Formulation and Evaluation of Microwave-Modified Chitosan-Curcumin Nanoparticles—A Promising Nanomaterials Platform for Skin Tissue Regeneration Applications Following Burn Wounds

 and
and 
Abstract
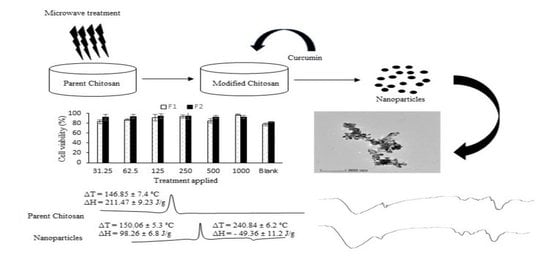
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Chitosan Molecular Weight Modification
2.3. Specific Viscosity (SV)
2.4. Degree of Deacetylation (DD)
2.5. Molecular Weight Analysis of Chitosan
2.6. LMW Chitosan-Curcumin Nanoparticles Preparation
2.7. Size and Zeta Potential
2.8. Entrapment Efficiency and Drug Content
2.9. Morphology
2.10. In Vitro Drug Release
2.11. Drug Release Kinetics
2.12. Vibrational Spectroscopic and Thermal Analysis of Nanoparticles
2.13. Antimicrobial Activity
2.14. Cell Viability
2.15. Cell Migration Analysis
2.16. Statistical Analysis
3. Results
3.1. Physicochemical Analysis of Parent and Modified Chitosan
3.2. Physicochemical Analysis of Modified Chitosan Curcumin Nanoparticles
3.3. Morphology
3.4. In Vitro Drug Release
3.5. Vibrational Spectroscopic Analysis
3.6. Thermal Analysis
3.7. Antimicrobial Analysis
3.8. Cell Viability and Cell Migration Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Burn. Available online: https://www.who.int/en/news-room/fact-sheets/detail/burns (accessed on 27 November 2019).
- Dumas, A.; Gaudin-Audrain, C.; Mabilleau, G.; Massin, P.; Hubert, L.; Baslé, M.F.; Chappard, D. The influence of processes for the purification of human bone allografts on the matrix surface and cytocompatibility. Biomaterials 2006, 27, 4204–4211. [Google Scholar] [CrossRef] [PubMed]
- Katayama, Y.; Montenegro, R.; Freier, T.; Midha, R.; Belkas, J.S.; Shoichet, M.S. Coil-reinforced hydrogel tubes promote nerve regeneration equivalent to that of nerve autografts. Biomaterials 2006, 27, 505–518. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, M.D.; Xie, C.; Zhang, X.; Benoit, D.S.W. The effect of mesenchymal stem cells delivered via hydrogel-based tissue engineered periosteum on bone allograft healing. Biomaterials 2013, 34, 8887–8898. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Santamaría, L.; Guerrero-Aspizua, S.; Del Río, M. Skin bioengineering: Preclinical and clinical applications. Actas Dermosifiliogr. 2012, 103, 5–11. [Google Scholar] [CrossRef]
- Tabata, Y. Tissue Regeneration Based on Drug Delivery Technology. In Topics in Tissue Engineering; Ashammakhi, N., Ferretti, P., Eds.; Oulu University: Oulu, Finland, 2003; pp. 1–32. [Google Scholar]
- Noori, S.; Kokabi, M.; Hassan, Z.M. Nanoclay Enhanced the Mechanical Properties of Poly (Vinyl Alcohol)/Chitosan/Montmorillonite Nanocomposite Hydrogel as Wound Dressing. Procedia Mater. Sci. 2015, 11, 152–156. [Google Scholar] [CrossRef]
- Huang, N.; Lin, J.; Li, S.; Deng, Y.; Kong, S.; Hong, P.; Yang, P.; Liao, M.; Hu, Z. Preparation and evaluation of squid ink polysaccharide-chitosan as a wound-healing sponge. Mater. Sci. Eng. C 2018, 82, 354–362. [Google Scholar] [CrossRef]
- Chen, S.A.; Chen, H.M.; Yao, Y.D.; Hung, C.F.; Tu, C.S.; Liang, Y.J. Topical treatment with anti-oxidants and Au nanoparticles promote healing of diabetic wound through receptor for advance glycation end-products. Eur. J. Pharm. Sci. 2012, 47, 875–883. [Google Scholar] [CrossRef]
- Xu, F.; Weng, B.; Gilkerson, R.; Materon, L.A.; Lozano, K. Development of tannic acid/chitosan/pullulan composite nanofibers from aqueous solution for potential applications as wound dressing. Carbohydr. Polym. 2015, 115, 16–24. [Google Scholar] [CrossRef]
- Wong, T.W.; Ramli, N.A. Carboxymethylcellulose film for bacterial wound infection control and healing. Carbohydr. Polym. 2014, 112, 367–375. [Google Scholar] [CrossRef]
- Jayakumar, R.; Prabaharan, M.; Reis, R.L.; Mano, J.F. Graft copolymerized chitosan—Present status and applications. Carbohydr. Polym. 2005, 62, 142–158. [Google Scholar] [CrossRef]
- Madhumathi, K.; Kumar, P.T.S.; Abhilash, S.; Sreeja, V.; Tamura, H.; Manzoor, K.; Nair, S.V.; Jayakumar, R. Development of novel chitin / nanosilver composite scaffolds for wound dressing applications. J. Mater. Sci. Mater. Med. 2010, 21, 807–813. [Google Scholar] [CrossRef] [PubMed]
- Jayakumar, R.; Nwe, N.; Tokura, S.; Tamura, H. Sulfated chitin and chitosan as novel biomaterials. Int. J. Biol. Macromol. 2007, 40, 175–181. [Google Scholar] [CrossRef]
- Wang, J.J.; Zeng, Z.W.; Xiao, R.Z.; Xie, T.; Zhou, G.L.; Zhan, X.R.; Wang, S.L. Recent advances of chitosan nanoparticles as drug carriers. Int. J. Nanomed. 2011, 6, 765–774. [Google Scholar]
- Jayakumar, R.; Chennazhi, K.P.; Muzzarelli, R.A.A.; Tamura, H.; Nair, S.V.; Selvamurugan, N. Chitosan conjugated DNA nanoparticles in gene therapy. Carbohydr. Polym. 2010, 79, 1–8. [Google Scholar] [CrossRef]
- Nagahama, H.; Nwe, N.; Jayakumar, R.; Koiwa, S.; Furuike, T.; Tamura, H. Novel biodegradable chitin membranes for tissue engineering applications. Carbohydr. Polym. 2008, 73, 295–302. [Google Scholar] [CrossRef]
- Jayakumar, R.; Rajkumar, M.; Freitas, H.; Selvamurugan, N.; Nair, S.V.; Furuike, T.; Tamura, H. Preparation, characterization, bioactive and metal uptake studies of alginate/phosphorylated chitin blend films. Int. J. Biol. Macromol. 2009, 44, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Shalumon, K.T.; Binulal, N.S.; Selvamurugan, N.; Nair, S.V.; Menon, D.; Furuike, T.; Tamura, H.; Jayakumar, R. Electrospinning of carboxymethyl chitin/poly(vinyl alcohol) nanofibrous scaffolds for tissue engineering applications. Carbohydr. Polym. 2009, 77, 863–869. [Google Scholar] [CrossRef]
- Jayakumar, R.; Reis, R.L.; Mano, J.F. Phosphorous containing chitosan beads for controlled oral drug delivery. J. Bioact. Compat. Polym. 2006, 21, 327–340. [Google Scholar] [CrossRef]
- Prabaharan, M.; Mano, J.F. Chitosan-based particles as controlled drug delivery systems. Drug Deliv. 2005, 12, 41–57. [Google Scholar] [CrossRef]
- Anitha, A.; Divya Rani, V.V.; Krishna, R.; Sreeja, V.; Selvamurugan, N.; Nair, S.V.; Tamura, H.; Jayakumar, R. Synthesis, characterization, cytotoxicity and antibacterial studies of chitosan, O-carboxymethyl, N,O-carboxymethyl chitosan nanoparticles. Carbohydr. Polym. 2009, 78, 672–677. [Google Scholar] [CrossRef]
- Peter, M.; Binulol, N.S.; Soumya, S.; Nair, S.V.; Tamura, H.; Jayakumar, R. Nanocomposite scaffolds of bioactive glass ceramic nanoparticles disseminated chitosan matrix for tissue engineering applications. Carbohydr. Polym. 2010, 20, 284–289. [Google Scholar] [CrossRef]
- Kazuaki, M.; Shingo, M.; Yusuke, Y.; Akira, F. In vitro degradation behavior of freeze-dried carboxymethyl-chitin sponges processed by vacuumheating and gamma irradiation. Polym. Degrad. Stab. 2003, 81, 327–332. [Google Scholar]
- Baldrick, P. The safety of chitosan as a pharmaceutical excipient. Regul. Toxicol. Pharmacol. 2010, 56, 290–299. [Google Scholar] [CrossRef]
- Paul, W.; Sharma, C.P. Chitin and alginates wound dressings: A short review. Trends Biomater. Artif. Organs 2004, 18, 18–23. [Google Scholar]
- Alsarra, I.A. Chitosan topical gel formulation in the management of burn wounds. Int. J. Biol. Macromol. 2009, 45, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.S.; Lee, S.B.; Hong, S.R.; Lee, Y.M.; Song, K.W.; Park, M.H. Studies on gelatin-based sponges. Part III: A comparative study of cross-linked gelatin/alginate, gelatin/hyaluronate and chitosan/hyaluronate sponges and their application as a wound dressing in full-thickness skin defect of rat. J. Mater. Sci. Mater. Med. 2001, 12, 67–73. [Google Scholar] [CrossRef]
- Joe, B.; Vijaykumar, M.; Lokesh, B.R. Biological Properties of Curcumin-Cellular and Molecular Mechanisms of Action. Crit. Rev. Food Sci. Nutr. 2004, 44, 97–111. [Google Scholar] [CrossRef]
- Krausz, A.E.; Adler, B.L.; Cabral, V.; Navati, M.; Doerner, J.; Charafeddine, R.A.; Chandra, D.; Liang, H.; Gunther, L.; Clendaniel, A.; et al. Curcumin-encapsulated nanoparticles as innovative antimicrobial and wound healing agent. Nanomed. Nanotechnol. Biol. Med. 2015, 11, 195–206. [Google Scholar] [CrossRef]
- Chereddy, K.K.; Coco, R.; Memvanga, P.B.; Ucakar, B.; Rieux, A.D.; Vandermeulen, G.; Préat, V. Combined effect of PLGA and curcumin on wound healing activity. J. Control. Release 2013, 171, 208–215. [Google Scholar] [CrossRef]
- Thomasa, L.; Zakira, F.; Aamir Mirzab, M.; Anwer, K.; Ahmad, F.J.; Iqbal, Z. Development of Curcumin loaded chitosan polymer based nanoemulsion gel: In vitro, ex vivo evaluation and in vivo wound healing studies. Int. J. Biol. Macromol. 2017, 101, 569–579. [Google Scholar] [CrossRef]
- Kanta, V.; Gopala, A.; Pathaka, N.N.; Kumarb, P.; Tandana, S.K.; Kumar, D. Antioxidant and anti-inflammatory potential of curcumin accelerated the cutaneous wound healing in streptozotocin-induced diabetic rats. Int. Immunopharmacol. 2014, 20, 322–330. [Google Scholar] [CrossRef]
- El-Refaie, W.M.; Elnaggar, Y.S.R.; El-Massik, M.A.; Abdallah, O.Y. Novel curcumin-loaded gel-core hyaluosomes with promising burn-wound healing potential: Development, in-vitro appraisal and in-vivo studies. Int. J. Pharm. 2015, 486, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Li, X.; Ye, X.; Qi, J.; Fan, R.; Gao, X.; Wu, Y.; Zhou, L.; Tong, A. EGF and curcumin co-encapsulated nanoparticle/hydrogel system as potent skin regeneration agent. Int. J. Nanomed. 2016, 11, 3993–4009. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.Y.; Wu, Q.J.; Wang, Y.J.; Zhang, D.D.; Luo, F.; Zhao, X.; Wei, Y.Q.; Qian, Z.Y. A biodegradable hydrogel system containing curcumin encapsulated in micelles for cutaneous wound healing. Biomaterials 2013, 34, 6377–6387. [Google Scholar] [CrossRef] [PubMed]
- Croisier, F.; Jérôme, C. Chitosan-based biomaterials for tissue engineering. Eur. Polym. J. 2013, 49, 780–792. [Google Scholar] [CrossRef]
- Shi, D.; Wang, F.; Lan, T.; Zhang, Y.; Shao, Z. Convenient fabrication of carboxymethyl cellulose electrospun nanofibers functionalized with silver nanoparticles. Cellulose 2016, 23, 1899–1909. [Google Scholar] [CrossRef]
- Arockianathan, P.M.; Sekar, S.; Kumaran, B.; Sastry, T.P. Preparation, characterization and evaluation of biocomposite films containing chitosan and sago starch impregnated with silver nanoparticles. Int. J. Biol. Macromol. 2012, 50, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Gao, J.; Wang, L.; Hu, X.Y. Composite chitosan/poly(ethylene oxide) electrospun nanofibrous mats as novel wound dressing matrixes for the controlled release of drugs. J. Appl. Polym. Sci. 2015, 132, 1–8. [Google Scholar] [CrossRef]
- Tømmeraas, K.; Vårum, K.M.; Christensen, B.E.; Smidsrød, O. Preparation and characterisation of oligosaccharides produced by nitrous acid depolymerisation of chitosans. Carbohydr. Res. 2001, 333, 137–144. [Google Scholar] [CrossRef]
- Sato, K.; Kishimoto, T.; Morimoto, M.; Saimoto, H.; Shigemasa, Y. Hydrolysis of acetals in water under hydrothermal conditions. Tetrahedron Lett. 2003, 44, 8623–8625. [Google Scholar] [CrossRef]
- Cravotto, G.; Tagliapietra, S.; Robaldo, B.; Trotta, M. Chemical modification of chitosan under high-intensity ultrasound. Ultrason. Sonochem. 2005, 12, 95–98. [Google Scholar] [CrossRef]
- Naveed, M.; Phil, L.; Sohail, M.; Hasnat, M.; Baig, M.M.F.A.; Ihsan, A.U.; Shumzaid, M.; Kakar, M.U.; Mehmood Khan, T.; Akabar, M.D.; et al. Chitosan oligosaccharide (COS): An overview. Int. J. Biol. Macromol. 2019, 129, 827–843. [Google Scholar] [CrossRef]
- Nawaz, A.; Wong, T.W. Microwave as skin permeation enhancer for transdermal drug delivery of chitosan-5-fluorouracil nanoparticles. Carbohydr. Polym. 2017, 157, 906–919. [Google Scholar] [CrossRef]
- Xia, W.; Liu, P.; Zhang, J.; Chen, J. Biological activities of chitosan and chitooligosaccharides. Food Hydrocoll. 2011, 25, 170–179. [Google Scholar] [CrossRef]
- Wong, T.W.; Nurulaini, H. Sustained-release alginate-chitosan pellets prepared by melt pelletization technique. Drug Dev. Ind. Pharm. 2012, 38, 1417–1427. [Google Scholar] [CrossRef] [PubMed]
- Bagheri-Khoulenjani, S.; Taghizadeh, S.M.; Mirzadeh, H. An investigation on the short-term biodegradability of chitosan with various molecular weights and degrees of deacetylation. Carbohydr. Polym. 2009, 78, 773–778. [Google Scholar] [CrossRef]
- Katas, H.; Wen, C.Y.; Siddique, M.I.; Hussain, Z.; Fadhil, F.H.M. Thermoresponsive curcumin/DsiRNA nanoparticle gels for the treatment of diabetic wounds: Synthesis and drug release. Ther. Deliv. 2017, 8, 137–150. [Google Scholar] [CrossRef] [PubMed]
- Chuah, L.H.; Billa, N.; Roberts, C.J.; Burley, J.C.; Manickam, S. Curcumin-containing chitosan nanoparticles as a potential mucoadhesive delivery system to the colon. Pharm. Dev. Technol. 2013, 18, 591–599. [Google Scholar] [CrossRef]
- Dash, S.; Murthy, P.N.; Nath, L.; Chowdhury, P. Kinetic modeling on drug release from controlled drug delivery systems. Acta Pol. Pharm.-Drug Res. 2010, 67, 217–223. [Google Scholar]
- Lai, J.C.Y.; Lai, H.Y.; Rao, N.K.; Ng, S.F. Treatment for diabetic ulcer wounds using a fern tannin optimized hydrogel formulation with antibacterial and antioxidative properties. J. Ethnopharmacol. 2016, 189, 277–289. [Google Scholar] [CrossRef]
- Andrews, J.M. Determination of minimum inhibitory concentrations. J. Antimicrob. Chemother. 2002, 49, 1049. [Google Scholar] [CrossRef]
- Khan, N.R.; Wong, T.W. 5-Fluorouracil ethosomes–skin deposition and melanoma permeation synergism with microwave. Artif. Cells, Nanomed. Biotechnol. 2018, 46, 568–577. [Google Scholar] [CrossRef] [PubMed]
- Sreekumar, S.; Goycoolea, F.M.; Moerschbacher, B.M.; Rivera-Rodriguez, G.R. Parameters influencing the size of chitosan-TPP nano- and microparticles. Sci. Rep. 2018, 8, 1–11. [Google Scholar]
- Nair, R.S.; Morris, A.; Billa, N.; Leong, C.-O. An Evaluation of Curcumin-Encapsulated Chitosan Nanoparticles for Transdermal Delivery. AAPS PharmSciTech 2019, 20, 68–69. [Google Scholar] [CrossRef] [PubMed]
- Martins, A.F.; de Oliveira, D.M.; Pereira, A.G.B.; Rubira, A.F.; Muniz, E.C. Chitosan/TPP microparticles obtained by microemulsion method applied in controlled release of heparin. Int. J. Biol. Macromol. 2012, 51, 1127–1133. [Google Scholar] [CrossRef] [PubMed]
- Stillwell, W. An Introduction to Biological Membranes from Bilayers to Rafts, 1st ed.; Elsevier B.V.: Oxford, UK, 2013; ISBN 9780444521538. [Google Scholar]
- Raja, M.A.G.; Katas, H.; Hamid, Z.A.; Razali, N.A. Physicochemical properties and in vitro cytotoxicity studies of chitosan as a potential carrier for dicer-substrate siRNA. J. Nanomater. 2013, 2013, 1–10. [Google Scholar] [CrossRef]
- Shu, X.Z.; Zhu, K.J. The influence of multivalent phosphate structure on the properties of ionically cross-linked chitosan films for controlled drug release. Eur. J. Pharm. Biopharm. 2002, 54, 235–243. [Google Scholar] [CrossRef]
- Villegas-Peralta, Y.; López-Cervantes, J.; Madera Santana, T.J.; Sánchez-Duarte, R.G.; Sánchez-Machado, D.I.; Martínez-Macías, M. del R.; Correa-Murrieta, M.A. Impact of the molecular weight on the size of chitosan nanoparticles: Characterization and its solid-state application. Polym. Bull. 2020. [Google Scholar] [CrossRef]
- Chen, Y.; Mohanraj, V.J.; Parkin, J.E. Chitosan-dextran sulfate nanoparticles for delivery of an anti-angiogenesis peptide. Lett. Pept. Sci. 2003, 10, 621–629. [Google Scholar] [CrossRef]
- Gan, Q.; Wang, T. Chitosan nanoparticle as protein delivery carrier-Systematic examination of fabrication conditions for efficient loading and release. Colloids Surf. B Biointerfaces 2007, 59, 24–34. [Google Scholar] [CrossRef]
- Mora-Huertas, C.E.; Fessi, H.; Elaissari, A. Polymer-based nanocapsules for drug delivery. Int. J. Pharm. 2010, 385, 113–142. [Google Scholar] [CrossRef] [PubMed]
- Preetz, C.; Rübe, A.; Reiche, I.; Hause, G.; Mäder, K. Preparation and characterization of biocompatible oil-loaded polyelectrolyte nanocapsules. Nanomed. Nanotechnol. Biol. Med. 2008, 4, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Dhawade, P.P.; Jagtap, R.N. Characterization of the glass transition temperature of chitosan and its oligomers by temperature modulated differential scanning calorimetry. Pelagia Res. Libr. Adv. Appl. Sci. Res. 2012, 3, 1372–1382. [Google Scholar]
- Rampino, A.; Borgogna, M.; Blasi, P.; Bellich, B.; Cesàro, A. Chitosan nanoparticles: Preparation, size evolution and stability. Int. J. Pharm. 2013, 455, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Parize, A.L.; Stulzer, H.K.; Laranjeira, M.C.M.; Brighente, I.M.D.C.; De Souza, T.C.R. Evaluation of chitosan microparticles containing curcumin and crosslinked with sodium tripolyphosphate produced by spray drying. Quim. Nova 2012, 35, 1127–1132. [Google Scholar] [CrossRef]
- Kittur, F.S.; Prashanth, K.V.H.; Sankar, K.U.; Tharanathan, R.N. Characterization of chitin, chitosan and their carboxymethyl derivatives by differential scanning calorimetry. Carbohydr. Polym. 2002, 49, 185–193. [Google Scholar] [CrossRef]
- Khezri, A.; Karimi, A.; Yazdian, F.; Jokar, M.; Mofradnia, S.R.; Rashedi, H.; Tavakoli, Z. Molecular dynamic of curcumin/chitosan interaction using a computational molecular approach: Emphasis on biofilm reduction. Int. J. Biol. Macromol. 2018, 114, 972–978. [Google Scholar] [CrossRef]
- Goy, R.C.; Morais, S.T.B.; Assis, O.B.G. Evaluation of the antimicrobial activity of chitosan and its quaternized derivative on E. Coli and S. aureus growth. Braz. J. Pharmacogn. 2016, 26, 122–127. [Google Scholar] [CrossRef]
- Kehinde, A.O.; Ademola, S.A.; Okeshola, O.A.; Oluwatosin, O.M.; Bakare, R.A. Pattern of Bacterial Pathogens in Burn Wound Infections in Ibadan, Nigeria. Ann. Burn. Fire Disasters 2004, 17, 12–15. [Google Scholar]
- Motayo, B.O.; Akinbo, J.A.; Ogiogwa, I.J.; Idowu, A.A.; Nwanze, J.C.; Onoh, C.C.; Okerentugba, P.O.; Innocent-Adiele, H.C.; Okonko, I.O. Bacteria Colonisation and Antibiotic Susceptibility Pattern of Wound Infections in a Hospital in Abeokuta. Front. Sci. 2013, 3, 43–48. [Google Scholar]
- Da Silva, A.C.; Santos, P.D.D.F.; Silva, J.T.D.P.; Leimann, F.V.; Bracht, L.; Gonçalves, O.H. Impact of curcumin nanoformulation on its antimicrobial activity. Trends Food Sci. Technol. 2018, 72, 74–82. [Google Scholar] [CrossRef]
- Benhabiles, M.S.; Salah, R.; Lounici, H.; Drouiche, N.; Goosen, M.F.A.; Mameri, N. Antibacterial activity of chitin, chitosan and its oligomers prepared from shrimp shell waste. Food Hydrocoll. 2012, 29, 48–56. [Google Scholar] [CrossRef]
- Howling, G.I.; Dettmar, P.W.; Goddard, P.A.; Hampson, F.C.; Dornish, M.; Wood, E.J. The effect of chitin and chitosan on the proliferation of human skin fibroblasts and keratinocytes in vitro. Biomaterials 2001, 22, 2959–2966. [Google Scholar] [CrossRef]
- Cianfruglia, L.; Minnelli, C.; Laudadio, E.; Scirè, A.; Armeni, T. Side effects of curcumin: Epigenetic and antiproliferative implications for normal dermal fibroblast and breast cancer cells. Antioxidants 2019, 8, 382. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | Chitosan (%) | Curcumin (µg) | Size (nm) | Zeta Potential (mV) | PDI | Drug Content (%) | Entrapment Efficiency (%) |
|---|---|---|---|---|---|---|---|
| Parent chitosan | - | - | 4123.7 ± 421.4 | 21.4 ± 2.3 | 1.0 ± 0.0 | - | - |
| LMW Chitosan | - | - | 667.7 ± 145.7 | 67.4 ± 6.2 | 0.72 ± 0.1 | - | - |
| F1 blank | 0.3 | - | 170.7 ± 2.9 | 46.4 ± 2.6 | 0.27 ± 0.05 | - | - |
| F2 blank | 0.4 | - | 223.7 ± 9.3 | 46.7 ± 1.6 | 0.5 ± 0.01 | - | - |
| F1 | 0.3 | 300 | 279.7 ± 20.3 | 52.4 ± 1.50 | 0.67 ± 0.05 | 99.99 ± 1.39 | 99.93 ± 3.43 |
| F2 | 0.4 | 400 | 259.2 ± 19.4 | 42.7 ± 1.53 | 0.54 ± 0.05 | 99.99 ± 0.34 | 99.96 ± 2.12 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Basit, H.M.; Mohd Amin, M.C.I.; Ng, S.-F.; Katas, H.; Shah, S.U.; Khan, N.R. Formulation and Evaluation of Microwave-Modified Chitosan-Curcumin Nanoparticles—A Promising Nanomaterials Platform for Skin Tissue Regeneration Applications Following Burn Wounds. Polymers 2020, 12, 2608. https://doi.org/10.3390/polym12112608
Basit HM, Mohd Amin MCI, Ng S-F, Katas H, Shah SU, Khan NR. Formulation and Evaluation of Microwave-Modified Chitosan-Curcumin Nanoparticles—A Promising Nanomaterials Platform for Skin Tissue Regeneration Applications Following Burn Wounds. Polymers. 2020; 12(11):2608. https://doi.org/10.3390/polym12112608
Chicago/Turabian StyleBasit, Hafiz Muhammad, Mohd Cairul Iqbal Mohd Amin, Shiow-Fern Ng, Haliza Katas, Shefaat Ullah Shah, and Nauman Rahim Khan. 2020. "Formulation and Evaluation of Microwave-Modified Chitosan-Curcumin Nanoparticles—A Promising Nanomaterials Platform for Skin Tissue Regeneration Applications Following Burn Wounds" Polymers 12, no. 11: 2608. https://doi.org/10.3390/polym12112608
APA StyleBasit, H. M., Mohd Amin, M. C. I., Ng, S.-F., Katas, H., Shah, S. U., & Khan, N. R. (2020). Formulation and Evaluation of Microwave-Modified Chitosan-Curcumin Nanoparticles—A Promising Nanomaterials Platform for Skin Tissue Regeneration Applications Following Burn Wounds. Polymers, 12(11), 2608. https://doi.org/10.3390/polym12112608