Conducting Polymers for Optoelectronic Devices and Organic Solar Cells: A Review

 , ,
, , 
Abstract
1. Introduction
2. Electronic Structure and Doping in Conjugated Polymers
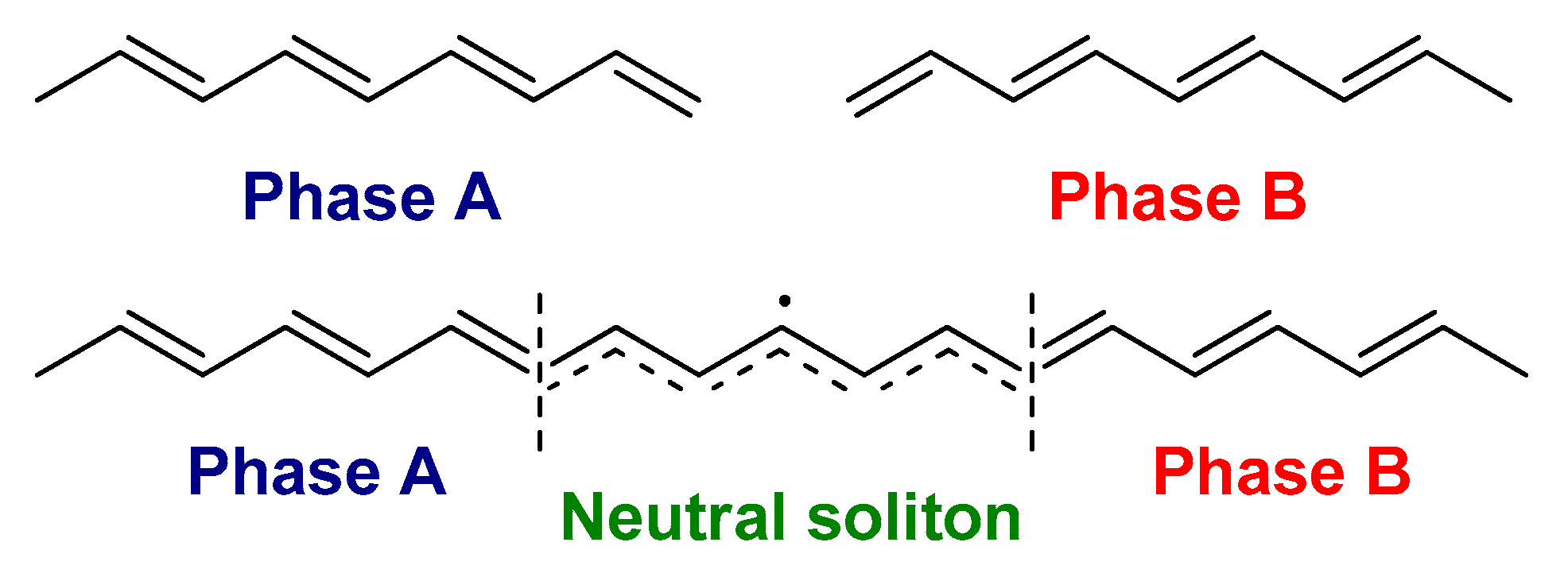
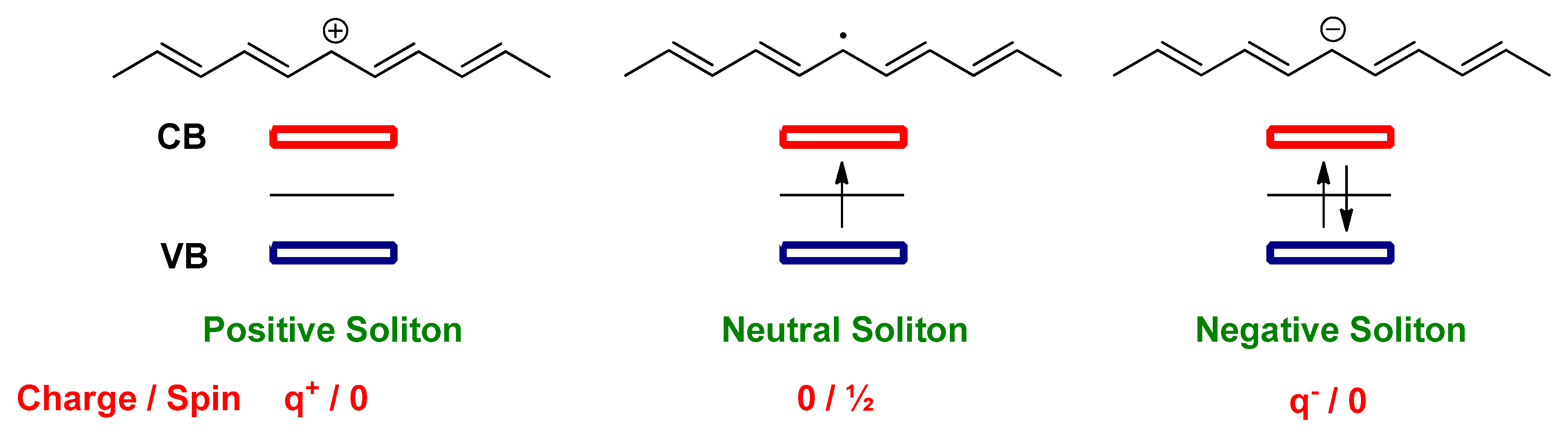
2.1. The Electronic Structure of Conjugated Polymers
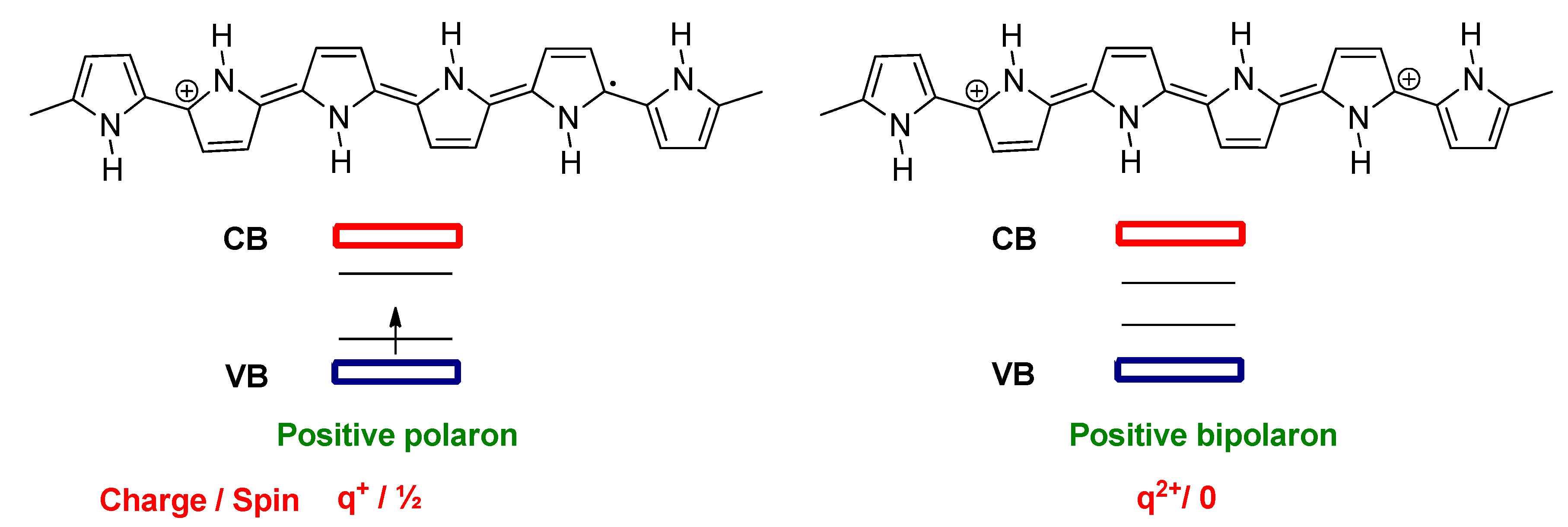
2.2. Doping in Conjugated Polymers
3. Methods for Preparation of Conjugated Polymers
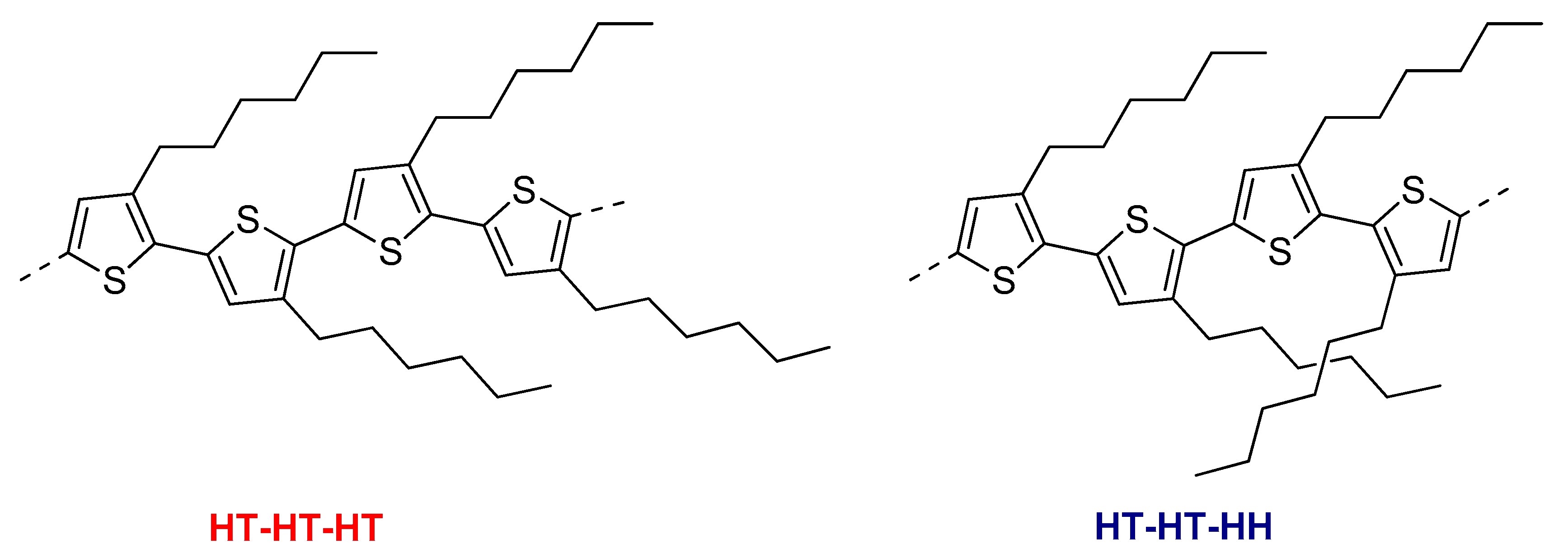
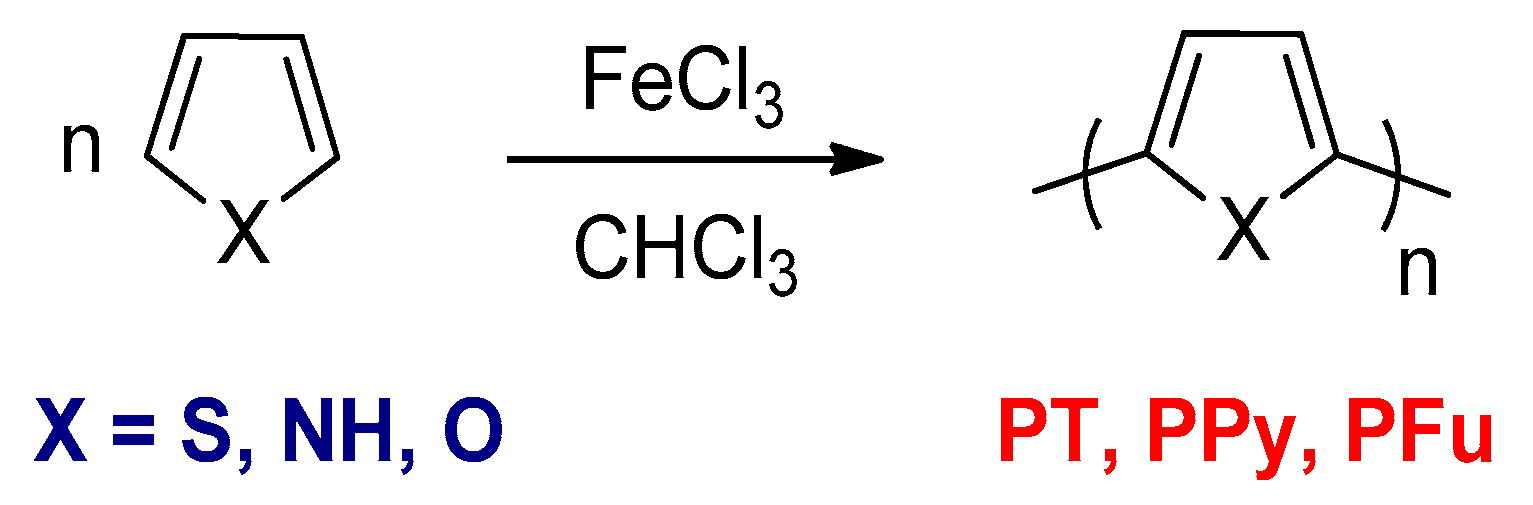
3.1. The Oxidative Polymerizations
3.1.1. Electrochemical Oxidative Polymerizations
3.1.2. The Chemical Oxidative Polymerizations
3.2. Transition Metal Catalyzed Cross-Coupling Polymerizations
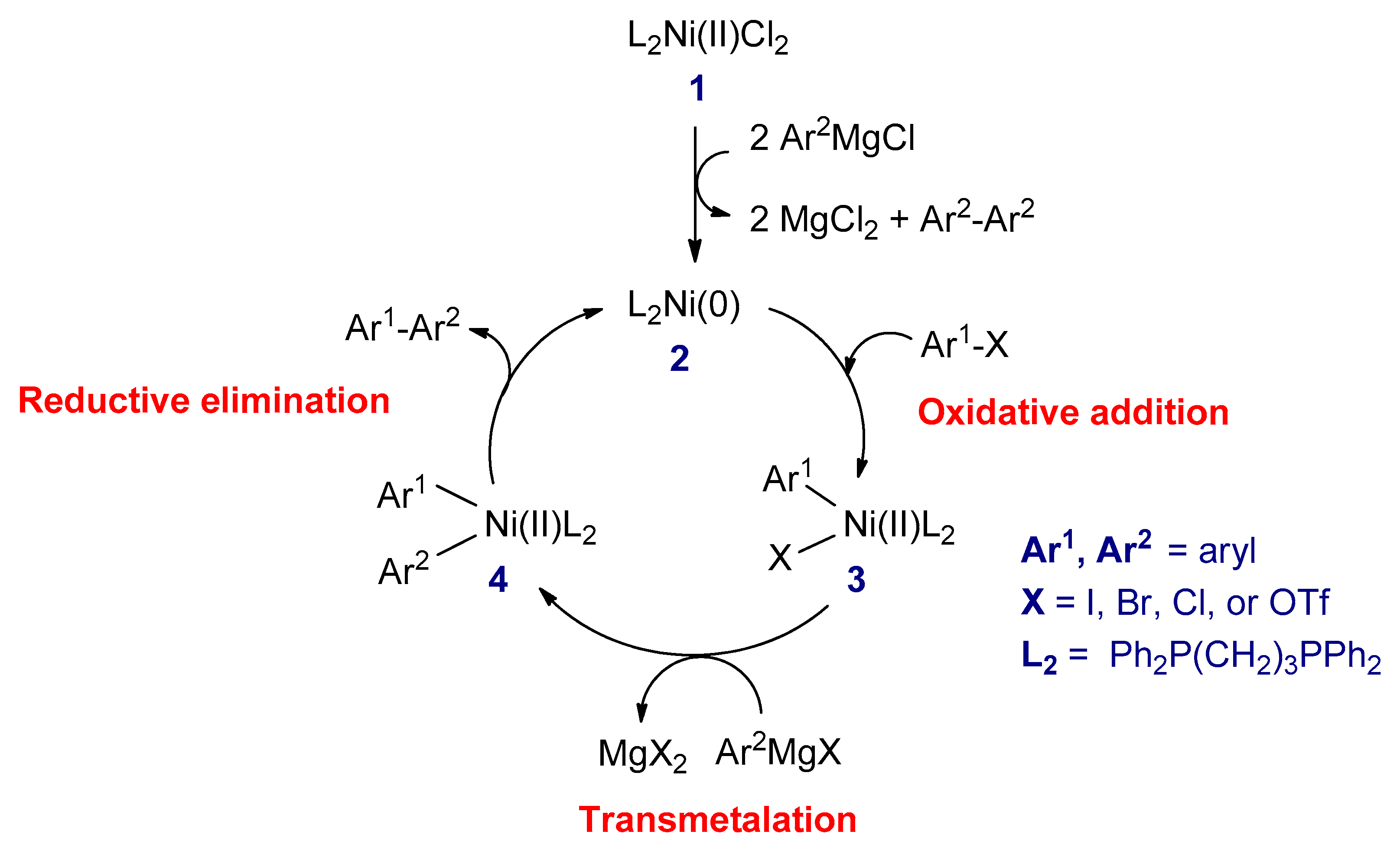
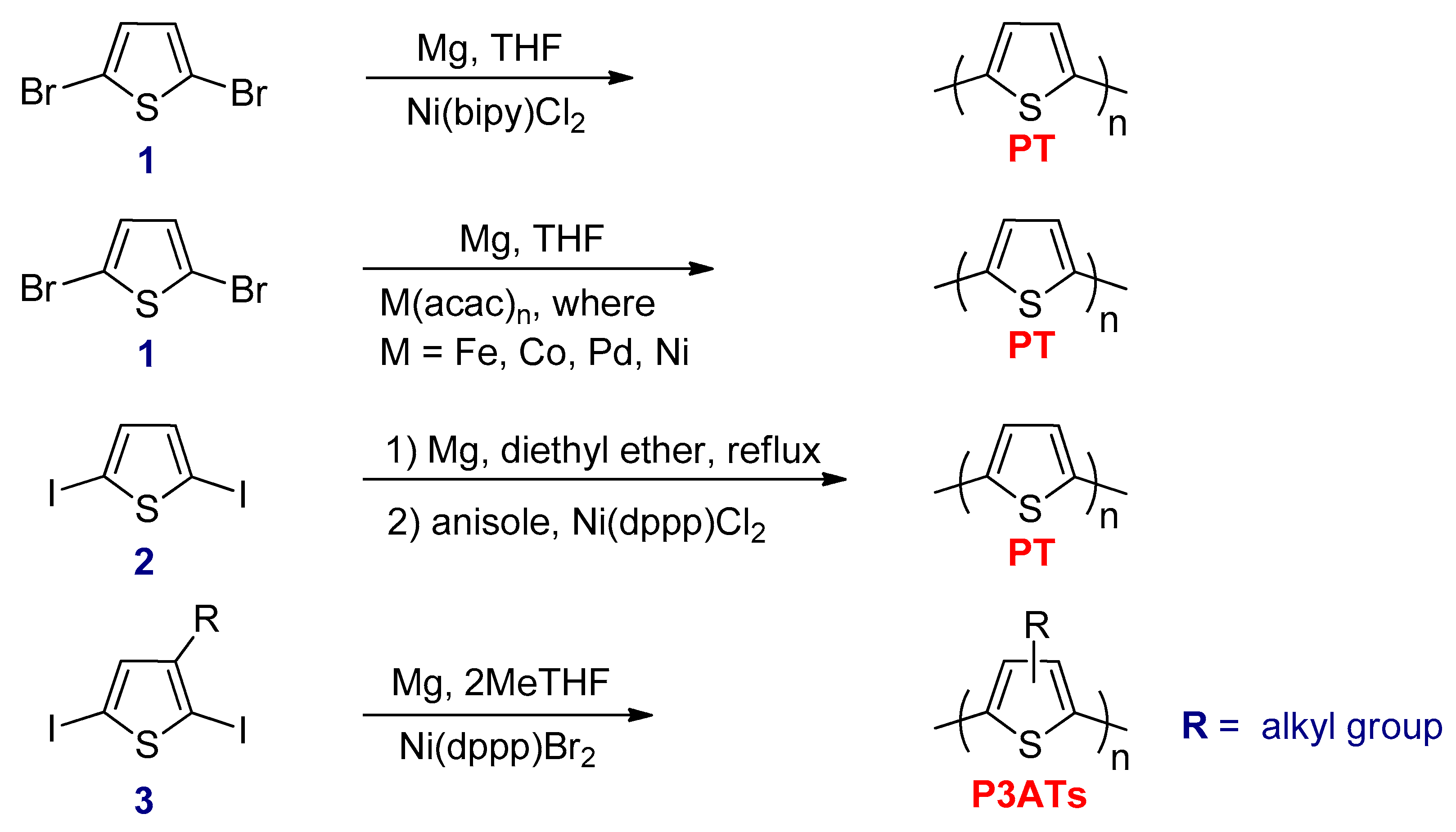
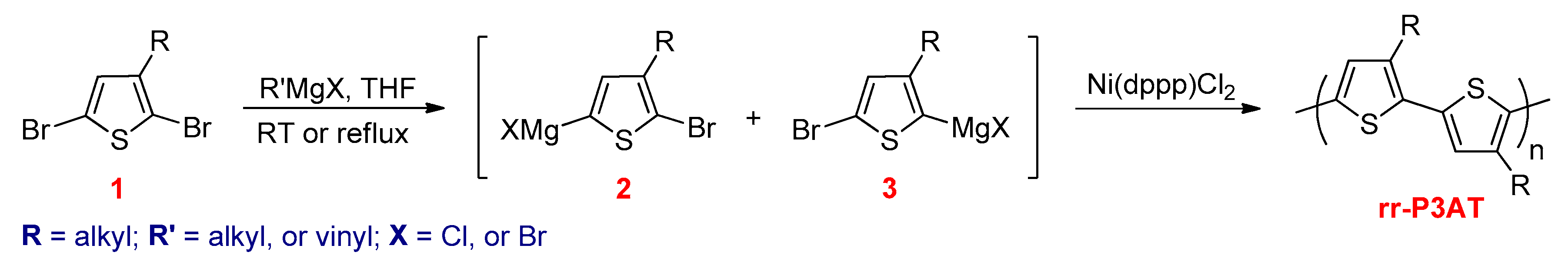
3.2.1. Kumada–Corriu Cross-Coupling Reactions
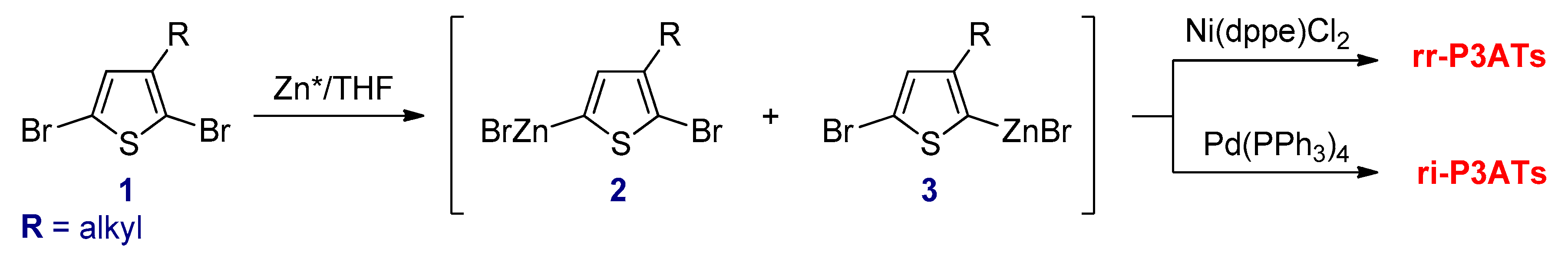
3.2.2. Negishi Cross-Coupling Reactions
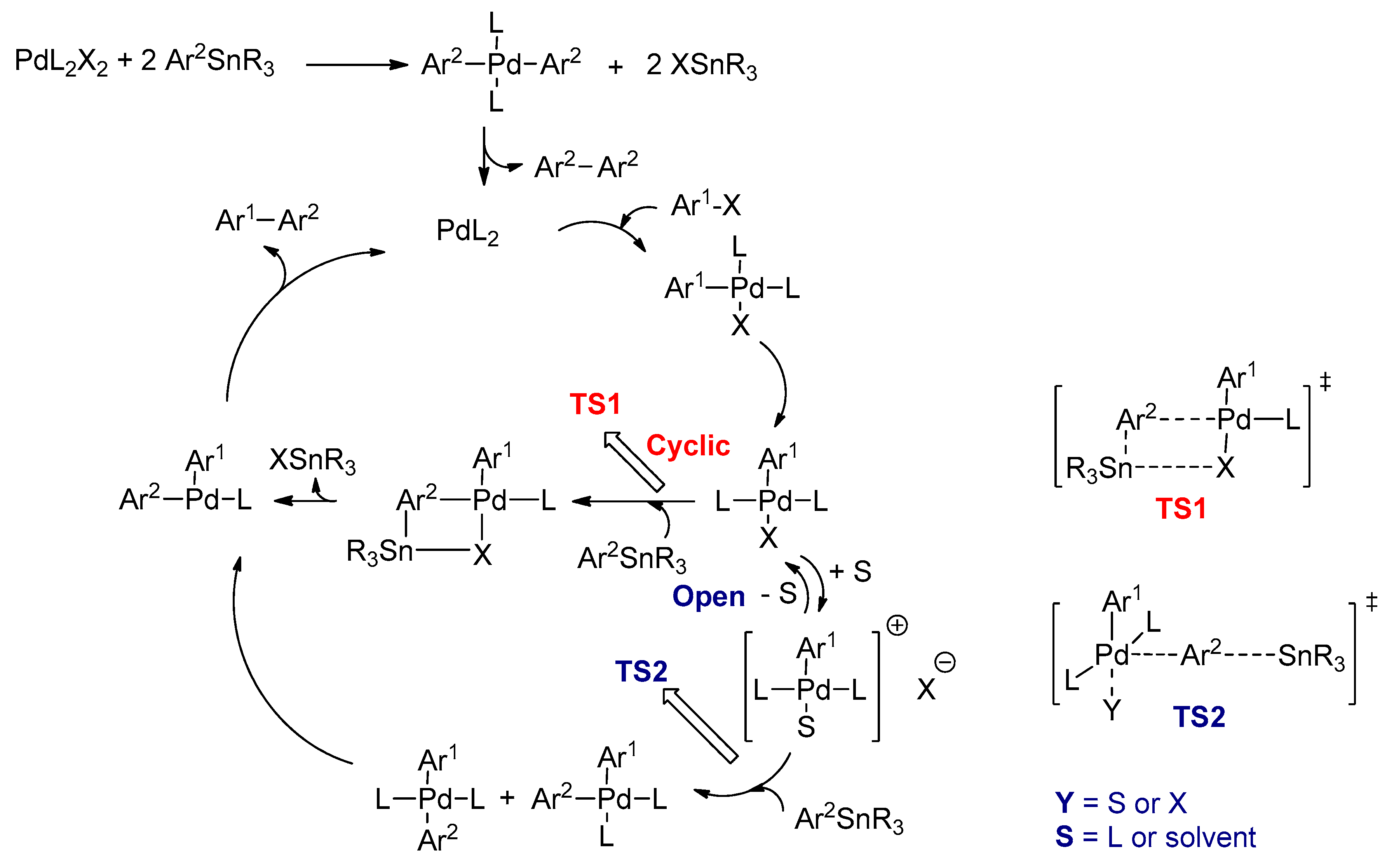
3.2.3. Stille Cross-Coupling Reactions
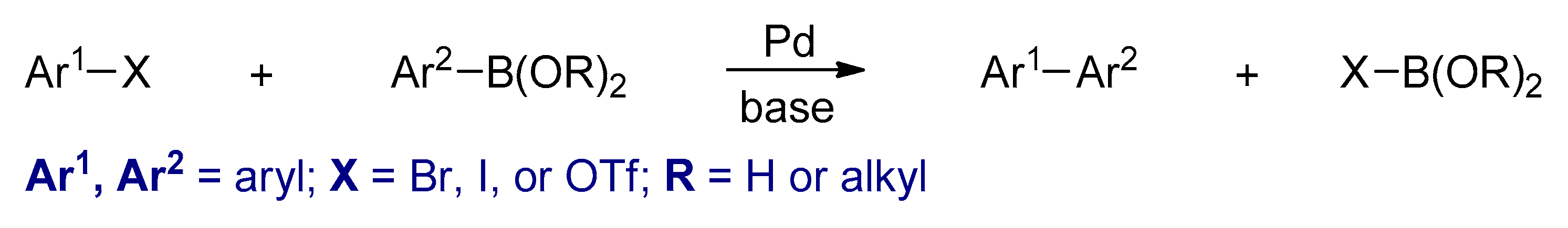
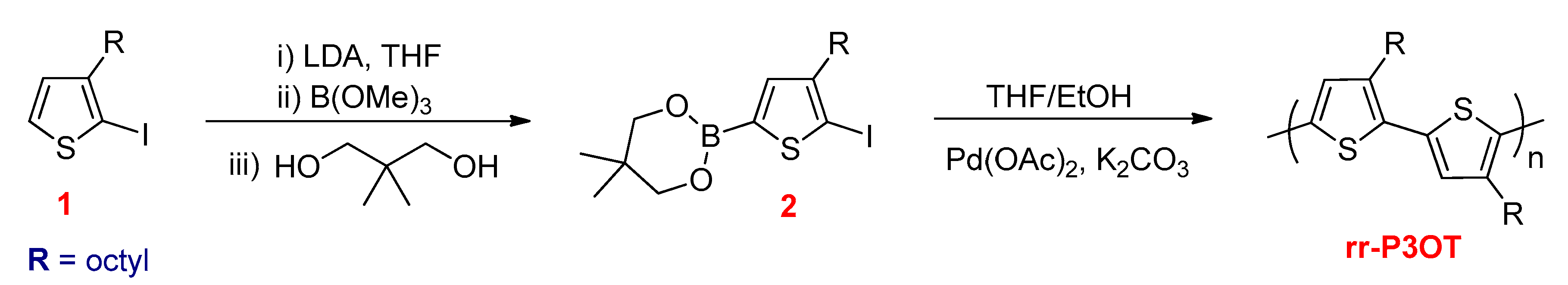
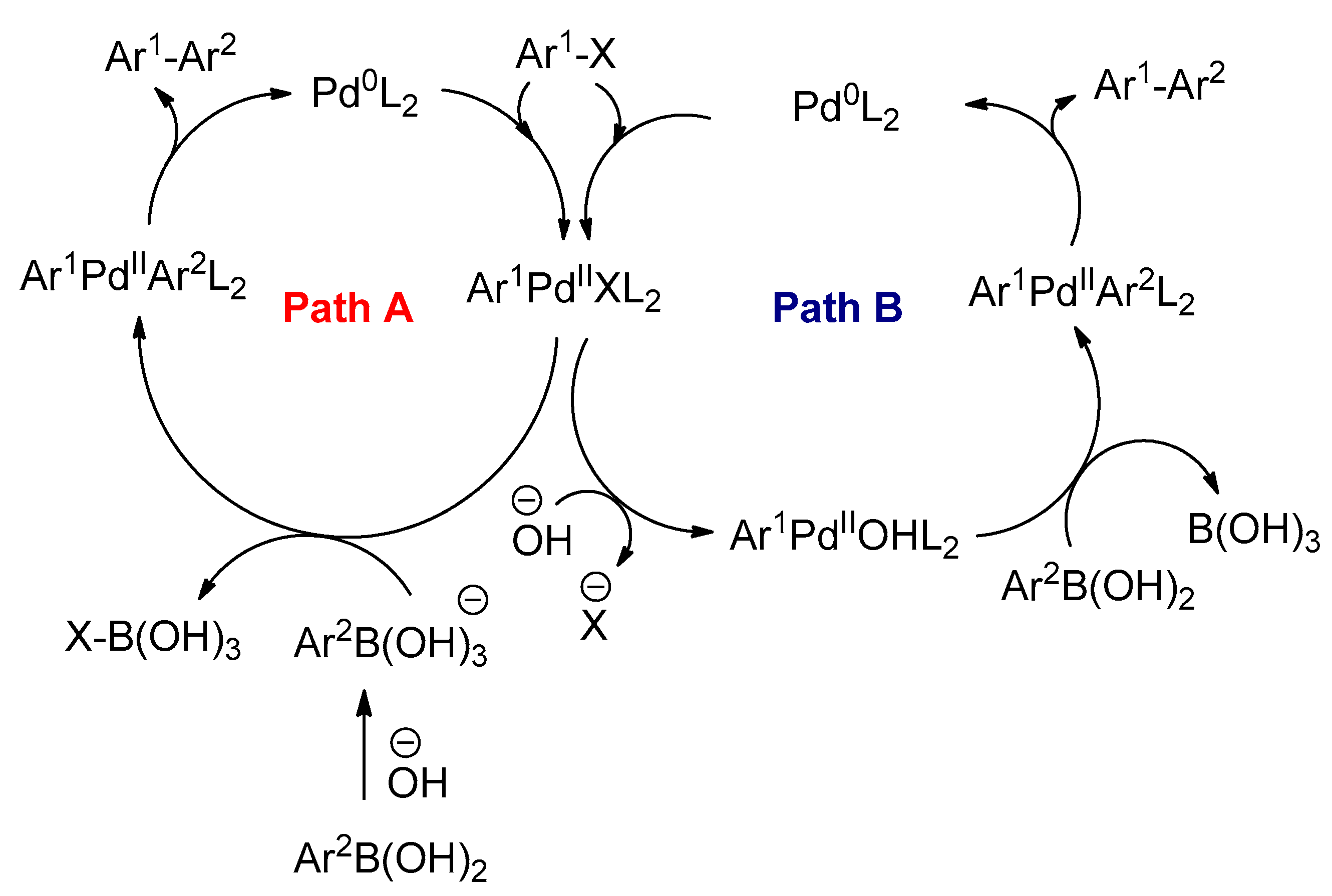
3.2.4. The Suzuki–Miyaura Reactions
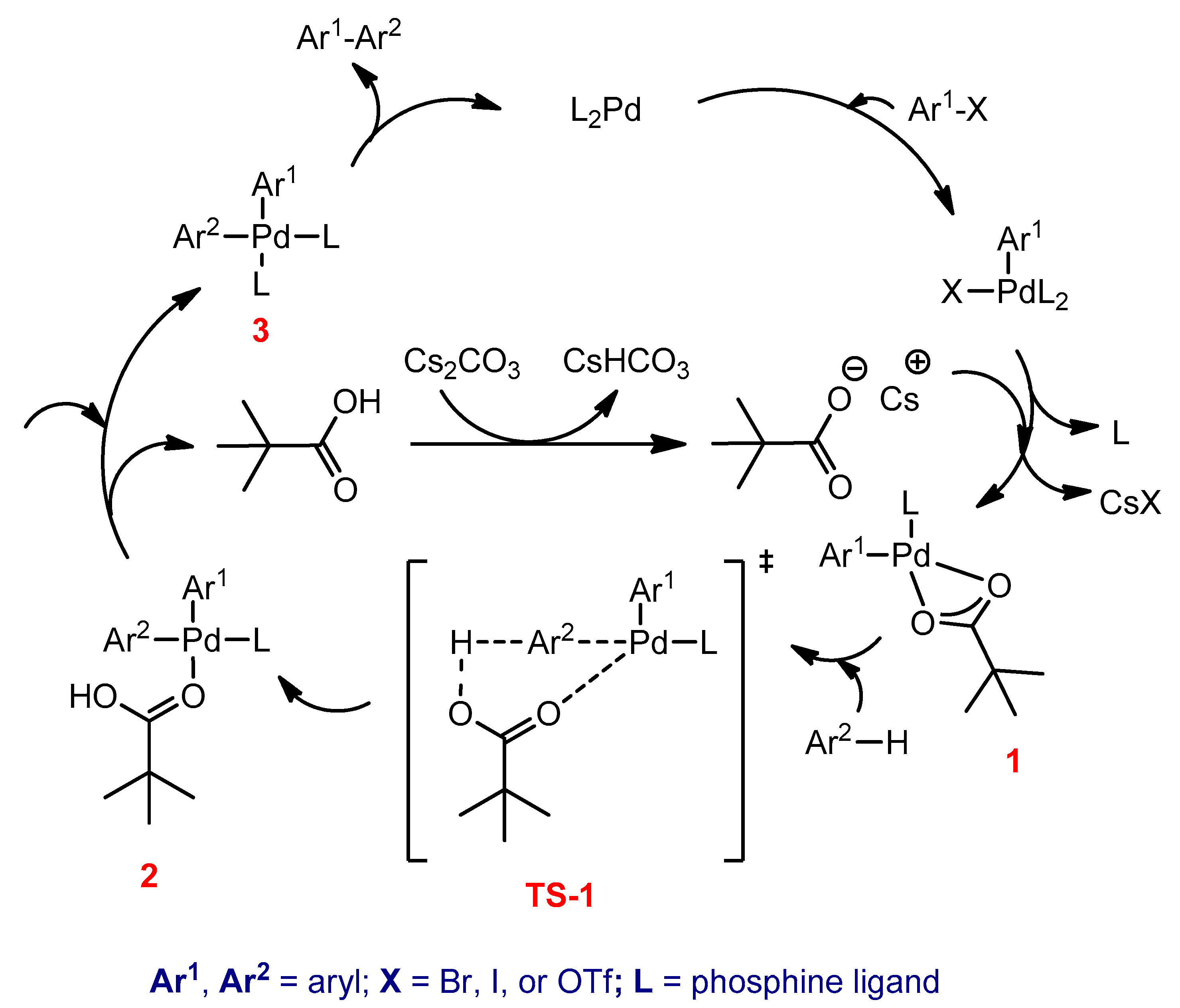
3.2.5. Direct Hetero (Arylation) Cross Coupling Reactions
3.3. Yamamoto Coupling Reactions
3.4. Condensation Polymerization Methods
4. Applications of Conjugated Polymers
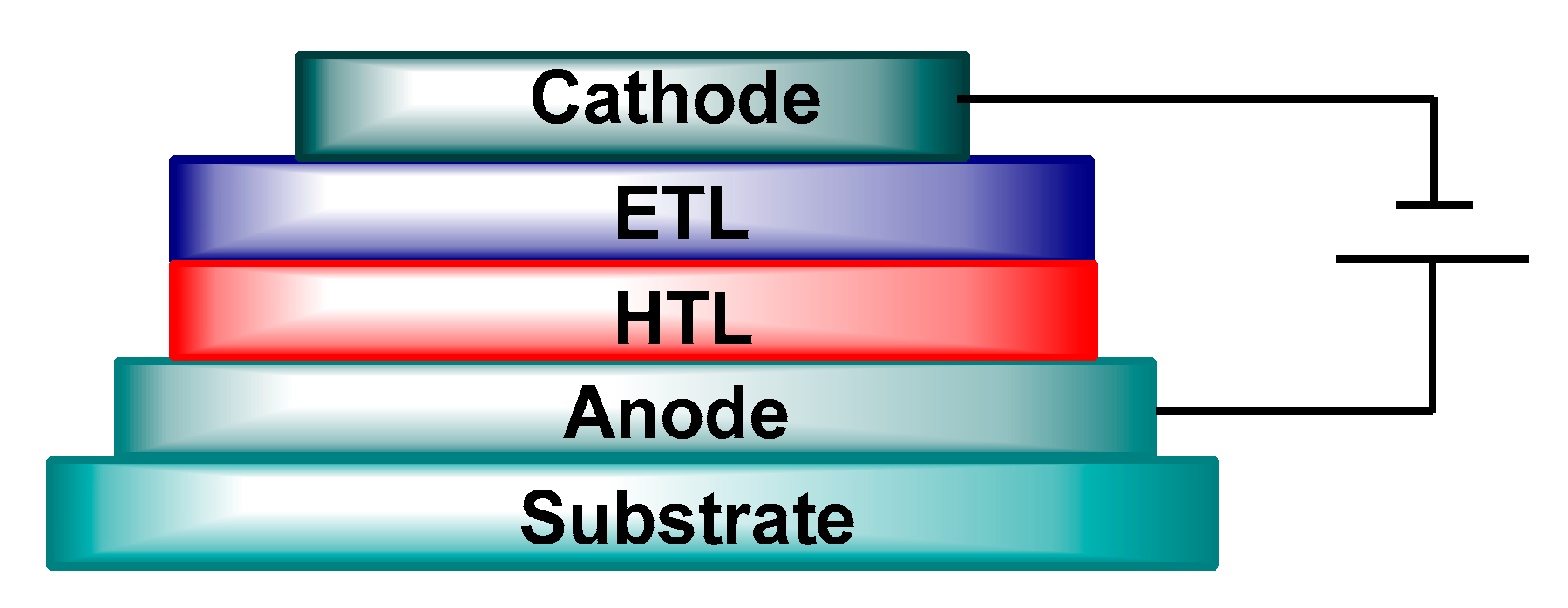
4.1. Organic Light Emitting Diodes (OLEDs)
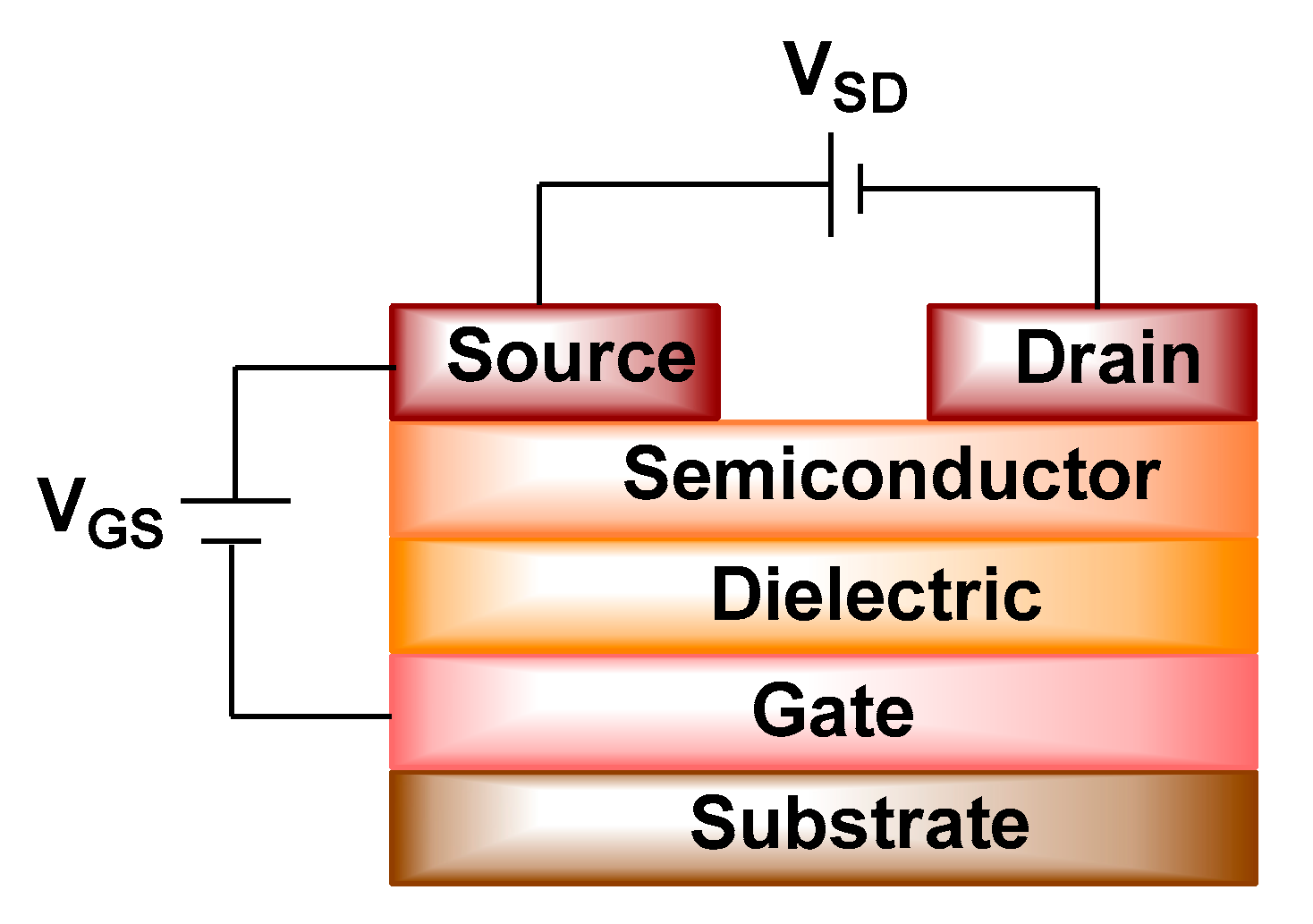
4.2. Organic Field Effect Transistors (OFETs)
4.3. Organic Photovoltaics (OPVs)
5. Architecture of Polymer Solar Cells
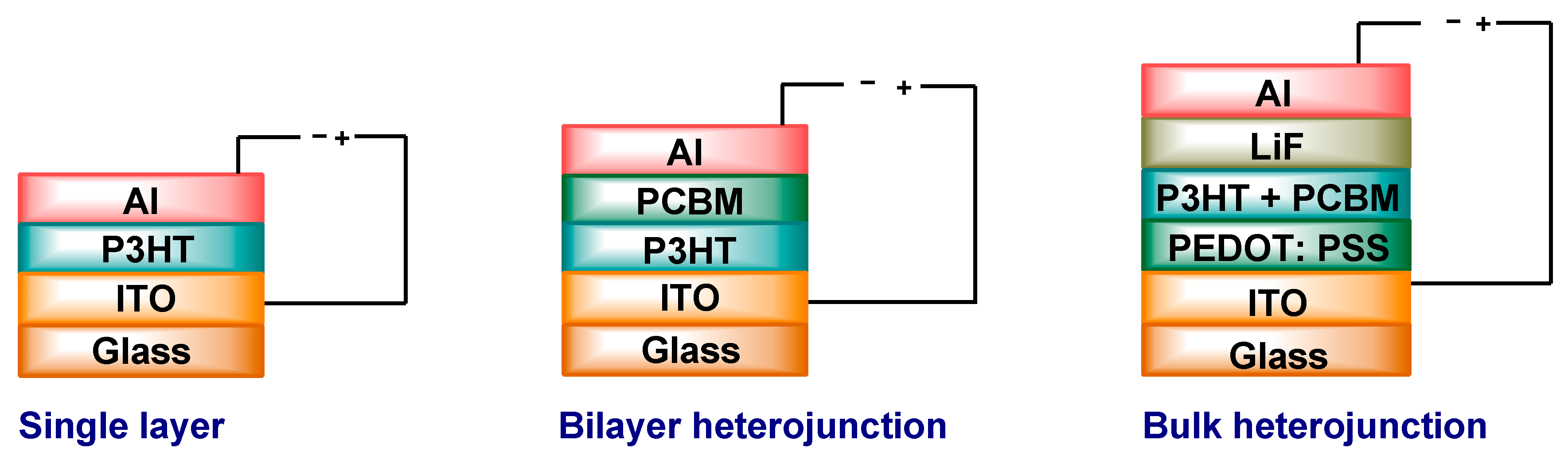
5.1. Single Layer
5.2. Bilayer Planar Heterojunction
5.3. Bulk Heterojunction
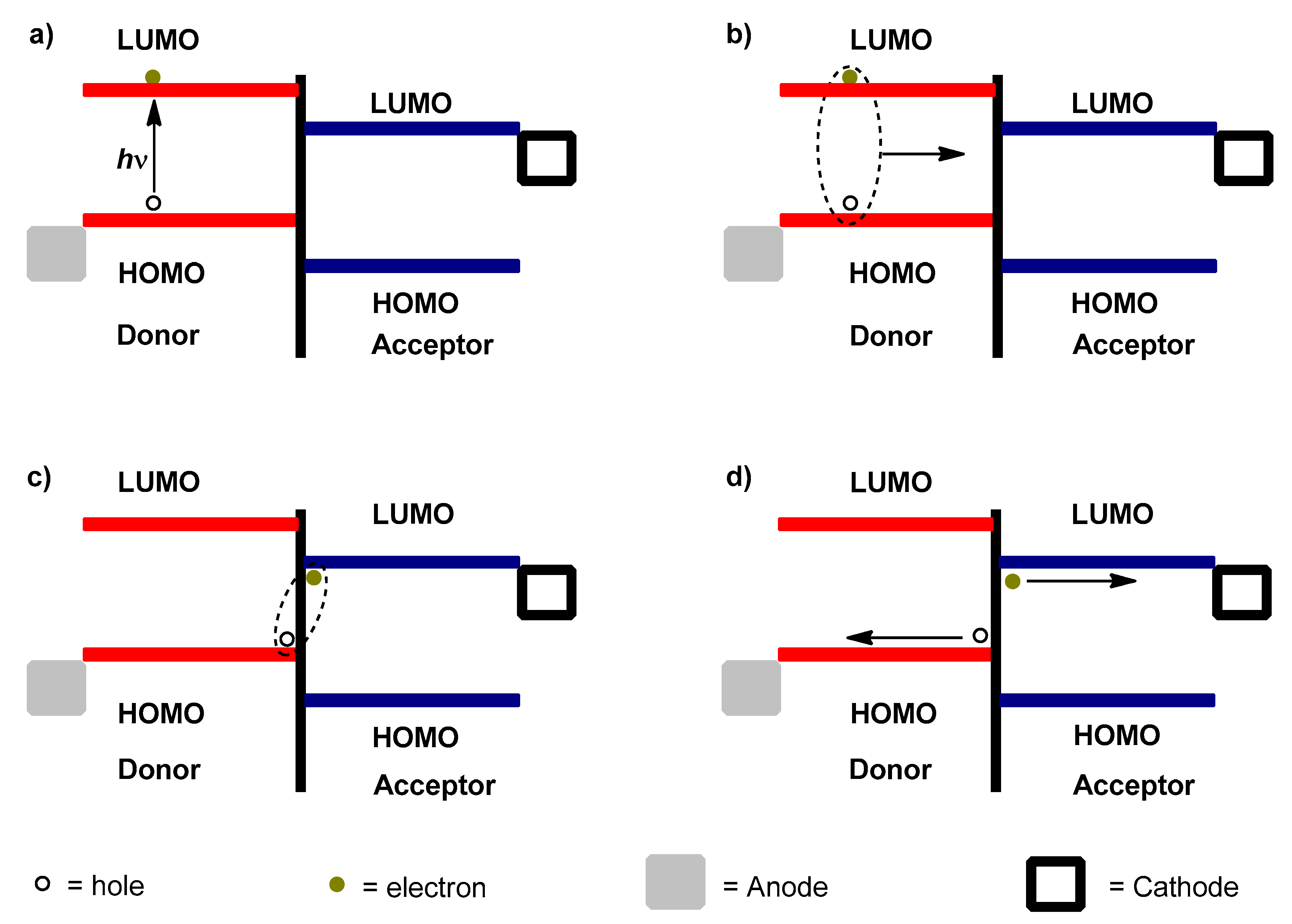
6. Principle Work of Polymer Solar Cells
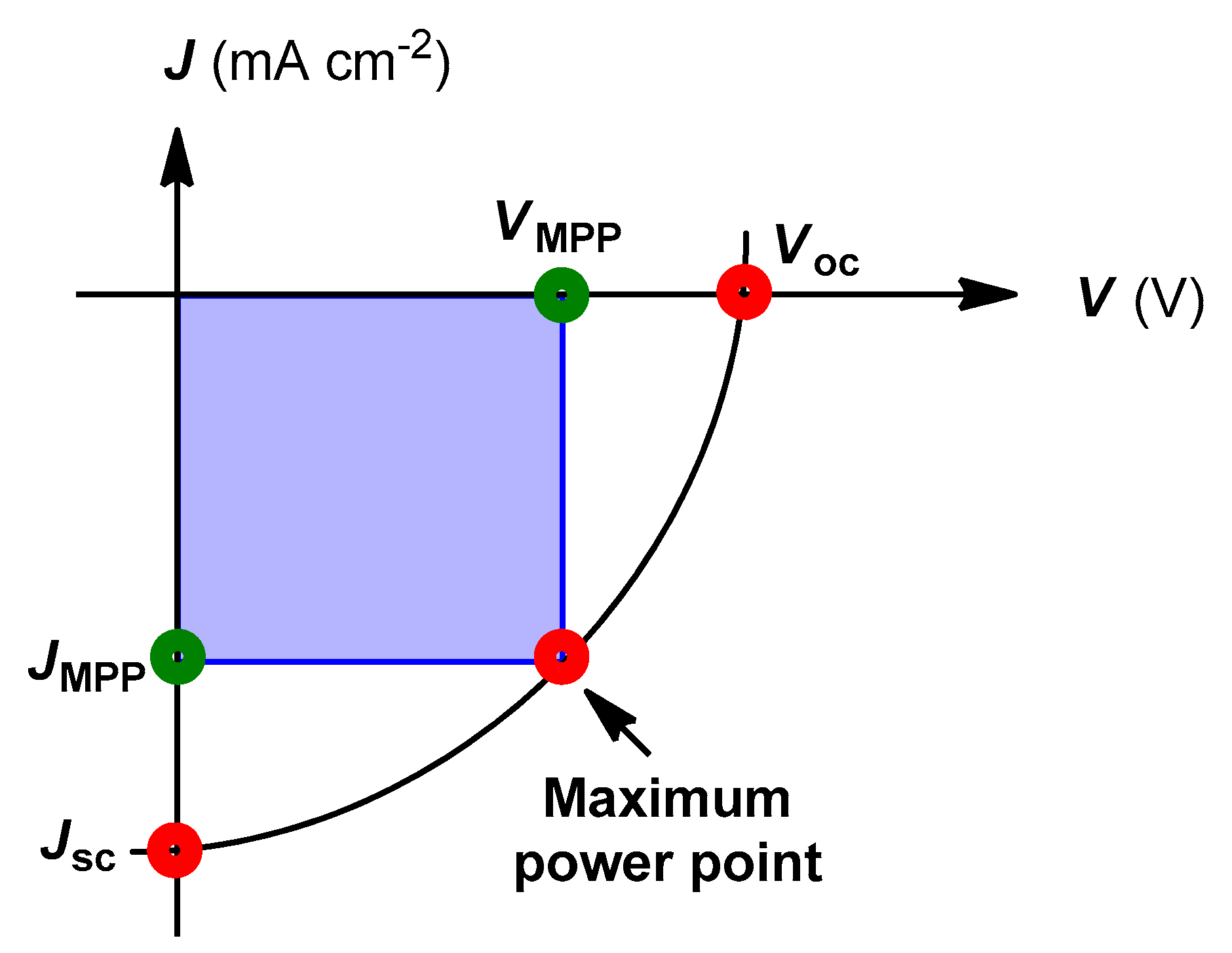
7. Characterization of Polymer Solar Cells
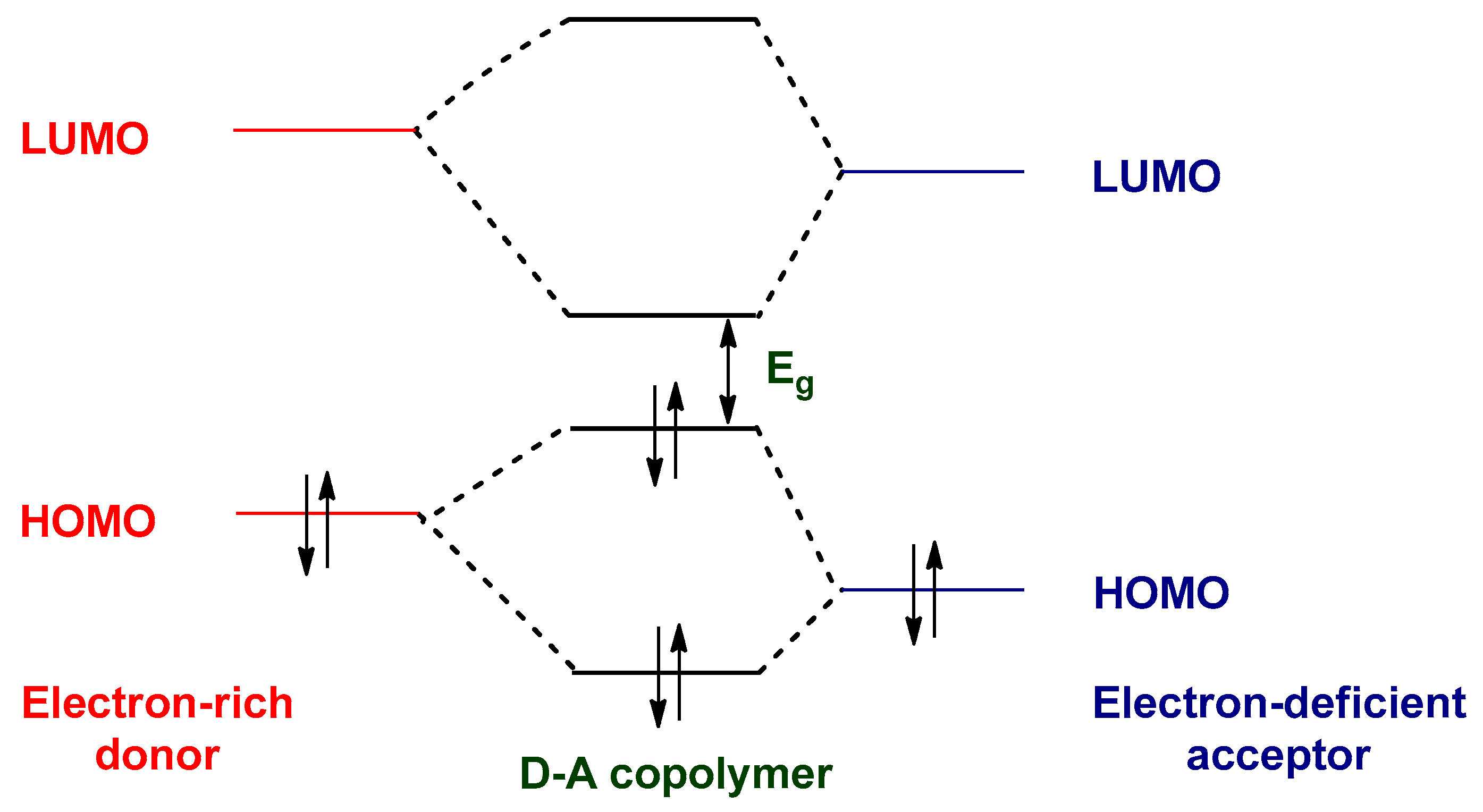
8. Designing Conjugated Polymers for Photovoltaic Applications
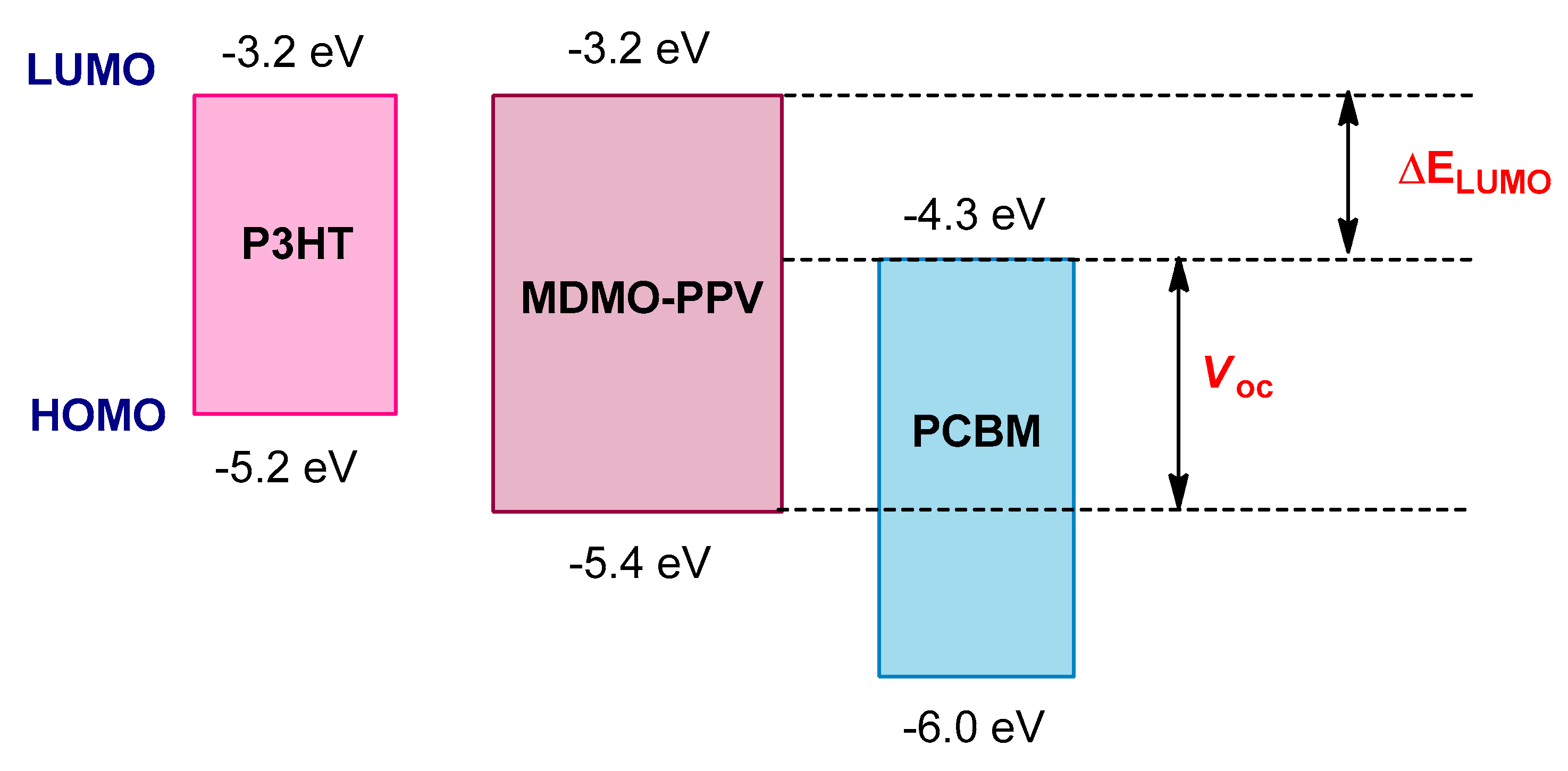
8.1. Optimization of HOMO–LUMO Energy Levels and Optical Band Gaps
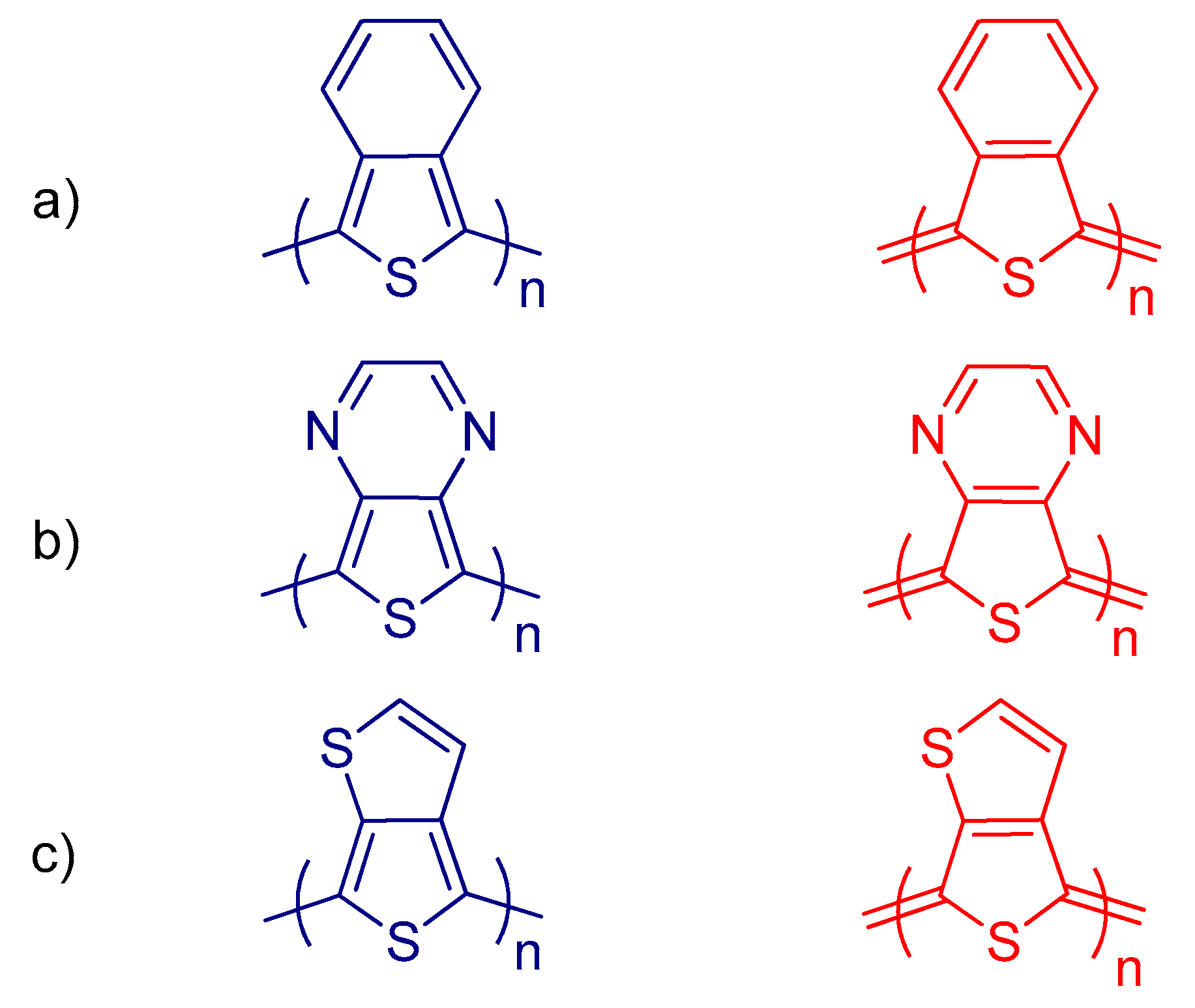
8.2. Strategies for Band Gap Tuning
9. Morphology
9.1. Choice of Solvent(s)
9.2. Thermal Annealing
9.3. Solvent Annealing
9.4. Solvent Additives
9.5. Blend Composition
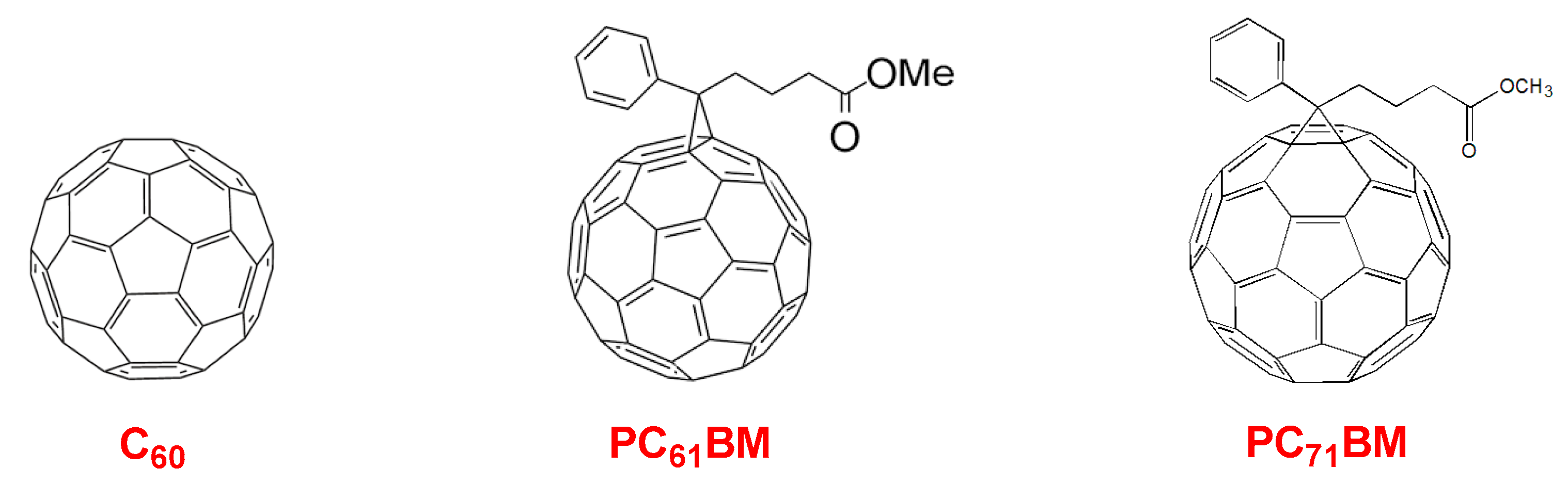
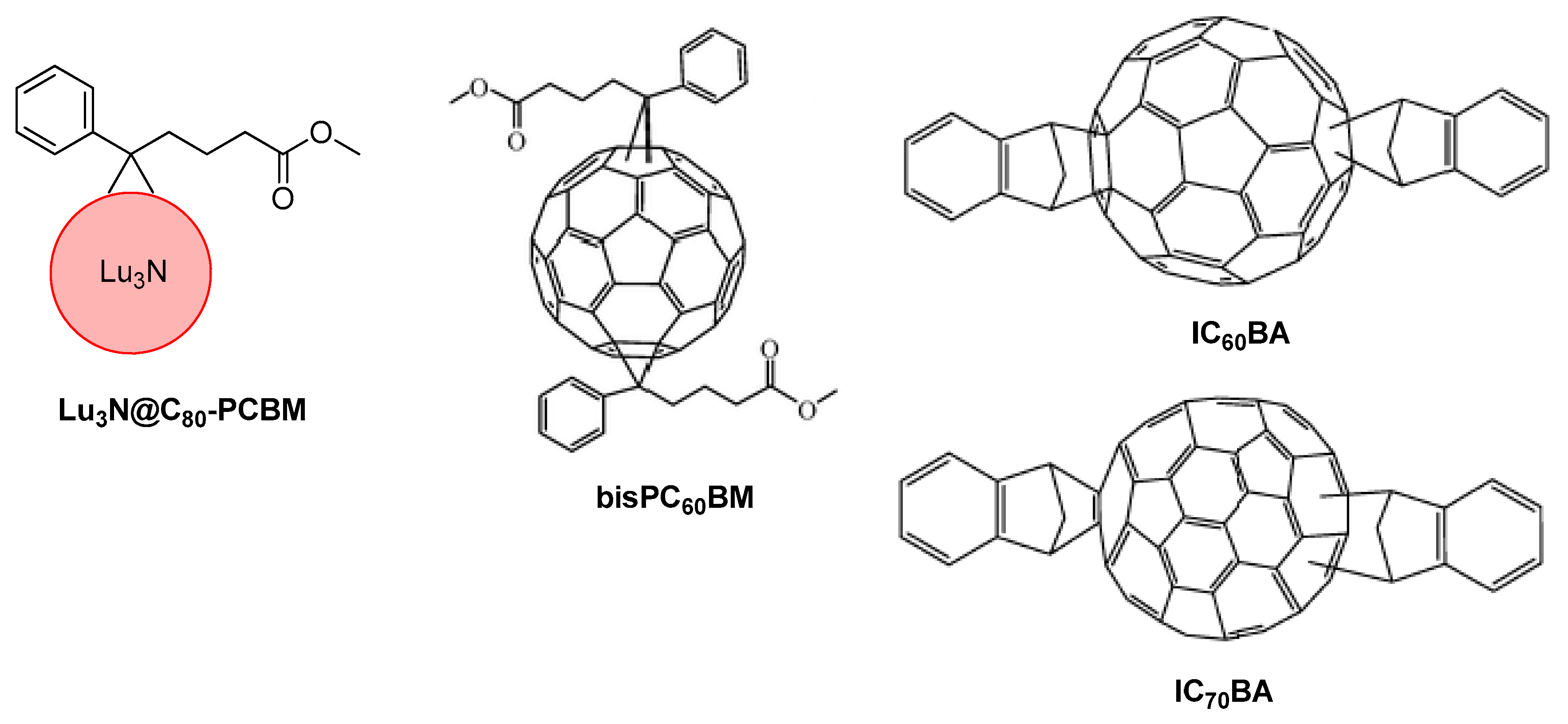
10. Novel Acceptor Materials
11. Summary and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Zulkifli, A.M.; Said, N.I.A.M.; Aziz, S.B.; Dannoun, E.M.A.; Hisham, S.; Shah, S.; Abu Bakar, A.; Abidin, Z.H.Z.; Tajuddin, H.A.; Hadi, J.M.; et al. Characteristics of Dye-Sensitized Solar Cell Assembled from Modified Chitosan-Based Gel Polymer Electrolytes Incorporated with Potassium Iodide. Molecules 2020, 25, 4115. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.-J.; Yang, S.-H.; Hsu, C.-S. Synthesis of Conjugated Polymers for Organic Solar Cell Applications. Chem. Rev. 2009, 109, 5868–5923. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.G.; Rao, K.S.R.K. Physics and chemistry of CdTe/CdS thin film heterojunction photovoltaic devices: Fundamental and critical aspects. Energy Environ. Sci. 2014, 7, 45–102. [Google Scholar] [CrossRef]
- Zulkifli, A.M. Electrochemical Characteristics of Phthaloyl Chitosan Based Gel Polymer Electrolyte for Dye Sensitized Solar Cell Application. Int. J. Electrochem. Sci. 2020, 15, 7434–7447. [Google Scholar] [CrossRef]
- Hamsan, M.H.; Shukur, M.F.; Aziz, S.B.; Yusof, Y.M.; Kadir, M.F.Z. Influence of Br as an ionic source on the structural/electrical properties of dextran-based biopolymer electrolytes and EDLC application. Bull. Mater. Sci. 2020, 43, 1–7. [Google Scholar] [CrossRef]
- Brza, M.; Aziz, S.; Anuar, H.; Ali, F. Structural, ion transport parameter and electrochemical properties of plasticized polymer composite electrolyte based on PVA: A novel approach to fabricate high performance EDLC devices. Polym. Test. 2020, 91, 106813. [Google Scholar] [CrossRef]
- Aziz, S.B.; Brevik, I.; Hamsan, M.H.; Brza, M.A.; Nofal, M.M.; Abdullah, A.M.; Rostam, S.; Al-Zangana, S.; Muzakir, S.K.; Kadir, M.F.Z. Compatible Solid Polymer Electrolyte Based on Methyl Cellulose for Energy Storage Application: Structural, Electrical, and Electrochemical Properties. Polymers 2020, 12, 2257. [Google Scholar] [CrossRef] [PubMed]
- Marf, A.S.; Aziz, S.B.; Abdullah, R.M. Plasticized H+ ion-conducting PVA:CS-based polymer blend electrolytes for energy storage EDLC application. J. Mater. Sci. Mater. Electron. 2020, 1–15. [Google Scholar] [CrossRef]
- Brza, M.A.; Aziz, S.B.; Anuar, H.; Dannoun, E.M.A.; Ali, F.B.; Abdulwahid, R.T.; Al-Zangana, S.; Kadir, M.F.Z. The Study of EDLC Device with High Electrochemical Performance Fabricated from Proton Ion Conducting PVA-Based Polymer Composite Electrolytes Plasticized with Glycerol. Polymers 2020, 12, 1896. [Google Scholar] [CrossRef]
- Azli, A.A.; Manan, N.S.A.; Aziz, S.B.; Kadir, M.F.Z. Structural, impedance and electrochemical double-layer capacitor characteristics of improved number density of charge carrier electrolytes employing potato starch blend polymers. Ionics 2020, 26, 5773–5804. [Google Scholar] [CrossRef]
- Hadi, J.M.; Aziz, S.B.; Nofal, M.M.; Hussein, S.A.; Hamsan, M.H.; Brza, M.A.; Abdulwahid, R.T.; Kadir, M.F.Z.; Woo, H.J. Electrical, Dielectric Property and Electrochemical Performances of Plasticized Silver Ion-Conducting Chitosan-Based Polymer Nanocomposites. Membranes 2020, 10, 151. [Google Scholar] [CrossRef]
- Hadi, J.M.; Aziz, S.B.; Mustafa, M.S.; Hamsan, M.H.; Abdulwahid, R.T.; Kadir, M.F.Z.; Ghareeb, H.O. Role of nano-capacitor on dielectric constant enhancement in PEO:NH4SCN:xCeO2 polymer nano-composites: Electrical and electrochemical properties. J. Mater. Res. Technol. 2020, 9, 9283–9294. [Google Scholar] [CrossRef]
- Fan, X.; Zhang, M.; Wang, X.; Yang, F.; Meng, X. Recent progress in organic–inorganic hybrid solar cells. J. Mater. Chem. A 2013, 1, 8694–8709. [Google Scholar] [CrossRef]
- Ostroverkhova, O. Organic Optoelectronic Materials: Mechanisms and Applications. Chem. Rev. 2016, 116, 13279–13412. [Google Scholar] [CrossRef]
- Dou, L.; Liu, Y.; Hong, Z.; Li, G.; Yang, Y. Low-Bandgap Near-IR Conjugated Polymers/Molecules for Organic Electronics. Chem. Rev. 2015, 115, 12633–12665. [Google Scholar] [CrossRef]
- Wang, C.; Dong, H.; Hu, W.; Liu, Y.; Zhu, D. Semiconducting π-Conjugated Systems in Field-Effect Transistors: A Material Odyssey of Organic Electronics. Chem. Rev. 2012, 112, 2208–2267. [Google Scholar] [CrossRef]
- Günes, S.; Neugebauer, H.; Sariciftci, N.S. Conjugated Polymer-Based Organic Solar Cells. Chem. Rev. 2007, 107, 1324–1338. [Google Scholar] [CrossRef] [PubMed]
- Grimsdale, A.C.; Chan, K.L.; Martin, R.E.; Jokisz, P.G.; Holmes, A.B. Synthesis of Light-Emitting Conjugated Polymers for Applications in Electroluminescent Devices. Chem. Rev. 2009, 109, 897–1091. [Google Scholar] [CrossRef]
- Liu, M.; Gao, Y.; Zhang, Y.; Liu, Z.; Zhao, L. Quinoxaline-based conjugated polymers for polymer solar cells. Polym. Chem. 2017, 8, 4613–4636. [Google Scholar] [CrossRef]
- Kumar, K.R.P.; Murali, M.G.; Udayakumar, D. Synthesis and study of optical properties of linear and hyperbranched conjugated polymers containing thiophene and riphenylamine units. Des. Monomers Polym. 2014, 17, 7–18. [Google Scholar] [CrossRef]
- Koyuncu, F.B.; Sefer, E.; Koyuncu, S.; Ozdemir, E. A new low band gap electrochromic polymer containing 2,5-bis-dithienyl-1H-pyrrole and 2,1,3-benzoselenadiazole moiety with high contrast ratio. Polymers 2011, 52, 5772–5779. [Google Scholar] [CrossRef]
- Huang, J.; Yin, Z.; Zheng, Q. Applications of ZnO in organic and hybrid solar cells. Energy Environ. Sci. 2011, 4, 3861–3877. [Google Scholar] [CrossRef]
- Mühlbacher, D.; Scharber, M.; Morana, M.; Zhu, Z.; Waller, D.; Gaudiana, R.; Brabec, C. High Photovoltaic Performance of a Low-Bandgap Polymer. Adv. Mater. 2006, 18, 2884–2889. [Google Scholar] [CrossRef]
- Dam, N.; Scurlock, R.D.; Wang, B.; Ma, L.; Sundahl, M.; Ogilby, P.R. Singlet Oxygen as a Reactive Intermediate in the Photodegradation of Phenylenevinylene Oligomers. Chem. Mater. 1999, 11, 1302–1305. [Google Scholar] [CrossRef]
- Park, S.H.; Roy, A.; Beaupré, S.; Cho, S.; Coates, N.E.; Moon, J.S.; Moses, D.; Leclerc, M.; Lee, K.; Heeger, A.J. Bulk heterojunction solar cells with internal quantum efficiency approaching 100%. Nat. Photonics 2009, 3, 297–302. [Google Scholar] [CrossRef]
- Winder, C.; Sariciftci, N.S. Low bandgap polymers for photon harvesting in bulk heterojunction solar cells. J. Mater. Chem. 2004, 14, 1077–1086. [Google Scholar] [CrossRef]
- Norrman, K.; Madsen, M.V.; Gevorgyan, S.A.; Krebs, F.C. Degradation Patterns in Water and Oxygen of an Inverted Polymer Solar Cell. J. Am. Chem. Soc. 2010, 132, 16883–16892. [Google Scholar] [CrossRef]
- Gusain, A.; Faria, R.M.; Miranda, P.B. Polymer Solar Cells—Interfacial Processes Related to Performance Issues. Front. Chem. 2019, 7, 61. [Google Scholar] [CrossRef]
- Meng, L.; Zhang, Y.; Wan, X.; Li, C.; Zhang, X.; Wang, Y.; Ke, X.; Xiao, Z.; Ding, L.; Xia, R.; et al. Organic and solution-processed tandem solar cells with 17.3% efficiency. Science 2018, 361, 1094–1098. [Google Scholar] [CrossRef]
- Tang, C.W. Two-layer organic photovoltaic cell. Appl. Phys. Lett. 1986, 48, 183–185. [Google Scholar] [CrossRef]
- Yu, G.; Gao, J.; Hummelen, J.C.; Wudl, F.; Heeger, A.J. Polymer Photovoltaic Cells: Enhanced Efficiencies via a Network of Internal Donor-Acceptor Heterojunctions. Science 1995, 270, 1789–1791. [Google Scholar] [CrossRef]
- Halls, J.J.M.; Walsh, C.A.; Greenham, N.C.; Marseglia, E.A.; Friend, R.H.; Moratti, S.C.; Holmes, A.B. Efficient photodiodes from interpenetrating polymer networks. Nat. Cell Biol. 1995, 376, 498–500. [Google Scholar] [CrossRef]
- Azmer, M.I.; Ahmad, Z.; Sulaiman, K.; Touati, F. Morphological and structural properties of VoPcPhO:P3HT composite thin films. Mater. Lett. 2016, 164, 605–608. [Google Scholar] [CrossRef]
- Zhang, G.; Zhang, K.; Yin, Q.; Jiang, X.-F.; Wang, Z.; Xin, J.; Ma, W.; Yan, H.; Huang, F.; Cao, Y. High-Performance Ternary Organic Solar Cell Enabled by a Thick Active Layer Containing a Liquid Crystalline Small Molecule Donor. J. Am. Chem. Soc. 2017, 139, 2387–2395. [Google Scholar] [CrossRef] [PubMed]
- Kan, B.; Feng, H.; Wan, X.; Liu, F.; Ke, X.; Wang, Y.; Wang, Y.; Zhang, H.; Li, C.; Hou, J.; et al. Small-Molecule Acceptor Based on the Heptacyclic Benzodi(cyclopentadithiophene) Unit for Highly Efficient Nonfullerene Organic Solar Cells. J. Am. Chem. Soc. 2017, 139, 4929–4934. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.; Zhao, F.; Zhang, Q.; Lau, T.-K.; Li, T.; Liu, K.; Ling, Q.; Wang, C.; Lu, X.; You, W.; et al. Fused Nonacyclic Electron Acceptors for Efficient Polymer Solar Cells. J. Am. Chem. Soc. 2017, 139, 1336–1343. [Google Scholar] [CrossRef]
- Srivastava, A.; Singh, V.; Aggarwal, P. Optical studies of insulating polymers for radiation dose monitoring. Indian J. Pure Appl. Phys. 2010, 48, 782–786. [Google Scholar]
- Cheng, P.; Yang, Y. Narrowing the Band Gap: The Key to High-Performance Organic Photovoltaics. Acc. Chem. Res. 2020, 53, 1218–1228. [Google Scholar] [CrossRef]
- Ahmad, Z.; Karimov, K.S.; Fatima, N.; Touati, F. Flexible organic photo-thermogalvanic cell for low power applications. J. Mater. Sci. Mater. Electron. 2015, 27, 2442–2447. [Google Scholar] [CrossRef]
- Mallajosyula, A.T.; Srivastava, N.; Iyer, S.S.K.; Mazhari, B. Characterization of matrix and isolated organic solar cells. Sol. Energy Mater. Sol. Cells 2010, 94, 1319–1323. [Google Scholar] [CrossRef]
- Lei, H.; Fang, G.; Cheng, F.; Ke, W.; Qin, P.; Song, Z.; Zheng, Q.; Fan, X.; Huang, H.; Zhao, X. Enhanced efficiency in organic solar cells via In Situ fabricated p-type copper sulfide as the hole transporting layer. Sol. Energy Mater. Sol. Cells 2014, 128, 77–84. [Google Scholar] [CrossRef]
- Ameri, T.; Li, N.; Brabec, C.J. Highly efficient organic tandem solar cells: A follow up review. Energy Environ. Sci. 2013, 6, 2390–2413. [Google Scholar] [CrossRef]
- Salaneck, W.; Friend, R.; Brédas, J. Electronic structure of conjugated polymers: Consequences of electron–lattice coupling. Phys. Rep. 1999, 319, 231–251. [Google Scholar] [CrossRef]
- Heeger, A.J. Semiconducting and metallic polymers: The fourth generation of polymeric materials (Nobel lecture). Angew. Chem. Int. Ed. 2001, 40, 2591–2611. [Google Scholar] [CrossRef]
- Moliton, A.; Hiorns, R.C. Review of electronic and optical properties of semiconductingπ-conjugated polymers: Applications in optoelectronics. Polym. Int. 2004, 53, 1397–1412. [Google Scholar] [CrossRef]
- Pron, A.; Rannou, P. Processible conjugated polymers: From organic semiconductors to organic metals and superconductors. Prog. Polym. Sci. 2002, 27, 135–190. [Google Scholar] [CrossRef]
- MacDiarmid, A.G. Synthetic metals: A novel role for organic polymers (Nobel lecture). Angew. Chemie Int. Ed. 2001, 40, 2581–2590. [Google Scholar] [CrossRef]
- Chiang, C.K.; Fincher, C.R.; Park, Y.W.; Heeger, A.J.; Shirakawa, H.; Louis, E.J.; Gau, S.C.; MacDiarmid, A.G. Electrical Conductivity in Doped Polyacetylene. Phys. Rev. Lett. 1977, 39, 1098–1101. [Google Scholar] [CrossRef]
- Su, W.P.; Schrieffer, J.; Heeger, A.J. Solitons in polyacetylene. Phys. Rev. Lett. 1979, 42, 1698. [Google Scholar] [CrossRef]
- Bredas, J.; Themans, B.; Andre, J.; Chance, R.; Silbey, R. The role of mobile organic radicals and ions (solitons, polarons and bipolarons) in the transport properties of doped conjugated polymers. Synth. Met. 1984, 9, 265–274. [Google Scholar] [CrossRef]
- Heeger, A.J.; Kivelson, S.A.; Schrieffer, J.R.; Su, W.-P. Solitons in conducting polymers. Rev. Mod. Phys. 1988, 60, 781–850. [Google Scholar] [CrossRef]
- Brabec, C.J.; Dyakonov, V.; Parisi, J.; Sariciftci, N.S. Organic Photovoltaics: Concepts and Realization; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Bredas, J.L.; Street, G.B. Polarons, bipolarons, and solitons in conducting polymers. Acc. Chem. Res. 1985, 18, 309–315. [Google Scholar] [CrossRef]
- Feast, W.; Tsibouklis, J.; Pouwer, K.; Groenendaal, L.; Meijer, E. Synthesis, processing and material properties of conjugated polymers. Polymers 1996, 37, 5017–5047. [Google Scholar] [CrossRef]
- Wei, Y.; Chan, C.C.; Tian, J.; Jang, G.W.; Hsueh, K.F. Electrochemical polymerization of thiophenes in the presence of bithiophene or terthiophene: Kinetics and mechanism of the polymerization. Chem. Mater. 1991, 3, 888–897. [Google Scholar] [CrossRef]
- Heinze, J.; Frontana-Uribe, B.A.; Ludwigs, S. Electrochemistry of Conducting Polymers—Persistent Models and New Concepts†. Chem. Rev. 2010, 110, 4724–4771. [Google Scholar] [CrossRef]
- Waltman, R.J.; Bargon, J. Electrically conducting polymers: A review of the electropolymerization reaction, of the effects of chemical structure on polymer film properties, and of applications towards technology. Can. J. Chem. 1986, 64, 76–95. [Google Scholar] [CrossRef]
- Hayakawa, T.; Fukukawa, K.I.; Morishima, M.; Takeuchi, K.; Asai, M.; Ando, S.; Ueda, M. Formation of regioregular head-to-tail poly[3-(4-butylphenyl)thiophene] by an oxidative coupling polymerization with vanadium acetylacetonate. J. Polym. Sci. Part A Polym. Chem. 2001, 39, 2287–2295. [Google Scholar] [CrossRef]
- Yoshino, K.; Nakajima, S.; Sugimoto, R.-I. Fusibility of Polythiophene Derivatives with Substituted Long Alkyl Chain and Their Properties. Jpn. J. Appl. Phys. 1987, 26, L1038–L1039. [Google Scholar] [CrossRef]
- Yoshino, K.; Hayashi, S.; Sugimoto, R.-I. Preparation and Properties of Conducting Heterocyclic Polymer Films by Chemical Method. Jpn. J. Appl. Phys. 1984, 23, L899–L900. [Google Scholar] [CrossRef]
- Mao, H.; Xu, B.; Holdcroft, S. Synthesis and structure-property relationships of regioirregular poly(3-hexylthiophenes). Macromolecules 1993, 26, 1163–1169. [Google Scholar] [CrossRef]
- Andersson, M.R.; Selse, D.; Berggren, M.; Jaervinen, H.; Hjertberg, T.; Inganaes, O.; Wennerstroem, O.; Oesterholm, J.-E. Regioselective polymerization of 3-(4-octylphenyl)thiophene with FeCl3. Macromolecules 1994, 27, 6503–6506. [Google Scholar] [CrossRef]
- Cheng, Y.-J.; Luh, T.-Y. Synthesizing optoelectronic heteroaromatic conjugated polymers by cross-coupling reactions. J. Organomet. Chem. 2004, 689, 4137–4148. [Google Scholar] [CrossRef]
- Tamao, K.; Sumitani, K.; Kumada, M. Selective carbon-carbon bond formation by cross-coupling of Grignard reagents with organic halides. Catalysis by nickel-phosphine complexes. J. Am. Chem. Soc. 1972, 94, 4374–4376. [Google Scholar] [CrossRef]
- Kumada, M. Nickel and palladium complex catalyzed cross-coupling reactions of organometallic reagents with organic halides. Pure Appl. Chem. 1980, 52, 669–679. [Google Scholar] [CrossRef]
- Tamao, K.; Kodama, S.; Nakajima, I.; Kumada, M.; Minato, A.; Suzuki, K. Nickel-phosphine complex-catalyzed Grignard coupling—II. Tetrahedron 1982, 38, 3347–3354. [Google Scholar] [CrossRef]
- Jana, R.; Pathak, T.P.; Sigman, M.S. Advances in Transition Metal (Pd,Ni,Fe)-Catalyzed Cross-Coupling Reactions Using Alkyl-organometallics as Reaction Partners. Chem. Rev. 2011, 111, 1417–1492. [Google Scholar] [CrossRef]
- Yamamoto, T.; Sanechika, K.; Yamamoto, A. Preparation of thermostable and electric-conducting poly(2,5-thienylene). J. Polym. Sci. Part C Polym. Lett. 1980, 18, 9–12. [Google Scholar] [CrossRef]
- Lin, J.W.-P.; Dudek, L.P. Synthesis and properties of poly(2,5-thienylene). J. Polym. Sci. Polym. Chem. Ed. 1980, 18, 2869–2873. [Google Scholar] [CrossRef]
- Kobayashi, M.; Chen, J.; Chung, T.-C.; Moraes, F.; Heeger, A.; Wudl, F. Synthesis and properties of chemically coupled poly(thiophene). Synth. Met. 1984, 9, 77–86. [Google Scholar] [CrossRef]
- Elsenbaumer, R.; Jen, K.; Oboodi, R. Processible and environmentally stable conducting polymers. Synth. Met. 1986, 15, 169–174. [Google Scholar] [CrossRef]
- Elsenbaumer, R.; Jen, A.K.-Y.; Miller, G.; Shacklette, L. Processible, environmentally stable, highly conductive forms of polythiophene. Synth. Met. 1987, 18, 277–282. [Google Scholar] [CrossRef]
- McCullough, R.D.; Lowe, R.D. Enhanced electrical conductivity in regioselectively synthesized poly(3-alkylthiophenes). J. Chem. Soc. Chem. Commun. 1992, 70–72. [Google Scholar] [CrossRef]
- McCullough, R.D. The chemistry of conducting polythiophenes. Adv. Mater. 1998, 10, 93–116. [Google Scholar] [CrossRef]
- McCullough, R.D.; Lowe, R.D.; Jayaraman, M.; Anderson, D.L. Design, synthesis, and control of conducting polymer architectures: Structurally homogeneous poly(3-alkylthiophenes). J. Org. Chem. 1993, 58, 904–912. [Google Scholar] [CrossRef]
- Loewe, R.S.; Khersonsky, S.M.; McCullough, R.D. A Simple Method to Prepare Head-to-Tail Coupled, Regioregular Poly(3-alkylthiophenes) Using Grignard Metathesis. Adv. Mater. 1999, 11, 250–253. [Google Scholar] [CrossRef]
- Loewe, R.S.; Ewbank, P.C.; Liu, J.; Zhai, A.L.; McCullough, R.D. Regioregular, Head-to-Tail Coupled Poly(3-alkylthiophenes) Made Easy by the GRIM Method: Investigation of the Reaction and the Origin of Regioselectivity. Macromolecules 2001, 34, 4324–4333. [Google Scholar] [CrossRef]
- Negishi, E. Palladium- or nickel-catalyzed cross coupling. A new selective method for carbon-carbon bond formation. Acc. Chem. Res. 1982, 15, 340–348. [Google Scholar] [CrossRef]
- Chen, T.A.; Rieke, R.D. The first regioregular head-to-tail poly(3-hexylthiophene-2,5-diyl) and a regiorandom isopolymer: Nickel versus palladium catalysis of 2(5)-bromo-5(2)-(bromozincio)-3-hexylthiophene polymerization. J. Am. Chem. Soc. 1992, 114, 10087–10088. [Google Scholar] [CrossRef]
- Chen, T.-A.; Wu, X.; Rieke, R.D. Regiocontrolled Synthesis of Poly(3-alkylthiophenes) Mediated by Rieke Zinc: Their Characterization and Solid-State Properties. J. Am. Chem. Soc. 1995, 117, 233–244. [Google Scholar] [CrossRef]
- Carsten, B.; He, F.; Son, H.J.; Xu, T.; Yu, L. Stille Polycondensation for Synthesis of Functional Materials. Chem. Rev. 2011, 111, 1493–1528. [Google Scholar] [CrossRef]
- Stille, J.K. The Palladium-Catalyzed Cross-Coupling Reactions of Organotin Reagents with Organic Electrophiles. Angew. Chem. Int. Ed. 1986, 25, 508–524. [Google Scholar] [CrossRef]
- Farina, V.; Krishnan, B. Large rate accelerations in the stille reaction with tri-2-furylphosphine and triphenylarsine as palladium ligands: Mechanistic and synthetic implications. J. Am. Chem. Soc. 1991, 113, 9585–9595. [Google Scholar] [CrossRef]
- Hassan, J.; Sévignon, M.; Gozzi, C.; Schulz, E.; Lemaire, M. Aryl−Aryl Bond Formation One Century after the Discovery of the Ullmann Reaction. Chem. Rev. 2002, 102, 1359–1470. [Google Scholar] [CrossRef]
- Stanforth, S.P. Catalytic cross-coupling reactions in biaryl synthesis. Tetrahedron 1998, 54, 263–303. [Google Scholar] [CrossRef]
- Bao, Z.; Chan, W.K.; Yu, L. Exploration of the Stille Coupling Reaction for the Synthesis of Functional Polymers. J. Am. Chem. Soc. 1995, 117, 12426–12435. [Google Scholar] [CrossRef]
- Iraqi, A.; Barker, G.W. Synthesis and characterisation of telechelic regioregular head-to-tail poly(3-alkylthiophenes). J. Mater. Chem. 1998, 8, 25–29. [Google Scholar] [CrossRef]
- Casado, A.L.; Espinet, P. Mechanism of the Stille reaction. 1. The transmetalation step. Coupling of R1I and R2SnBu3 catalyzed by trans-[PdR1IL2](R1 = C6Cl2F3; R2 = vinyl, 4-methoxyphenyl; L = AsPh3). J. Am. Chem. Soc. 1998, 120, 8978–8985. [Google Scholar] [CrossRef]
- Espinet, P.; Echavarren, A.M. The mechanisms of the Stille reaction. Angew. Chem. Int. Ed. 2004, 43, 4704–4734. [Google Scholar]
- Casado, A.L.; Espinet, P.; Gallego, A.M.; Martínez-Ilarduya, J.M. Snapshots of a Stille reaction. Chem. Commun. 2001, 339–340. [Google Scholar] [CrossRef]
- Nova, A.; Ujaque, G.; Maseras, F.; Lledós, A.; Espinet, P. A Critical Analysis of the Cyclic and Open Alternatives of the Transmetalation Step in the Stille Cross-Coupling Reaction. J. Am. Chem. Soc. 2006, 128, 14571–14578. [Google Scholar] [CrossRef]
- Casado, A.L.; Espinet, P.; Gallego, A.M. Mechanism of the Stille Reaction. 2. Couplings of Aryl Triflates with Vinyltributyltin. Observation of Intermediates. A More Comprehensive Scheme. J. Am. Chem. Soc. 2000, 122, 11771–11782. [Google Scholar] [CrossRef]
- Suzuki, A. Recent advances in the cross-coupling reactions of organoboron derivatives with organic electrophiles, 1995–1998. J. Organomet. Chem. 1999, 576, 147–168. [Google Scholar] [CrossRef]
- Suzuki, A. Cross-Coupling Reactions of Organoboranes: An Easy Way to Construct C-C Bonds (Noble Lecture). Angew. Chem. Int. Ed. 2011, 50, 6722–6737. [Google Scholar] [CrossRef] [PubMed]
- Miyaura, N. Cross-coupling reaction of organoboron compounds via base-assisted transmetalation to palladium (II) complexes. J. Organomet. Chem. 2002, 653, 54–57. [Google Scholar] [CrossRef]
- Miyaura, N.; Suzuki, A. Palladium-Catalyzed Cross-Coupling Reactions of Organoboron Compounds. Chem. Rev. 1995, 95, 2457–2483. [Google Scholar] [CrossRef]
- Schlüter, A. The tenth anniversary of Suzuki polycondensation (SPC). J. Polym. Sci. Part A Polym. Chem. 2001, 39, 1533–1556. [Google Scholar] [CrossRef]
- Guillerez, S.; Bidan, G. New convenient synthesis of highly regioregular poly(3-octylthiophene) based on the Suzuki coupling reaction. Synth. Met. 1998, 93, 123–126. [Google Scholar] [CrossRef]
- Amatore, C.; Jutand, A. Anionic Pd(0) and Pd(II) Intermediates in Palladium-Catalyzed Heck and Cross-Coupling Reactions. Acc. Chem. Res. 2000, 33, 314–321. [Google Scholar] [CrossRef]
- Casado, A.L.; Espinet, P. On the configuration resulting from oxidative addition of RX to Pd(PPh3)4 and the mechanism of the cis-to-trans isomerization of [PdRX(PPh3)2] complexes (R= aryl, X= halide). Organometallics 1998, 17, 954–959. [Google Scholar] [CrossRef]
- Braga, A.A.C.; Ujaque, G.; Maseras, F. A DFT Study of the Full Catalytic Cycle of the Suzuki−Miyaura Cross-Coupling on a Model System. Organometallics 2006, 25, 3647–3658. [Google Scholar] [CrossRef]
- Braga, A.A.; Morgon, N.H.; Ujaque, G.; Maseras, F. Computational Characterization of the Role of the Base in the Suzuki−Miyaura Cross-Coupling Reaction. J. Am. Chem. Soc. 2005, 127, 9298–9307. [Google Scholar] [CrossRef]
- Braga, A.A.; Morgon, N.H.; Ujaque, G.; Lledós, A.; Maseras, F. Computational study of the transmetalation process in the Suzuki–Miyaura cross-coupling of aryls. J. Organomet. Chem. 2006, 691, 4459–4466. [Google Scholar] [CrossRef]
- Matos, K.; Soderquist, J.A. Alkylboranes in the Suzuki−Miyaura Coupling: Stereochemical and Mechanistic Studies. J. Org. Chem. 1998, 63, 461–470. [Google Scholar] [CrossRef]
- Carrow, B.P.; Hartwig, J.F. Distinguishing Between Pathways for Transmetalation in Suzuki−Miyaura Reactions. J. Am. Chem. Soc. 2011, 133, 2116–2119. [Google Scholar] [CrossRef]
- Martin, R.; Buchwald, S.L. Palladium-Catalyzed Suzuki−Miyaura Cross-Coupling Reactions Employing Dialkylbiaryl Phosphine Ligands. Acc. Chem. Res. 2008, 41, 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- Aliprantis, A.O.; Canary, J.W. Observation of Catalytic Intermediates in the Suzuki Reaction by Electrospray Mass Spectrometry. J. Am. Chem. Soc. 1994, 116, 6985–6986. [Google Scholar] [CrossRef]
- Xue, L.; Lin, Z. Theoretical aspects of palladium-catalysed carbon–carbon cross-coupling reactions. Chem. Soc. Rev. 2010, 39, 1692–1705. [Google Scholar] [CrossRef]
- Morin, P.-O.; Bura, T.; Sun, B.; Gorelsky, S.I.; Li, Y.; Leclerc, M. Conjugated Polymers à la Carte from Time-Controlled Direct (Hetero)Arylation Polymerization. ACS Macro Lett. 2014, 4, 21–24. [Google Scholar] [CrossRef]
- Facchetti, A.; Vaccaro, L.; Marrocchi, A. Semiconducting Polymers Prepared by Direct Arylation Polycondensation. Angew. Chem. Int. Ed. 2012, 51, 3520–3523. [Google Scholar] [CrossRef]
- Liu, S.; Shi, M.; Huang, J.-C.; Jin, Z.-N.; Hu, X.-L.; Pan, J.-Y.; Li, H.; Jen, A.K.-Y.; Chen, H.-Z. C–H activation: Making diketopyrrolopyrrole derivatives easily accessible. J. Mater. Chem. A 2013, 1, 2795. [Google Scholar] [CrossRef]
- Se, M.; Papillon, J.; Schulz, E.; Lemaire, M. New synthetic method for the polymerization of alkylthiophenes. Tetrahedron Lett. 1999, 40, 5873–5876. [Google Scholar] [CrossRef]
- Wang, Q.; Takita, R.; Kikuzaki, Y.; Ozawa, F. Palladium-Catalyzed Dehydrohalogenative Polycondensation of 2-Bromo-3-hexylthiophene: An Efficient Approach to Head-to-Tail Poly(3-hexylthiophene). J. Am. Chem. Soc. 2010, 132, 11420–11421. [Google Scholar] [CrossRef]
- Gorelsky, S.I.; Lapointe, D.; Fagnou, K. Analysis of the Concerted Metalation-Deprotonation Mechanism in Palladium-Catalyzed Direct Arylation Across a Broad Range of Aromatic Substrates. J. Am. Chem. Soc. 2008, 130, 10848–10849. [Google Scholar] [CrossRef] [PubMed]
- Lapointe, D.; Fagnou, K. Overview of the Mechanistic Work on the Concerted Metallation–Deprotonation Pathway. Chem. Lett. 2010, 39, 1118–1126. [Google Scholar] [CrossRef]
- Ackermann, L. Carboxylate-Assisted Transition-Metal-Catalyzed C−H Bond Functionalizations: Mechanism and Scope. Chem. Rev. 2011, 111, 1315–1345. [Google Scholar] [CrossRef]
- Mercier, L.G.; Leclerc, M. Direct (Hetero)Arylation: A New Tool for Polymer Chemists. Acc. Chem. Res. 2013, 46, 1597–1605. [Google Scholar] [CrossRef]
- Lafrance, M.; Fagnou, K. Palladium-Catalyzed Benzene Arylation: Incorporation of Catalytic Pivalic Acid as a Proton Shuttle and a Key Element in Catalyst Design. J. Am. Chem. Soc. 2006, 128, 16496–16497. [Google Scholar] [CrossRef]
- Yamamoto, T.; Morita, A.; Maruyama, T.; Zhou, Z.-H.; Kanbara, T.; Sanechika, K. New Method for the Preparation of Poly(2,5-thienylene), Poly(p-phenylene), and Related Polymers. Polym. J. 1990, 22, 187–190. [Google Scholar] [CrossRef]
- Zhang, Z.-B.; Fujiki, M.; Tang, H.-Z.; Motonaga, M.; Torimitsu, K. The First High Molecular Weight Poly(N-alkyl-3,6-carbazole)s. Macromolecules 2002, 35, 1988–1990. [Google Scholar] [CrossRef]
- Akcelrud, L. Electroluminescent polymers. Prog. Polym. Sci. 2003, 28, 875–962. [Google Scholar] [CrossRef]
- Kraft, A.; Grimsdale, A.C.; Holmes, A.B. Electroluminescent Conjugated Polymers—Seeing Polymers in a New Light. Angew. Chem. Int. Ed. 1998, 37, 402–428. [Google Scholar] [CrossRef]
- Burroughes, J.H.; Bradley, D.D.C.; Brown, A.R.; Marks, R.N.; Mackay, K.D.; Friend, R.H.; Burns, P.L.; Holmes, A.B. Light-emitting diodes based on conjugated polymers. Nat. Cell Biol. 1990, 347, 539–541. [Google Scholar] [CrossRef]
- Allard, S.; Forster, M.; Souharce, B.; Thiem, H.; Scherf, U. Organic Semiconductors for Solution-Processable Field-Effect Transistors (OFETs). Angew. Chem. Int. Ed. 2008, 47, 4070–4098. [Google Scholar] [CrossRef]
- Marinov, O.; Deen, M.J.; Zschieschang, U.; Klauk, H. Organic Thin-Film Transistors: Part I—Compact DC Modeling. IEEE Trans. Electron Devices 2009, 56, 2952–2961. [Google Scholar] [CrossRef]
- Brabec, C.J.; Gowrisanker, S.; Halls, J.J.M.; Laird, D.; Jia, S.; Williams, S.P. Polymer-Fullerene Bulk-Heterojunction Solar Cells. Adv. Mater. 2010, 22, 3839–3856. [Google Scholar] [CrossRef]
- Lu, L.; Zheng, T.; Wu, Q.; Schneider, A.M.; Zhao, D.; Yu, L. Recent Advances in Bulk Heterojunction Polymer Solar Cells. Chem. Rev. 2015, 115, 12666–12731. [Google Scholar] [CrossRef]
- Pope, M.; Kallmann, H.P.; Magnante, P.C. Electroluminescence in Organic Crystals. J. Chem. Phys. 1963, 38, 2042–2043. [Google Scholar] [CrossRef]
- Tang, C.W.; VanSlyke, S.A. Organic electroluminescent diodes. Appl. Phys. Lett. 1987, 51, 913–915. [Google Scholar]
- Brown, A.R.; Bradley, D.D.C.; Burroughes, J.H.; Friend, R.H.; Greenham, N.C.; Burn, P.L.; Holmes, A.B.; Kraft, A. Poly(p-phenylenevinylene) light-emitting diodes: Enhanced electroluminescent efficiency through charge carrier confinement. Appl. Phys. Lett. 1992, 61, 2793–2795. [Google Scholar] [CrossRef]
- Friend, R.H.; Gymer, R.W.; Holmes, A.B.; Burroughes, J.H.; Marks, R.N.; Taliani, C.; Bradley, D.D.C.; Dos Santos, D.A.; Brédas, J.L.; Lögdlund, M.; et al. Electroluminescence in conjugated polymers. Nat. Cell Biol. 1999, 397, 121–128. [Google Scholar] [CrossRef]
- Yin, Z.; Wei, J.; Zheng, Q. Interfacial Materials for Organic Solar Cells: Recent Advances and Perspectives. Adv. Sci. 2016, 3, 1500362. [Google Scholar] [CrossRef]
- Grem, G.; Ullrich, B. Realization of a blue-light-emitting device using poly(p-phenylene). Adv. Mater. 1992, 4, 36–37. [Google Scholar] [CrossRef]
- Andersson, M.R.; Berggren, M.; Inganaes, O.; Gustafsson, G.; Gustafsson-Carlberg, J.C.; Selse, D.; Hjertberg, T.; Wennerstroem, O. Electroluminescence from Substituted Poly(thiophenes): From Blue to Near-Infrared. Macromolecules 1995, 28, 7525–7529. [Google Scholar] [CrossRef]
- Perepichka, I.F.; Meng, H.; Wudl, F. Light-Emitting Polythiophenes. Adv. Mater. 2005, 17, 2281–2305. [Google Scholar] [CrossRef]
- Morin, J.-F.; Leclerc, M. 2,7-Carbazole-Based Conjugated Polymers for Blue, Green, and Red Light Emission. Macromolecules 2002, 35, 8413–8417. [Google Scholar] [CrossRef]
- Grice, A.W.; Bradley, D.D.C.; Bernius, M.T.; Inbasekaran, M.; Wu, W.W.; Woo, E.P. High brightness and efficiency blue light-emitting polymer diodes. Appl. Phys. Lett. 1998, 73, 629–631. [Google Scholar] [CrossRef]
- Dimitrakopoulos, C.D.; Malenfant, P.R. Organic thin film transistors for large area electronics. Adv. Mater. 2002, 14, 99–117. [Google Scholar]
- Facchetti, A. Semiconductors for organic transistors. Mater. Today 2007, 10, 28–37. [Google Scholar] [CrossRef]
- Horowitz, G. Organic field-effect transistors. Adv. Mater. 1998, 10, 365–377. [Google Scholar]
- Tsumura, A.; Koezuka, H.; Ando, T. Macromolecular electronic device: Field-effect transistor with a polythiophene thin film. Appl. Phys. Lett. 1986, 49, 1210–1212. [Google Scholar] [CrossRef]
- Bao, Z.; Dodabalapur, A.; Lovinger, A.J. Soluble and processable regioregular poly(3-hexylthiophene) for thin film field-effect transistor applications with high mobility. Appl. Phys. Lett. 1996, 69, 4108–4110. [Google Scholar] [CrossRef]
- Assadi, A.H.; Svensson, C.; Willander, M.; Inganas, O. Field-effect mobility of poly(3-hexylthiophene). Appl. Phys. Lett. 1988, 53, 195–197. [Google Scholar] [CrossRef]
- Sirringhaus, H.; Brown, P.J.; Friend, R.H.; Nielsen, M.M.; Bechgaard, K.; Langeveld-Voss, B.M.W.; Spiering, A.J.H.; Janssen, R.A.J.; Meijer, E.W.; Herwig, P.T.; et al. Two-dimensional charge transport in self-organized, high-mobility conjugated polymers. Nat. Cell Biol. 1999, 401, 685–688. [Google Scholar] [CrossRef]
- Lewis, N.S.; Nocera, D.G. Powering the planet: Chemical challenges in solar energy utilization. Proc. Natl. Acad. Sci. USA 2006, 103, 15729–15735. [Google Scholar] [CrossRef]
- Solomon, S.; Plattner, G.-K.; Knutti, R.; Friedlingstein, P. Irreversible climate change due to carbon dioxide emissions. Proc. Natl. Acad. Sci. USA 2009, 106, 1704–1709. [Google Scholar] [CrossRef]
- Lewis, N.S.; Crabtree, G.; Nozik, A.J.; Wasielewski, M.R.; Alivisatos, P.; Kung, H.; Tsao, J.; Chandler, E.; Walukiewicz, W.; Spitler, M.; et al. Basic Research Needs for Solar Energy Utilization. Report of the Basic Energy Sciences Workshop on Solar Energy Utilization, April 18–21, 2005; DOESC (USDOE Office of Science (SC)): Washington, DC, USA, 2005; pp. 1–276.
- Smalley, R.E. Future Global Energy Prosperity: The Terawatt Challenge. MRS Bull. 2005, 30, 412–417. [Google Scholar] [CrossRef]
- Lewis, N.S. Basic research needs for solar energy utilization: Report of the basic energy sciences workshop on solar energy utilization. Science 2007, 315, 798–801. [Google Scholar]
- Morton, O. Solar energy: A new day dawning? Silicon Valley sunrise. Nature 2006, 443, 19–22. [Google Scholar] [PubMed]
- Chapin, D.M.; Fuller, C.S.; Pearson, G.L. A New Silicon p-n Junction Photocell for Converting Solar Radiation into Electrical Power. J. Appl. Phys. 1954, 25, 676–677. [Google Scholar] [CrossRef]
- Ameri, T.; Dennler, G.; Lungenschmied, C.; Brabec, C.J. Organic tandem solar cells: A review. Energy Environ. Sci. 2009, 2, 347–363. [Google Scholar] [CrossRef]
- Green, M.A.; Emery, K.; Hishikawa, Y.; Warta, W.; Dunlop, E.D. Characterizing electrical output of bifacial photovoltaic modules by altering reflective materials. Prog. Photovolt. Res. Appl. 2015, 23, 1–9. [Google Scholar] [CrossRef]
- Shah, A.; Meier, J.; Vallat-Sauvain, E.; Droz, C.; Kroll, U.; Wyrsch, N.; Guillet, J.; Graf, U. Microcrystalline silicon and ‘micromorph’tandem solar cells. Thin Solid Film. 2002, 403, 179–187. [Google Scholar] [CrossRef]
- Kippelen, B.; Brédas, J.-L. Organic photovoltaics. Energy Environ. Sci. 2009, 2, 251–261. [Google Scholar] [CrossRef]
- Green, M.A.; Emery, K.; Hishikawa, Y.; Warta, W. Solar cell efficiency tables (Version 36). Prog. Photovolt. Res. Appl. 2010, 18, 346–352. [Google Scholar] [CrossRef]
- Wu, X. High-efficiency polycrystalline CdTe thin-film solar cells. Sol. Energy 2004, 77, 803–814. [Google Scholar] [CrossRef]
- Ward, J.S.; Ramanathan, K.; Hasoon, F.S.; Coutts, T.J.; Keane, J.; Contreras, M.A.; Moriarty, T.; Noufi, R. A 21.5% efficient Cu (In,Ga)Se2 thin-film concentrator solar cell. Prog. Photovolt. Res. Appl. 2002, 10, 41–46. [Google Scholar] [CrossRef]
- Chen, Y.; Wan, X.; Long, G. High Performance Photovoltaic Applications Using Solution-Processed Small Molecules. Acc. Chem. Res. 2013, 46, 2645–2655. [Google Scholar] [CrossRef]
- Sun, Y.; Welch, G.C.; Leong, W.L.; Takacs, C.J.; Bazan, G.C.; Heeger, A.J. Solution-processed small-molecule solar cells with 6.7% efficiency. Nat. Mater. 2012, 11, 44–48. [Google Scholar] [CrossRef]
- Bijleveld, J.C.; Zoombelt, A.P.; Mathijssen, S.G.J.; Wienk, M.M.; Turbiez, M.; De Leeuw, D.M.; Janssen, R.A.J. Poly(diketopyrrolopyrrole−terthiophene) for Ambipolar Logic and Photovoltaics. J. Am. Chem. Soc. 2009, 131, 16616–16617. [Google Scholar] [CrossRef] [PubMed]
- Cartwright, L.; Iraqi, A.; Zhang, Y.; Wang, T.; Lidzey, D.G. Impact of fluorine substitution upon the photovoltaic properties of benzothiadiazole-fluorene alternate copolymers. RSC Adv. 2015, 5, 46386–46394. [Google Scholar] [CrossRef]
- Grätzel, M. Photoelectrochemical cells. Nature 2001, 414, 338–344. [Google Scholar] [CrossRef]
- Bai, Y.; Cao, Y.; Zhang, J.; Wang, M.; Li, R.; Wang, P.; Zakeeruddin, S.M.; Grätzel, M. High-performance dye-sensitized solar cells based on solvent-free electrolytes produced from eutectic melts. Nat. Mater. 2008, 7, 626–630. [Google Scholar] [CrossRef]
- Krebs, F.C. Fabrication and processing of polymer solar cells: A review of printing and coating techniques. Sol. Energy Mater. Sol. Cells 2009, 93, 394–412. [Google Scholar] [CrossRef]
- Krebs, F.C. Polymer solar cell modules prepared using roll-to-roll methods: Knife-over-edge coating, slot-die coating and screen printing. Sol. Energy Mater. Sol. Cells 2009, 93, 465–475. [Google Scholar] [CrossRef]
- Krebs, F.C. Roll-to-roll fabrication of monolithic large-area polymer solar cells free from indium-tin-oxide. Sol. Energy Mater. Sol. Cells 2009, 93, 1636–1641. [Google Scholar] [CrossRef]
- Scharber, M.C.; Mühlbacher, D.; Koppe, M.; Denk, P.; Waldauf, C.; Heeger, A.J.; Brabec, C.J. Design Rules for Donors in Bulk-Heterojunction Solar Cells—Towards 10 % Energy-Conversion Efficiency. Adv. Mater. 2006, 18, 789–794. [Google Scholar] [CrossRef]
- Brédas, J.-L.; Norton, J.E.; Cornil, J.; Coropceanu, V. Molecular Understanding of Organic Solar Cells: The Challenges. Acc. Chem. Res. 2009, 42, 1691–1699. [Google Scholar] [CrossRef]
- Jørgensen, M.; Norrman, K.; Krebs, F.C. Stability/degradation of polymer solar cells. Sol. Energy Mater. Sol. Cells 2008, 92, 686–714. [Google Scholar] [CrossRef]
- Jørgensen, M.; Norrman, K.; Gevorgyan, S.A.; Tromholt, T.; Andreasen, B.; Krebs, F.C. Stability of Polymer Solar Cells. Adv. Mater. 2012, 24, 580–612. [Google Scholar] [CrossRef]
- Nelson, J. Organic photovoltaic films. Mater. Today 2002, 5, 20–27. [Google Scholar] [CrossRef]
- Brabec, C.J.; Sariciftci, N.S.; Hummelen, J.C. Plastic solar cells. Adv. Funct. Mater. 2001, 11, 15–26. [Google Scholar] [CrossRef]
- Marks, R.N.; Halls, J.J.M.; Bradley, D.D.C.; Friend, R.H.; Holmes, A.B. The photovoltaic response in poly(p-phenylene vinylene) thin-film devices. J. Phys. Condens. Matter 1994, 6, 1379–1394. [Google Scholar] [CrossRef]
- Blom, P.W.M.; Mihailetchi, V.D.; Koster, L.J.A.; Markov, D.E. Device Physics of Polymer: Fullerene Bulk Heterojunction Solar Cells. Adv. Mater. 2007, 19, 1551–1566. [Google Scholar] [CrossRef]
- Spanggaard, H.; Krebs, F.C. A brief history of the development of organic and polymeric photovoltaics. Sol. Energy Mater. Sol. Cells 2004, 83, 125–146. [Google Scholar] [CrossRef]
- Coakley, K.M.; McGehee, M.D. Conjugated polymer photovoltaic cells. Chem. Mater. 2004, 16, 4533–4542. [Google Scholar] [CrossRef]
- Halls, J.; Pichler, K.; Friend, R.; Moratti, S.; Holmes, A. Exciton diffusion and dissociation in a poly(p-phenylenevinylene)/C60 heterojunction photovoltaic cell. Appl. Phys. Lett. 1996, 68, 3120–3122. [Google Scholar] [CrossRef]
- Savenije, T.J.; Warman, J.M.; Goossens, A. Visible light sensitisation of titanium dioxide using a phenylene vinylene polymer. Chem. Phys. Lett. 1998, 287, 148–153. [Google Scholar] [CrossRef]
- Markov, D.E.; Amsterdam, E.; Blom, P.W.M.; Sieval, A.B.; Hummelen, J.C. Accurate Measurement of the Exciton Diffusion Length in a Conjugated Polymer Using a Heterostructure with a Side-Chain Cross-Linked Fullerene Layer. J. Phys. Chem. A 2005, 109, 5266–5274. [Google Scholar] [CrossRef]
- Allemand, P.M.; Koch, A.; Wudl, F.; Rubin, Y.; Diederich, F.; Alvarez, M.M.; Anz, S.J.; Whetten, R.L. Two different fullerenes have the same cyclic voltammetry. J. Am. Chem. Soc. 1991, 113, 1050–1051. [Google Scholar] [CrossRef]
- Martín, N.; Sánchez, L.; Illescas, B.; Pérez, I. C60-Based Electroactive Organofullerenes. Chem. Rev. 1998, 98, 2527–2548. [Google Scholar] [CrossRef]
- Sariciftci, N.S.; Smilowitz, L.; Heeger, A.J.; Wudl, F. Photoinduced Electron Transfer from a Conducting Polymer to Buckminsterfullerene. Science 1992, 258, 1474–1476. [Google Scholar] [CrossRef]
- Morita, S.; Zakhidov, A.; Yoshino, K. Doping effect of buckminsterfullerene in conducting polymer: Change of absorption spectrum and quenching of luminescene. Solid State Commun. 1992, 82, 249–252. [Google Scholar] [CrossRef]
- Singh, T.; Marjanovic, N.; Matt, G.; Güneş, S.; Sariciftci, N.; Ramil, A.M.; Andreev, A.; Sitter, H.; Schwödiauer, R.; Bauer, S. High-mobility n-channel organic field-effect transistors based on epitaxially grown C60 films. Org. Electron. 2005, 6, 105–110. [Google Scholar] [CrossRef]
- Sariciftci, N.; Smilowitz, L.; Heeger, A.; Wudl, F. Semiconducting polymers (as donors) and buckminsterfullerene (as acceptor): Photoinduced electron transfer and heterojunction devices. Synth. Met. 1993, 59, 333–352. [Google Scholar] [CrossRef]
- Sariciftci, N.S.; Braun, D.; Zhang, C.; Srdanov, V.I.; Heeger, A.J.; Stucky, G.; Wudl, F. Semiconducting polymer-buckminsterfullerene heterojunctions: Diodes, photodiodes, and photovoltaic cells. Appl. Phys. Lett. 1993, 62, 585–587. [Google Scholar] [CrossRef]
- Hoppe, H.; Sariciftci, N.S. Organic solar cells: An overview. J. Mater. Res. 2004, 19, 1924–1945. [Google Scholar] [CrossRef]
- Dennler, G.; Scharber, M.C.; Brabec, C.J. Polymer-Fullerene Bulk-Heterojunction Solar Cells. Adv. Mater. 2009, 21, 1323–1338. [Google Scholar] [CrossRef]
- Po, R.; Maggini, M.; Camaioni, N. Polymer Solar Cells: Recent Approaches and Achievements. J. Phys. Chem. C 2009, 114, 695–706. [Google Scholar] [CrossRef]
- Son, H.J.; Carsten, B.; Jung, I.H.; Yu, L. Overcoming efficiency challenges in organic solar cells: Rational development of conjugated polymers. Energy Environ. Sci. 2012, 5, 8158–8170. [Google Scholar] [CrossRef]
- Groenendaal, L.; Jonas, F.; Freitag, D.; Pielartzik, H.; Reynolds, J.R. Poly(3,4-ethylenedioxythiophene) and its derivatives: Past, present, and future. Adv. Mater. 2000, 12, 481–494. [Google Scholar] [CrossRef]
- Kirchmeyer, S.; Reuter, K. Scientific importance, properties and growing applications of poly(3,4-ethylenedioxythiophene). J. Mater. Chem. 2005, 15, 2077–2088. [Google Scholar] [CrossRef]
- Benanti, T.L.; Venkataraman, D. Organic Solar Cells: An Overview Focusing on Active Layer Morphology. Photosynth. Res. 2006, 87, 73–81. [Google Scholar] [CrossRef]
- Cai, W.; Gong, X.; Cao, Y. Polymer solar cells: Recent development and possible routes for improvement in the performance. Sol. Energy Mater. Sol. Cells 2010, 94, 114–127. [Google Scholar] [CrossRef]
- Son, H.J.; He, F.; Carsten, B.; Yu, L. Are we there yet? Design of better conjugated polymers for polymer solar cells. J. Mater. Chem. 2011, 21, 18934–18945. [Google Scholar] [CrossRef]
- Thompson, B.C.; Fréchet, J.M.J. Polymer–Fullerene Composite Solar Cells. Angew. Chem. Int. Ed. 2008, 47, 58–77. [Google Scholar] [CrossRef]
- Yeh, N.; Yeh, P. Organic solar cells: Their developments and potentials. Renew. Sustain. Energy Rev. 2013, 21, 421–431. [Google Scholar] [CrossRef]
- Wudl, F.; Srdanov, G. US Patent 5189136 (1990). Chem. Abstr. 1993, 118, 255575. [Google Scholar]
- Shrotriya, V.; Wu, E.H.-E.; Li, G.; Yao, Y.; Yang, Y. Efficient light harvesting in multiple-device stacked structure for polymer solar cells. Appl. Phys. Lett. 2006, 88, 64104. [Google Scholar] [CrossRef]
- Shaheen, S.E.; Brabec, C.J.; Sariciftci, N.S.; Padinger, F.; Fromherz, T.; Hummelen, J.C. 2.5% efficient organic plastic solar cells. Appl. Phys. Lett. 2001, 78, 841–843. [Google Scholar] [CrossRef]
- Wienk, M.M.; Kroon, J.M.; Verhees, W.J.H.; Knol, J.; Hummelen, J.C.; Van Hal, P.A.; Janssen, R.A.J. Efficient Methano[70]fullerene/MDMO-PPV Bulk Heterojunction Photovoltaic Cells. Angew. Chem. 2003, 115, 3493–3497. [Google Scholar] [CrossRef]
- Melzer, C.; Koop, E.J.; Mihailetchi, V.D.; Blom, P.W. Hole transport in poly(phenylene vinylene)/methanofullerene bulk-heterojunction solar cells. Adv. Funct. Mater. 2004, 14, 865–870. [Google Scholar] [CrossRef]
- Brabec, C.J.; Shaheen, S.E.; Winder, C.; Sariciftci, N.S.; Denk, P. Effect of LiF/metal electrodes on the performance of plastic solar cells. Appl. Phys. Lett. 2002, 80, 1288–1290. [Google Scholar] [CrossRef]
- Sirringhaus, H.; Tessler, N.; Friend, R.H. Integrated Optoelectronic Devices Based on Conjugated Polymers. Science 1998, 280, 1741–1744. [Google Scholar] [CrossRef]
- Schilinsky, P.; Waldauf, C.; Brabec, C.J. Recombination and loss analysis in polythiophene based bulk heterojunction photodetectors. Appl. Phys. Lett. 2002, 81, 3885–3887. [Google Scholar] [CrossRef]
- Li, G.; Shrotriya, V.; Huang, J.; Yao, Y.; Moriarty, T.; Emery, K.; Yang, Y. High-efficiency solution processable polymer photovoltaic cells by self-organization of polymer blends. Nat. Mater. 2005, 4, 864–868. [Google Scholar] [CrossRef]
- Reyes-Reyes, M.; Kim, K.; Carroll, D.L. High-efficiency photovoltaic devices based on annealed poly(3-hexylthiophene) and 1-(3-methoxycarbonyl)-propyl-1- phenyl-(6,6)C61 blends. Appl. Phys. Lett. 2005, 87, 083506. [Google Scholar] [CrossRef]
- Irwin, M.D.; Buchholz, D.B.; Hains, A.W.; Chang, R.P.H.; Marks, T.J. p-Type semiconducting nickel oxide as an efficiency-enhancing anode interfacial layer in polymer bulk-heterojunction solar cells. Proc. Natl. Acad. Sci. USA 2008, 105, 2783–2787. [Google Scholar] [CrossRef]
- Ma, W.; Yang, C.; Gong, X.; Lee, K.; Heeger, A.J. Thermally Stable, Efficient Polymer Solar Cells with Nanoscale Control of the Interpenetrating Network Morphology. Adv. Funct. Mater. 2005, 15, 1617–1622. [Google Scholar] [CrossRef]
- Dou, L.; You, J.; Hong, Z.; Xu, Z.; Li, G.; Street, R.A.; Yang, Y. 25th Anniversary Article: A Decade of Organic/Polymeric Photovoltaic Research. Adv. Mater. 2013, 25, 6642–6671. [Google Scholar] [CrossRef]
- Thompson, B.C.; Kim, Y.-G.; McCarley, T.D.; Reynolds, J.R. Soluble Narrow Band Gap and Blue Propylenedioxythiophene-Cyanovinylene Polymers as Multifunctional Materials for Photovoltaic and Electrochromic Applications. J. Am. Chem. Soc. 2006, 128, 12714–12725. [Google Scholar] [CrossRef]
- Hou, J.; Tan, Z.; Yan, Y.; He, Y.; Yang, C.; Li, Y. Synthesis and Photovoltaic Properties of Two-Dimensional Conjugated Polythiophenes with Bi(thienylenevinylene) Side Chains. J. Am. Chem. Soc. 2006, 128, 4911–4916. [Google Scholar] [CrossRef]
- Li, Y. Molecular Design of Photovoltaic Materials for Polymer Solar Cells: Toward Suitable Electronic Energy Levels and Broad Absorption. Acc. Chem. Res. 2012, 45, 723–733. [Google Scholar] [CrossRef]
- Zhuravleva, T.; Vannikov, A. Polymer Solar Cells. Mater. Sci. Forum 1991, 21, 203. [Google Scholar] [CrossRef]
- Brabec, C.J.; Cravino, A.; Meissner, D.; Sariciftci, N.S.; Fromherz, T.; Rispens, M.T.; Sanchez, L.; Hummelen, J.C. Origin of the open circuit voltage of plastic solar cells. Adv. Funct. Mater. 2001, 11, 374–380. [Google Scholar] [CrossRef]
- Kooistra, F.B.; Knol, J.; Kastenberg, F.; Popescu, L.M.; Verhees, W.J.H.; Kroon, A.J.M.; Hummelen, J.C. Increasing the Open Circuit Voltage of Bulk-Heterojunction Solar Cells by Raising the LUMO Level of the Acceptor. Org. Lett. 2007, 9, 551–554. [Google Scholar] [CrossRef]
- Gadisa, A.; Svensson, M.; Andersson, M.R.; Inganäs, O. Correlation between oxidation potential and open-circuit voltage of composite solar cells based on blends of polythiophenes/ fullerene derivative. Appl. Phys. Lett. 2004, 84, 1609–1611. [Google Scholar] [CrossRef]
- Kroon, R.; Lenes, M.; Hummelen, J.C.; Blom, P.W.M.; De Boer, B. Small Bandgap Polymers for Organic Solar Cells (Polymer Material Development in the Last 5 Years). Polym. Rev. 2008, 48, 531–582. [Google Scholar] [CrossRef]
- Koster, L.J.A.; Mihailetchi, V.D.; Blom, P.W.M. Ultimate efficiency of polymer/fullerene bulk heterojunction solar cells. Appl. Phys. Lett. 2006, 88, 093511. [Google Scholar] [CrossRef]
- Blouin, N.; Michaud, A.; Gendron, D.; Wakim, S.; Blair, E.; Neagu-Plesu, R.; Belletete, M.; Durocher, G.; Tao, Y.; Leclerc, M. Toward a Rational Design of Poly(2,7-Carbazole) Derivatives for Solar Cells. J. Am. Chem. Soc. 2008, 130, 732–742. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.; Zhu, D. Conjugated polymers for high-efficiency organic photovoltaics. Polym. Chem. 2010, 1, 409. [Google Scholar] [CrossRef]
- Dang, M.T.; Hirsch, L.; Wantz, G.; Wuest, J.D. Controlling the Morphology and Performance of Bulk Heterojunctions in Solar Cells. Lessons Learned from the Benchmark Poly(3-hexylthiophene):[6,6]-Phenyl-C61-butyric Acid Methyl Ester System. Chem. Rev. 2013, 113, 3734–3765. [Google Scholar] [CrossRef]
- Roncali, J. Molecular Engineering of the Band Gap of π-Conjugated Systems: Facing Technological Applications. Macromol. Rapid Commun. 2007, 28, 1761–1775. [Google Scholar] [CrossRef]
- Roncali, J. Synthetic Principles for Bandgap Control in Linear π-Conjugated Systems. Chem. Rev. 1997, 97, 173–206. [Google Scholar] [CrossRef] [PubMed]
- Osaka, I.; McCullough, R.D. Advances in Molecular Design and Synthesis of Regioregular Polythiophenes. Acc. Chem. Res. 2008, 41, 1202–1214. [Google Scholar] [CrossRef]
- Zhang, Q.; Kelly, M.A.; Bauer, N.; You, W. The Curious Case of Fluorination of Conjugated Polymers for Solar Cells. Acc. Chem. Res. 2017, 50, 2401–2409. [Google Scholar] [CrossRef] [PubMed]
- Ko, S.; Hoke, E.T.; Pandey, L.; Hong, S.; Mondal, R.; Risko, C.; Yi, Y.; Noriega, R.; McGehee, M.D.; Brédas, J.-L.; et al. Controlled Conjugated Backbone Twisting for an Increased Open-Circuit Voltage while Having a High Short-Circuit Current in Poly(hexylthiophene) Derivatives. J. Am. Chem. Soc. 2012, 134, 5222–5232. [Google Scholar] [CrossRef]
- Van Mullekom, H.R.; Vekemans, J.; Havinga, E.E.; Meijer, E.B. Developments in the chemistry and band gap engineering of donor–acceptor substituted conjugated polymers. Mater. Sci. Eng. R Rep. 2001, 32, 1–40. [Google Scholar] [CrossRef]
- Wudl, F.; Kobayashi, M.; Heeger, A.J. Poly(isothianaphthene). J. Org. Chem. 1984, 49, 3382–3384. [Google Scholar] [CrossRef]
- Colaneri, N.; Kobayashi, M.; Heeger, A.; Wudl, F. Electrochemical and opto-electrochemical properties of poly(isothianaphthene). Synth. Met. 1986, 14, 45–52. [Google Scholar] [CrossRef]
- Pomerantz, M.; Chaloner-Gill, B.; Harding, L.O.; Tseng, J.J.; Pomerantz, W.J. Poly(2,3-dihexylthieno[3,4-b]pyrazine). A new processable low band-gap polyheterocycle. J. Chem. Soc. Chem. Commun. 1992, 1672. [Google Scholar] [CrossRef]
- Hong, S.Y.; Marynick, D.S. Understanding the conformational stability and electronic structures of modified polymers based on polythiophene. Macromolecules 1992, 25, 4652–4657. [Google Scholar] [CrossRef]
- Sotzing, G.A.; Lee, K. Poly(thieno[3,4-b]thiophene): A p and n-Dopable Polythiophene Exhibiting High Optical Transparency in the Semiconducting State. Macromolecules 2002, 35, 7281–7286. [Google Scholar] [CrossRef]
- Zhou, H.; Yang, L.; You, W. Rational Design of High Performance Conjugated Polymers for Organic Solar Cells. Macromolecules 2012, 45, 607–632. [Google Scholar] [CrossRef]
- Ajayaghosh, A. Donor-acceptor type low band gap polymers: Polysquaraines and related systems. Chem. Soc. Rev. 2003, 32, 181–191. [Google Scholar]
- Havinga, E.E.; Hoeve, W.T.; Wynberg, H. A new class of small band gap organic polymer conductors. Polym. Bull. 1992, 29, 119–126. [Google Scholar] [CrossRef]
- Brabec, C.J.; Lane, P.A.; Kafafi, Z.H. Special Section Guest Editorial: Guest Editorial: Special Section on Organic Photovoltaics. Mater. Today 2012, 2, 021099. [Google Scholar] [CrossRef]
- Zhang, Q.T.; Tour, J.M. Low Optical Bandgap Polythiophenes by an Alternating Donor/Acceptor Repeat Unit Strategy. J. Am. Chem. Soc. 1997, 119, 5065–5066. [Google Scholar] [CrossRef]
- Chochos, C.L.; Choulis, S.A. How the structural deviations on the backbone of conjugated polymers influence their optoelectronic properties and photovoltaic performance. Prog. Polym. Sci. 2011, 36, 1326–1414. [Google Scholar] [CrossRef]
- Duan, C.; Huang, F.; Cao, Y. Recent development of push–pull conjugated polymers for bulk-heterojunction photovoltaics: Rational design and fine tailoring of molecular structures. J. Mater. Chem. 2012, 22, 10416–10434. [Google Scholar] [CrossRef]
- Huo, L.; Hou, J.; Zhang, S.; Chen, H.-Y.; Yang, Y. A Polybenzo[1,2-b:4,5-b′]dithiophene Derivative with Deep HOMO Level and Its Application in High-Performance Polymer Solar Cells. Angew. Chem. Int. Ed. 2010, 49, 1500–1503. [Google Scholar] [CrossRef]
- Liang, Y.; Wu, Y.; Feng, D.; Tsai, S.-T.; Son, H.-J.; Li, G.; Yu, L. Development of New Semiconducting Polymers for High Performance Solar Cells. J. Am. Chem. Soc. 2009, 131, 56–57. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Feng, D.; Wu, Y.; Tsai, S.-T.; Li, G.; Ray, C.; Yu, L. Highly Efficient Solar Cell Polymers Developed via Fine-Tuning of Structural and Electronic Properties. J. Am. Chem. Soc. 2009, 131, 7792–7799. [Google Scholar] [CrossRef]
- Liang, Y.; Xu, Z.; Xia, J.; Tsai, S.T.; Wu, Y.; Li, G.; Ray, C.; Yu, L. For the bright future-bulk heterojunction polymer solar cells with power conversion efficiency of 7.4%. Adv. Mater. 2010, 22, E135–E138. [Google Scholar] [CrossRef]
- Liang, Y.; Yu, L. A New Class of Semiconducting Polymers for Bulk Heterojunction Solar Cells with Exceptionally High Performance. Acc. Chem. Res. 2010, 43, 1227–1236. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-Y.; Hou, J.; Zhang, S.; Liang, Y.; Yang, G.; Yang, Y.; Yu, L.; Wu, Y.; Li, G. Polymer solar cells with enhanced open-circuit voltage and efficiency. Nat. Photonics 2009, 3, 649–653. [Google Scholar] [CrossRef]
- Hou, J.; Chen, H.-Y.; Zhang, S.; Chen, R.I.; Yang, Y.; Wu, Y.; Li, G. Synthesis of a Low Band Gap Polymer and Its Application in Highly Efficient Polymer Solar Cells. J. Am. Chem. Soc. 2009, 131, 15586–15587. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Najari, A.; Berrouard, P.; Beaupré, B.; Réda, A.; Tao, Y.; Leclerc, M. A Thieno[3,4-c]pyrrole-4,6-dione-Based Copolymer for Efficient Solar Cells. J. Am. Chem. Soc. 2010, 132, 5330–5331. [Google Scholar] [CrossRef]
- Zhang, Y.; Hau, S.K.; Yip, H.-L.; Sun, Y.; Acton, O.; Jen, A.K.-Y. Efficient Polymer Solar Cells Based on the Copolymers of Benzodithiophene and Thienopyrroledione. Chem. Mater. 2010, 22, 2696–2698. [Google Scholar] [CrossRef]
- Piliego, C.; Holcombe, T.W.; Douglas, J.D.; Woo, C.H.; Beaujuge, P.M.; Fréchet, J.M.J. Synthetic Control of Structural Order in N-Alkylthieno[3,4-c]pyrrole-4,6-dione-Based Polymers for Efficient Solar Cells. J. Am. Chem. Soc. 2010, 132, 7595–7597. [Google Scholar] [CrossRef]
- Zhang, G.; Fu, Y.; Zhang, Q.; Xie, Z. Benzo[1,2-b:4,5-b′]dithiophene-dioxopyrrolothiophen copolymers for high performance solar cells. Chem. Commun. 2010, 46, 4997. [Google Scholar] [CrossRef]
- Cabanetos, C.; El Labban, A.; Bartelt, J.A.; Douglas, J.D.; Mateker, W.R.; Fréchet, J.M.J.; McGehee, M.D.; Beaujuge, P.M. Linear Side Chains in Benzo[1,2-b:4,5-b′]dithiophene–Thieno[3,4-c]pyrrole-4,6-dione Polymers Direct Self-Assembly and Solar Cell Performance. J. Am. Chem. Soc. 2013, 135, 4656–4659. [Google Scholar] [CrossRef]
- Peet, J.; Heeger, A.J.; Bazan, G.C. “Plastic” Solar Cells: Self-Assembly of Bulk Heterojunction Nanomaterials by Spontaneous Phase Separation. Acc. Chem. Res. 2009, 42, 1700–1708. [Google Scholar] [CrossRef]
- Brabec, C.J.; Heeney, M.; McCulloch, I.; Nelson, J. Influence of blend microstructure on bulk heterojunction organic photovoltaic performance. Chem. Soc. Rev. 2011, 40, 1185–1199. [Google Scholar] [CrossRef]
- Hoppe, H.; Niggemann, M.; Winder, C.; Kraut, J.; Hiesgen, R.; Hinsch, A.; Meissner, D.; Sariciftci, N. Nanoscale Morphology of Conjugated Polymer/Fullerene-Based Bulk- Heterojunction Solar Cells. Adv. Funct. Mater. 2004, 14, 1005–1011. [Google Scholar] [CrossRef]
- Yang, X.; van Duren, J.K.; Janssen, R.A.; Michels, M.A.; Loos, J. Morphology and thermal stability of the active layer in poly (p-phenylenevinylene)/methanofullerene plastic photovoltaic devices. Macromolecules 2004, 37, 2151–2158. [Google Scholar] [CrossRef]
- Liu, J.; Shi, Y.; Yang, Y. Solvation-Induced Morphology Effects on the Performance of Polymer-Based Photovoltaic Devices. Adv. Funct. Mater. 2001, 11, 420–424. [Google Scholar] [CrossRef]
- Rispens, M.T.; Meetsma, A.; Rittberger, R.; Brabec, C.J.; Sariciftci, N.S.; Hummelen, J.C. Influence of the solvent on the crystal structure of PCBM and the efficiency of MDMO-PPV: PCBM ‘plastic’ solar cells. Chem. Commun. 2003, 2116–2118. [Google Scholar] [CrossRef]
- Zhang, F.; Jespersen, K.G.; Björström, C.; Svensson, M.; Andersson, M.R.; Sundström, V.; Magnusson, K.; Moons, E.; Yartsev, A.; Inganäs, O. Influence of Solvent Mixing on the Morphology and Performance of Solar Cells Based on Polyfluorene Copolymer/Fullerene Blends. Adv. Funct. Mater. 2006, 16, 667–674. [Google Scholar] [CrossRef]
- Wienk, M.M.; Turbiez, M.M.; Gilot, J.J.; Janssen, R.A.J. Narrow-Bandgap Diketo-Pyrrolo-Pyrrole Polymer Solar Cells: The Effect of Processing on the Performance. Adv. Mater. 2008, 20, 2556–2560. [Google Scholar] [CrossRef]
- Liu, F.; Gu, Y.; Wang, C.; Zhao, W.; Chen, D.; Briseno, A.L.; Russell, T.P. Efficient Polymer Solar Cells Based on a Low Bandgap Semi-crystalline DPP Polymer-PCBM Blends. Adv. Mater. 2012, 24, 3947–3951. [Google Scholar] [CrossRef]
- Dittmer, J.J.; Marseglia, E.A.; Friend, R.H. Electron Trapping in Dye/Polymer Blend Photovoltaic Cells. Adv. Mater. 2000, 12, 1270–1274. [Google Scholar] [CrossRef]
- Camaioni, N.; Ridolfi, G.; Casalbore-Miceli, G.; Possamai, G.; Maggini, M. The Effect of a Mild Thermal Treatment on the Performance of Poly(3-alkylthiophene)/Fullerene Solar Cells. Adv. Mater. 2002, 14, 1735–1738. [Google Scholar] [CrossRef]
- Padinger, F.; Rittberger, R.; Sariciftci, N. Effects of Postproduction Treatment on Plastic Solar Cells. Adv. Funct. Mater. 2003, 13, 85–88. [Google Scholar] [CrossRef]
- Chirvase, D.; Parisi, J.; Hummelen, J.C.; Dyakonov, V. Influence of nanomorphology on the photovoltaic action of polymer–fullerene composites. Nanotechnology 2004, 15, 1317–1323. [Google Scholar] [CrossRef]
- Mihailetchi, V.D.; Xie, H.; De Boer, B.; Koster, L.J.A.; Blom, P.W.M. Charge Transport and Photocurrent Generation in Poly(3-hexylthiophene): Methanofullerene Bulk-Heterojunction Solar Cells. Adv. Funct. Mater. 2006, 16, 699–708. [Google Scholar] [CrossRef]
- Erb, T.; Zhokhavets, U.; Gobsch, G.; Raleva, S.; Stühn, B.; Schilinsky, P.; Waldauf, C.; Brabec, C.J. Correlation Between Structural and Optical Properties of Composite Polymer/Fullerene Films for Organic Solar Cells. Adv. Funct. Mater. 2005, 15, 1193–1196. [Google Scholar] [CrossRef]
- Yang, X.; Loos, J.; Veenstra, S.C.; Verhees, W.J.; Wienk, M.M.; Kroon, J.M.; Michels, M.A.; Janssen, R.A. Nanoscale Morphology of High-Performance Polymer Solar Cells. Nano Lett. 2005, 5, 579–583. [Google Scholar] [CrossRef] [PubMed]
- Hwang, I.-W.; Soci, C.; Moses, D.; Zhu, Z.; Waller, D.; Gaudiana, R.; Brabec, C.J.; Heeger, A.J. Ultrafast Electron Transfer and Decay Dynamics in a Small Band Gap Bulk Heterojunction Material. Adv. Mater. 2007, 19, 2307–2312. [Google Scholar] [CrossRef]
- Park, J.K.; Jo, J.; Seo, J.H.; Moon, J.S.; Park, Y.D.; Lee, K.; Heeger, A.J.; Bazan, G.C. End-Capping Effect of a Narrow Bandgap Conjugated Polymer on Bulk Heterojunction Solar Cells. Adv. Mater. 2011, 23, 2430–2435. [Google Scholar] [CrossRef]
- Zhao, Y.; Xie, Z.; Qu, Y.; Geng, Y.; Wang, L. Solvent-vapor treatment induced performance enhancement of poly(3-hexylthiophene): Methanofullerene bulk-heterojunction photovoltaic cells. Appl. Phys. Lett. 2007, 90, 43504. [Google Scholar] [CrossRef]
- Miller, S.; Fanchini, G.; Lin, Y.-Y.; Li, C.; Chen, C.-W.; Su, W.-F.; Chhowalla, M. Investigation of nanoscale morphological changes in organic photovoltaics during solvent vapor annealing. J. Mater. Chem. 2008, 18, 306–312. [Google Scholar] [CrossRef]
- Li, G.; Yao, Y.; Yang, H.; Shrotriya, V.; Yang, G.; Yang, Y. “Solvent Annealing” Effect in Polymer Solar Cells Based on Poly(3-hexylthiophene) and Methanofullerenes. Adv. Funct. Mater. 2007, 17, 1636–1644. [Google Scholar] [CrossRef]
- Mihailetchi, V.D.; Xie, H.; De Boer, B.; Popescu, L.M.; Hummelen, J.C.; Blom, P.W.M.; Koster, L.J.A. Origin of the enhanced performance in poly(3-hexylthiophene): [6,6]-phenyl C61-butyric acid methyl ester solar cells upon slow drying of the active layer. Appl. Phys. Lett. 2006, 89, 012107. [Google Scholar] [CrossRef]
- Chu, C.-W.; Yang, H.; Hou, W.-J.; Huang, J.; Li, G.; Yang, Y. Control of the nanoscale crystallinity and phase separation in polymer solar cells. Appl. Phys. Lett. 2008, 92, 103306. [Google Scholar] [CrossRef]
- Shrotriya, V.; Yao, Y.; Li, G.; Yang, Y. Effect of self-organization in polymer/fullerene bulk heterojunctions on solar cell performance. Appl. Phys. Lett. 2006, 89, 063505. [Google Scholar] [CrossRef]
- Peet, J.; Soci, C.; Coffin, R.C.; Nguyen, T.Q.; Mikhailovsky, A.; Moses, D.; Bazan, G.C. Method for increasing the photoconductive response in conjugated polymer/fullerene composites. Appl. Phys. Lett. 2006, 89, 252105. [Google Scholar] [CrossRef]
- Peet, J.; Kim, J.Y.; Coates, N.E.; Ma, W.L.; Moses, D.; Heeger, A.J.; Bazan, G.C. Efficiency enhancement in low-bandgap polymer solar cells by processing with alkane dithiols. Nat. Mater. 2007, 6, 497–500. [Google Scholar] [CrossRef]
- Lee, J.K.; Ma, W.L.; Brabec, C.J.; Yuen, J.; Moon, J.S.; Kim, J.Y.; Lee, K.; Bazan, A.G.C.; Heeger, A.J. Processing Additives for Improved Efficiency from Bulk Heterojunction Solar Cells. J. Am. Chem. Soc. 2008, 130, 3619–3623. [Google Scholar] [CrossRef]
- Yao, Y.; Hou, J.; Xu, Z.; Li, G.; Yang, Y. Effects of Solvent Mixtures on the Nanoscale Phase Separation in Polymer Solar Cells. Adv. Funct. Mater. 2008, 18, 1783–1789. [Google Scholar] [CrossRef]
- Hwang, I.-W.; Cho, S.; Kim, J.Y.; Lee, K.; Coates, N.E.; Moses, D.; Heeger, A.J. Carrier generation and transport in bulk heterojunction films processed with 1,8-octanedithiol as a processing additive. J. Appl. Phys. 2008, 104, 033706. [Google Scholar] [CrossRef]
- Martens, T.; D’Haen, J.; Munters, T.; Beelen, Z.; Goris, L.; Manca, J.; D’Olieslaeger, M.; Vanderzande, D.; De Schepper, L.; Andriessen, R. Disclosure of the nanostructure of MDMO-PPV: PCBM bulk hetero-junction organic solar cells by a combination of SPM and TEM. Synth. Met. 2003, 138, 243–247. [Google Scholar] [CrossRef]
- Van Duren, J.K.; Yang, X.; Loos, J.; Bulle-Lieuwma, C.W.; Sieval, A.B.; Hummelen, J.C.; Janssen, R.A. Relating the morphology of poly(p-phenylene vinylene)/methanofullerene blends to solar-cell performance. Adv. Funct. Mater. 2004, 14, 425–434. [Google Scholar] [CrossRef]
- Shrotriya, V.; Ouyang, J.; Tseng, R.J.; Li, G.; Yang, Y. Absorption spectra modification in poly(3-hexylthiophene): Methanofullerene blend thin films. Chem. Phys. Lett. 2005, 411, 138–143. [Google Scholar] [CrossRef]
- Ross, R.B.; Cardona, C.M.; Guldi, D.M.; Sankaranarayanan, S.G.; Reese, M.O.; Kopidakis, N.; Peet, J.; Walker, B.; Bazan, G.C.; Van Keuren, E.; et al. Endohedral fullerenes for organic photovoltaic devices. Nat. Mater. 2009, 8, 208–212. [Google Scholar] [CrossRef]
- Lenes, M.; Wetzelaer, G.-J.A.H.; Kooistra, F.B.; Veenstra, S.C.; Hummelen, J.C.; Blom, P.W.M. Fullerene Bisadducts for Enhanced Open-Circuit Voltages and Efficiencies in Polymer Solar Cells. Adv. Mater. 2008, 20, 2116–2119. [Google Scholar] [CrossRef]
- He, Y.; Chen, H.-Y.; Hou, J.; Li, Y. Indene−C60Bisadduct: A New Acceptor for High-Performance Polymer Solar Cells. J. Am. Chem. Soc. 2010, 132, 1377–1382. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Zhao, G.; Peng, B.; Li, Y. High-Yield Synthesis and Electrochemical and Photovoltaic Properties of Indene-C70 Bisadduct. Adv. Funct. Mater. 2010, 20, 3383–3389. [Google Scholar] [CrossRef]
- Zhao, G.; He, Y.; Li, Y. 6.5% Efficiency of Polymer Solar Cells Based on poly(3-hexylthiophene) and Indene-C60 Bisadduct by Device Optimization. Adv. Mater. 2010, 22, 4355–4358. [Google Scholar] [CrossRef]
- Sun, Y.; Cui, C.; Wang, H.; Li, Y. Efficiency Enhancement of Polymer Solar Cells Based on Poly(3-hexylthiophene)/Indene-C70 Bisadduct via Methylthiophene Additive. Adv. Energy Mater. 2011, 1, 1058–1061. [Google Scholar] [CrossRef]
- Guo, X.; Cui, C.; Zhang, M.; Huo, L.; Huang, Y.; Hou, J.; Li, Y. High efficiency polymer solar cells based on poly(3-hexylthiophene)/indene-C70 bisadduct with solvent additive. Energy Environ. Sci. 2012, 5, 7943–7949. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, J. Structures and properties of conjugated Donor–Acceptor copolymers for solar cell applications. J. Mater. Chem. 2012, 22, 4178–4187. [Google Scholar] [CrossRef]













































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
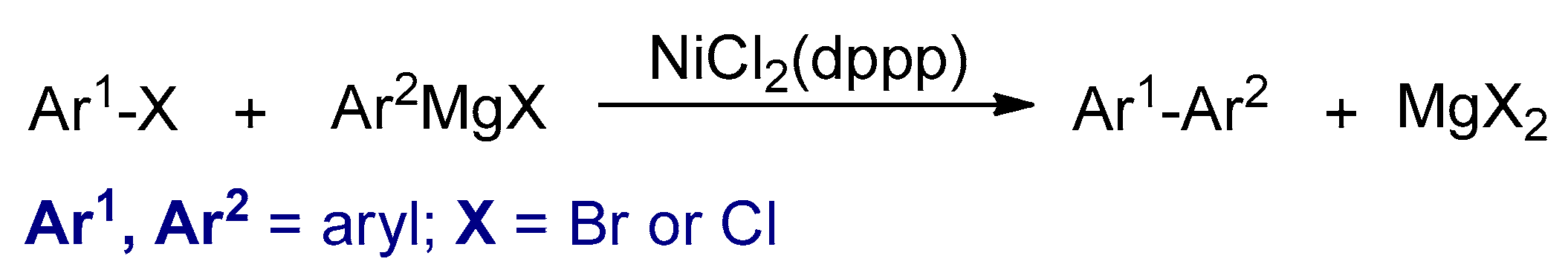{kind=link}
{kind=link}
{kind=link}
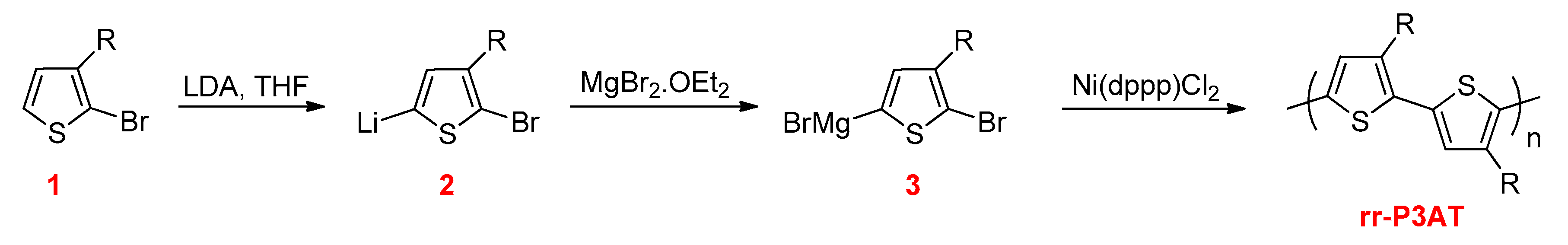{kind=link}
{kind=link}
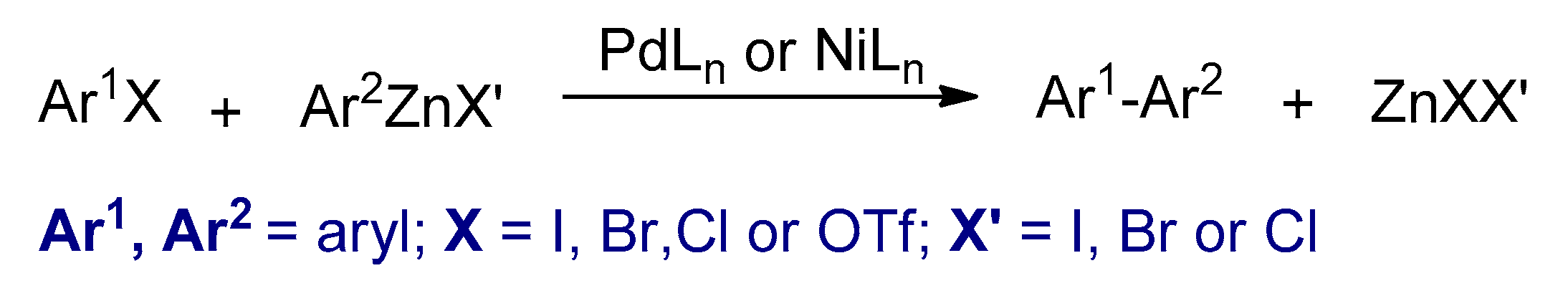{kind=link}
{kind=link}
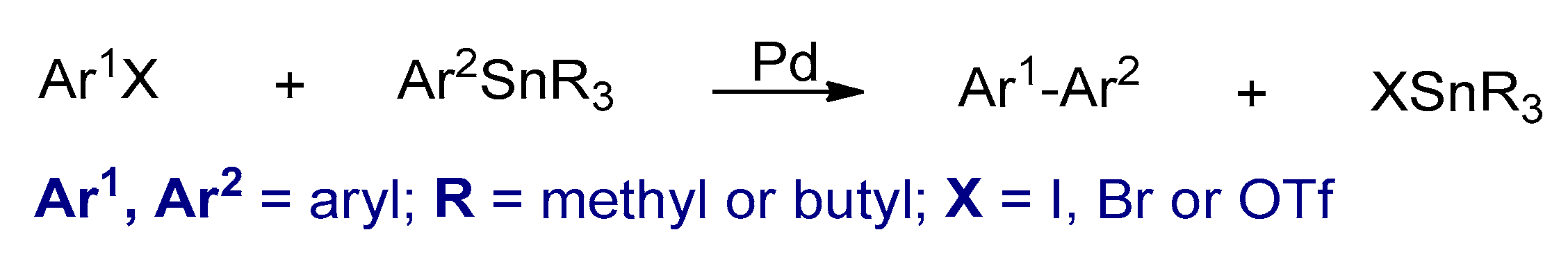{kind=link}
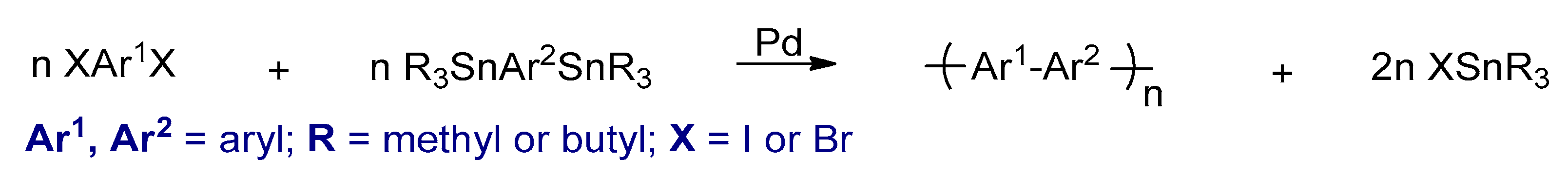{kind=link}
{kind=link}
{kind=link}
{kind=link}
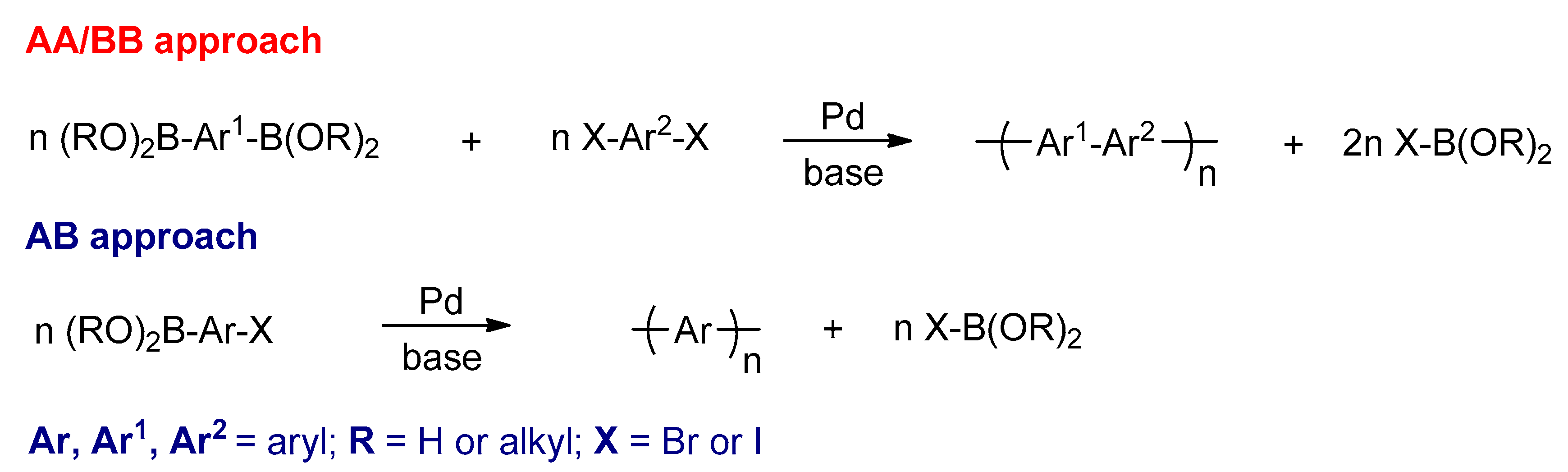{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
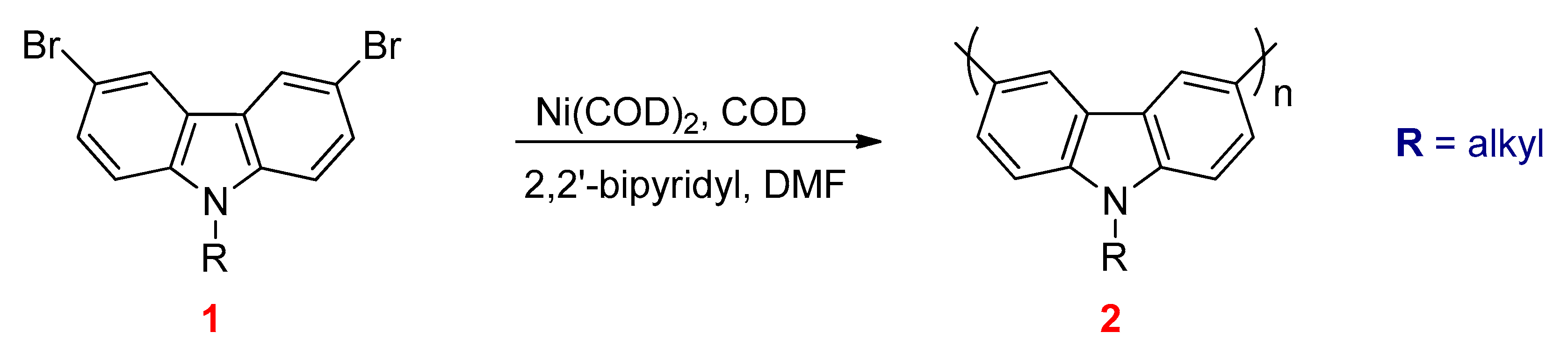{kind=link}
{kind=link}
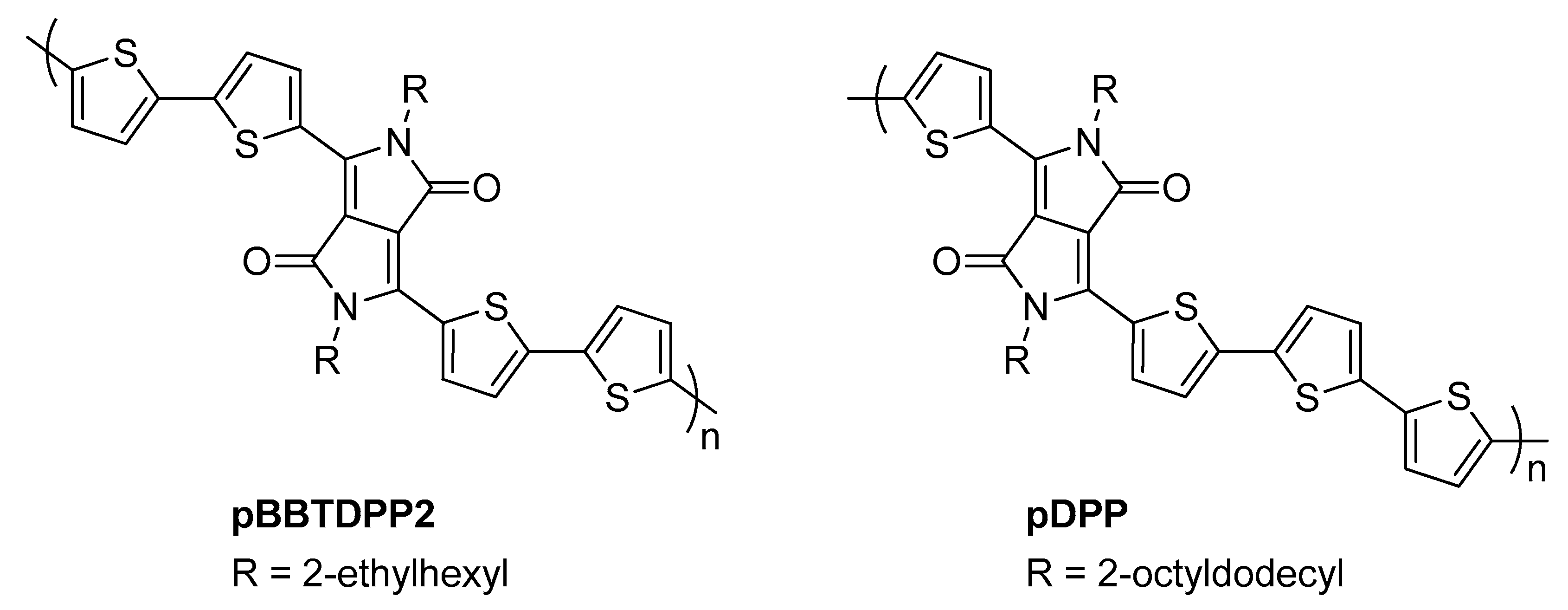{kind=link}
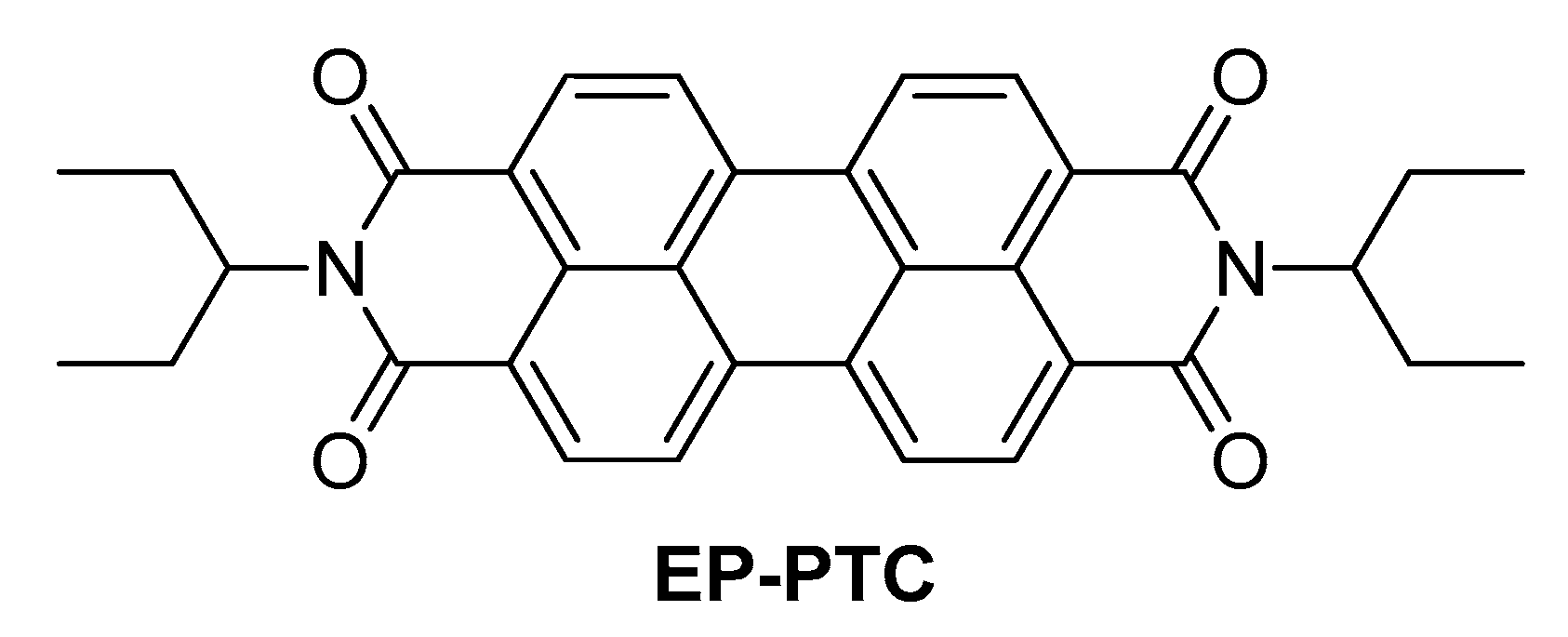{kind=link}
| Polymer | Voc (V) | Jsc (mA cm−2) | FF (%) | PCE (%) | Ref. |
|---|---|---|---|---|---|
PBDTT–DTBT R = octyl | 0.92 | 10.70 | 57.5 | 5.66 | [242] |
PTB1 X = H, R1 = octyloxy, R2 = dodecyl | 0.58 | 12.50 | 65.4 | 4.76 | [243] |
| PTB3: X = H, R1 = octyl, R2 = 2-ethylhexyl | 0.72 | 13.90 | 58.5 | 5.85 | [244] |
| PTB4: X = F, R1 = 2-ethylhexyloxy, R2 = octyl | 0.74 | 13.00 | 61.4 | 6.1 | [244] |
| PTB7: X = F, R1, R2 = 2-ethylhexyl | 0.74 | 14.50 | 68.9 | 7.4 | [243,246] |
PBDTTT–CF R1 = 2-ethylhexyloxy, R2 = heptyl | 0.76 | 15.20 | 66.9 | 7.7 | [247] |
| Polymer | Voc (V) | Jsc (mA cm−2) | FF (%) | PCE (%) | Ref. |
|---|---|---|---|---|---|
PBDTTPD R1 = 2-ethylhexyloxy, R2 = octyl | 0.85 0.84 0.85 | 9.81 9.80 11.50 | 66.0 49.5 68.0 | 5.5 a 4.1 b 6.8 c | [249] [250] [251] |
| PBDTTPD1 R1 = dodecyloxy, R2 = 2-octyldodecyl | 0.93 | 6.58 | 56.0 | 3.42 d | [252] |
| PBDTTPD2 R1 = 2-ethylhexyloxy, R2 = 2-octyldodecyl | 0.91 | 10.34 | 51.0 | 4.79 d | [252] |
| PBDTTPD R1 = 2-ethylhexyloxy, R2 = heptyl | 0.97 | 12.60 | 70.0 | 8.5 e | [253] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
R. Murad, A.; Iraqi, A.; Aziz, S.B.; N. Abdullah, S.; Brza, M.A. Conducting Polymers for Optoelectronic Devices and Organic Solar Cells: A Review. Polymers 2020, 12, 2627. https://doi.org/10.3390/polym12112627
R. Murad A, Iraqi A, Aziz SB, N. Abdullah S, Brza MA. Conducting Polymers for Optoelectronic Devices and Organic Solar Cells: A Review. Polymers. 2020; 12(11):2627. https://doi.org/10.3390/polym12112627
Chicago/Turabian StyleR. Murad, Ary, Ahmed Iraqi, Shujahadeen B. Aziz, Sozan N. Abdullah, and Mohamad A. Brza. 2020. "Conducting Polymers for Optoelectronic Devices and Organic Solar Cells: A Review" Polymers 12, no. 11: 2627. https://doi.org/10.3390/polym12112627
APA StyleR. Murad, A., Iraqi, A., Aziz, S. B., N. Abdullah, S., & Brza, M. A. (2020). Conducting Polymers for Optoelectronic Devices and Organic Solar Cells: A Review. Polymers, 12(11), 2627. https://doi.org/10.3390/polym12112627