] [CrossRef]
- Kaur, S.; Weerasekare, G.M.; Stewart, R.J. Multiphase Adhesive Coacervates Inspired by the Sandcastle Worm. ACS Appl. Mater. Interfaces 2011, 3, 941–944. [Google Scholar] [CrossRef]
- Shao, H.; Stewart, R.J. Biomimetic Underwater Adhesives with Environmentally Triggered Setting Mechanisms. Adv. Mater. 2010, 22, 729–733. [Google Scholar] [CrossRef]
- Dompé, M.; Cedano-Serrano, F.J.; Heckert, O.; van den Heuvel, N.; van der Gucht, J.; Tran, Y.; Hourdet, D.; Creton, C.; Kamperman, M. Thermoresponsive Complex Coacervate-Based Underwater Adhesive. Adv. Mater. 2019, 31, 1808179. [Google Scholar] [CrossRef]
- Dompé, M.; Cedano-Serrano, F.J.; Vahdati, M.; Westerveld, L.; Hourdet, D.; Creton, C.; Van der Gucht, J.; Kodger, T.; Kamperman, M. Underwater Adhesion of Multiresponsive Complex Coacervates. Adv. Mater. Interfaces 2019, 1901785. [Google Scholar] [CrossRef]
- Heskins, M.; Guillet, J.E. Solution Properties of Poly(N-isopropylacrylamide). J. Macromol. Sci. A 1968, 2, 1441–1455. [Google Scholar] [CrossRef]
- Sun, T.L.; Kurokawa, T.; Kuroda, S.; Ihsan, A.B.; Akasaki, T.; Sato, K.; Haque, M.A.; Nakajima, T.; Gong, J.P. Physical hydrogels composed of polyampholytes demonstrate high toughness and viscoelasticity. Nat. Mater. 2013, 12, 932–937. [Google Scholar] [CrossRef]
- Shadlou, S.; Ahmadi-Moghadam, B.; Taheri, F. Nano-Enhanced Adhesives. In Progress in Adhesion and Adhesives; Mittal, K., Ed.; Scrivener Publishing LCC.: Salem, MA, USA, 2015; pp. 357–396. [Google Scholar]
- Yu, S.; Tong, M.N.; Critchlow, G. Use of carbon nanotubes reinforced epoxy as adhesives to join aluminum plates. Mater. Des. 2010, 31, S126–S129. [Google Scholar] [CrossRef]
- Lilei, Y.; Zonghe, L.; Johan, L.; Tholen, A. Effect of Ag particle size on electrical conductivity of isotropically conductive adhesives. IEEE Trans. Electron. Packag. Manuf. 1999, 22, 299–302. [Google Scholar] [CrossRef]
- Kim, B.C.; Park, S.W.; Lee, D.G. Fracture toughness of the nano-particle reinforced epoxy composite. Compos. Struct. 2008, 86, 69–77. [Google Scholar] [CrossRef]
- Rosso, P.; Ye, L.; Friedrich, K.; Sprenger, S. A toughened epoxy resin by silica nanoparticle reinforcement. J. Appl. Polym. Sci. 2006, 100, 1849–1855. [Google Scholar] [CrossRef]
- Creton, C. 50th Anniversary Perspective: Networks and Gels: Soft but Dynamic and Tough. Macromolecules 2017, 50, 8297–8316. [Google Scholar] [CrossRef]
- Petit, L.; Bouteiller, L.; Brûlet, A.; Lafuma, F.; Hourdet, D. Responsive Hybrid Self-Assemblies in Aqueous Media. Langmuir 2007, 23, 147–158. [Google Scholar] [CrossRef]
- Portehault, D.; Petit, L.; Pantoustier, N.; Ducouret, G.; Lafuma, F.; Hourdet, D. Hybrid thickeners in aqueous media. Colloids Surf. A 2006, 278, 26–32. [Google Scholar] [CrossRef]
- Haraguchi, K.; Takehisa, T. Nanocomposite Hydrogels: A Unique Organic–Inorganic Network Structure with Extraordinary Mechanical, Optical, and Swelling/De-swelling Properties. Adv. Mater. 2002, 14, 1120–1124. [Google Scholar] [CrossRef]
- Durme, K.V.; Van Mele, B.; Loos, W.; Du Prez, F.E. Introduction of silica into thermo-responsive poly(N-isopropyl acrylamide) hydrogels: A novel approach to improve response rates. Polymer 2005, 46, 9851–9862. [Google Scholar] [CrossRef]
- Lin, W.-C.; Fan, W.; Marcellan, A.; Hourdet, D.; Creton, C. Large Strain and Fracture Properties of Poly(dimethylacrylamide)/Silica Hybrid Hydrogels. Macromolecules 2010, 43, 2554–2563. [Google Scholar] [CrossRef]
- Rose, S.; Dizeux, A.; Narita, T.; Hourdet, D.; Marcellan, A. Time Dependence of Dissipative and Recovery Processes in Nanohybrid Hydrogels. Macromolecules 2013, 46, 4095–4104. [Google Scholar] [CrossRef]
- Rose, S.; Prevoteau, A.; Elzière, P.; Hourdet, D.; Marcellan, A.; Leibler, L. Nanoparticle solutions as adhesives for gels and biological tissues. Nature 2013, 505, 382–392. [Google Scholar] [CrossRef]
- Meddahi-Pellé, A.; Legrand, A.; Marcellan, A.; Louedec, L.; Letourneur, D.; Leibler, L. Organ repair, hemostasis, and in vivo bonding of medical devices by aqueous solutions of nanoparticles. Angew. Chem. Int. Ed. 2014, 53, 6369–6373. [Google Scholar] [CrossRef]
- Durand, A.; Hourdet, D. Synthesis and thermoassociative properties in aqueous solution of graft copolymers containing poly(N-isopropylacrylamide) side chains. Polymer 1999, 40, 4941–4951. [Google Scholar] [CrossRef]
- Petit, L.; Karakasyan, C.; Pantoustier, N.; Hourdet, D. Synthesis of graft polyacrylamide with responsive self-assembling properties in aqueous media. Polymer 2007, 48, 7098–7112. [Google Scholar] [CrossRef]
- Sudre, G.; Olanier, L.; Tran, Y.; Hourdet, D.; Creton, C. Reversible adhesion between a hydrogel and a polymer brush. Soft Matter 2012, 8, 8184–8193. [Google Scholar] [CrossRef]
- Chollet, B.; Li, M.; Martwong, E.; Bresson, B.; Fretigny, C.; Tabeling, P.; Tran, Y. Multiscale Surface-Attached Hydrogel Thin Films with Tailored Architecture. ACS Appl. Mater. Interfaces 2016, 8, 11729–11738. [Google Scholar] [CrossRef]
- Li, L.; Srivastava, S.; Andreev, M.; Marciel, A.B.; de Pablo, J.J.; Tirrell, M.V. Phase Behavior and Salt Partitioning in Polyelectrolyte Complex Coacervates. Macromolecules 2018, 51, 2988–2995. [Google Scholar] [CrossRef]
- Spruijt, E.; Cohen Stuart, M.A.; van der Gucht, J. Linear Viscoelasticity of Polyelectrolyte Complex Coacervates. Macromolecules 2013, 46, 1633–1641. [Google Scholar] [CrossRef]
- Agrawal, S.K.; Sanabria-DeLong, N.; Tew, G.N.; Bhatia, S.R. Nanoparticle-Reinforced Associative Network Hydrogels. Langmuir 2008, 24, 13148–13154. [Google Scholar] [CrossRef]
- Portehault, D.; Petit, L.; Hourdet, D. Synthesis and self assembly processes of aqueous thermoresponsive hybrid formulations. Soft Matter 2010, 6, 2178–2186. [Google Scholar] [CrossRef]
- Ghostine, R.A.; Shamoun, R.F.; Schlenoff, J.B. Doping and Diffusion in an Extruded Saloplastic Polyelectrolyte Complex. Macromolecules 2013, 46, 4089–4094. [Google Scholar] [CrossRef]
- Flory, P.J. Principles of Polymer Chemistry; Cornell University Press: Ithaca, NY, USA, 1953. [Google Scholar]
- Wang, T.; Lei, C.-H.; Dalton, A.B.; Creton, C.; Lin, Y.; Fernando, K.A.S.; Sun, Y.-P.; Manea, M.; Asua, J.M.; Keddie, J.L. Waterborne, Nanocomposite Pressure-Sensitive Adhesives with High Tack Energy, Optical Transparency, and Electrical Conductivity. Adv. Mater. 2006, 18, 2730–2734. [Google Scholar] [CrossRef]
- Li, X.; Rombouts, W.; van der Gucht, J.; de Vries, R.; Dijksman, J.A. Mechanics of composite hydrogels approaching phase separation. PLoS ONE 2019, 14, e0211059. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | PNIPAM/Total Polymer Molar Ratio (%) | Mn Graft Copolymer (kg/mol) | PNIPAM Chains per Backbone | PDI |
|---|---|---|---|---|
| PAA-g-PNIPAM | 42 | 588 | 51 | - |
| PDMAPAA-g-PNIPAM | 26 | 248 | 7 | 4.41 |
| Sample Name | [SiO2] in Mixture (w/w %) | [SiO2] in Complex Coacervate Phase (w/w %) | [SiO2] in Dilute Phase (w/w %) | Percentage of SiO2 Ending in Complex Coacervate Phase (w/w %) |
|---|---|---|---|---|
| CC0 | 0 | 0 | 0 | 0 |
| CC1 | 0.1 | 0.97 | 0.01 | 87 |
| CC3.5 | 0.5 | 3.55 | 0.2 | 64 |
| CC6 | 1 | 6.07 | 0.49 | 55 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dompé, M.; Cedano-Serrano, F.J.; Vahdati, M.; Hourdet, D.; van der Gucht, J.; Kamperman, M.; Kodger, T.E. Hybrid Complex Coacervate. Polymers 2020, 12, 320. https://doi.org/10.3390/polym12020320
Dompé M, Cedano-Serrano FJ, Vahdati M, Hourdet D, van der Gucht J, Kamperman M, Kodger TE. Hybrid Complex Coacervate. Polymers. 2020; 12(2):320. https://doi.org/10.3390/polym12020320
Chicago/Turabian StyleDompé, Marco, Francisco Javier Cedano-Serrano, Mehdi Vahdati, Dominique Hourdet, Jasper van der Gucht, Marleen Kamperman, and Thomas E. Kodger. 2020. "Hybrid Complex Coacervate" Polymers 12, no. 2: 320. https://doi.org/10.3390/polym12020320
APA StyleDompé, M., Cedano-Serrano, F. J., Vahdati, M., Hourdet, D., van der Gucht, J., Kamperman, M., & Kodger, T. E. (2020). Hybrid Complex Coacervate. Polymers, 12(2), 320. https://doi.org/10.3390/polym12020320