Basic Approaches to the Design of Intrinsic Self-Healing Polymers for Triboelectric Nanogenerators

 ,
,  ,
,  and
and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
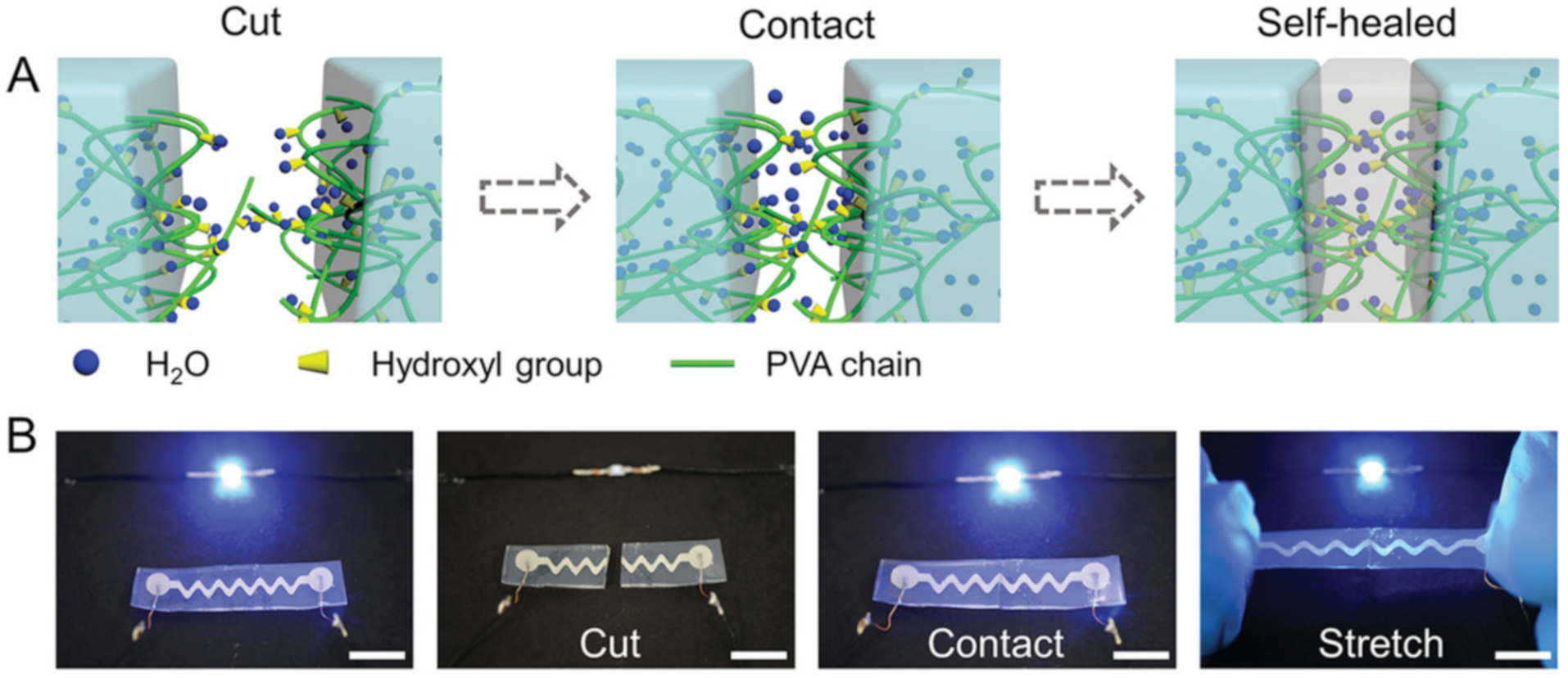{kind=link}
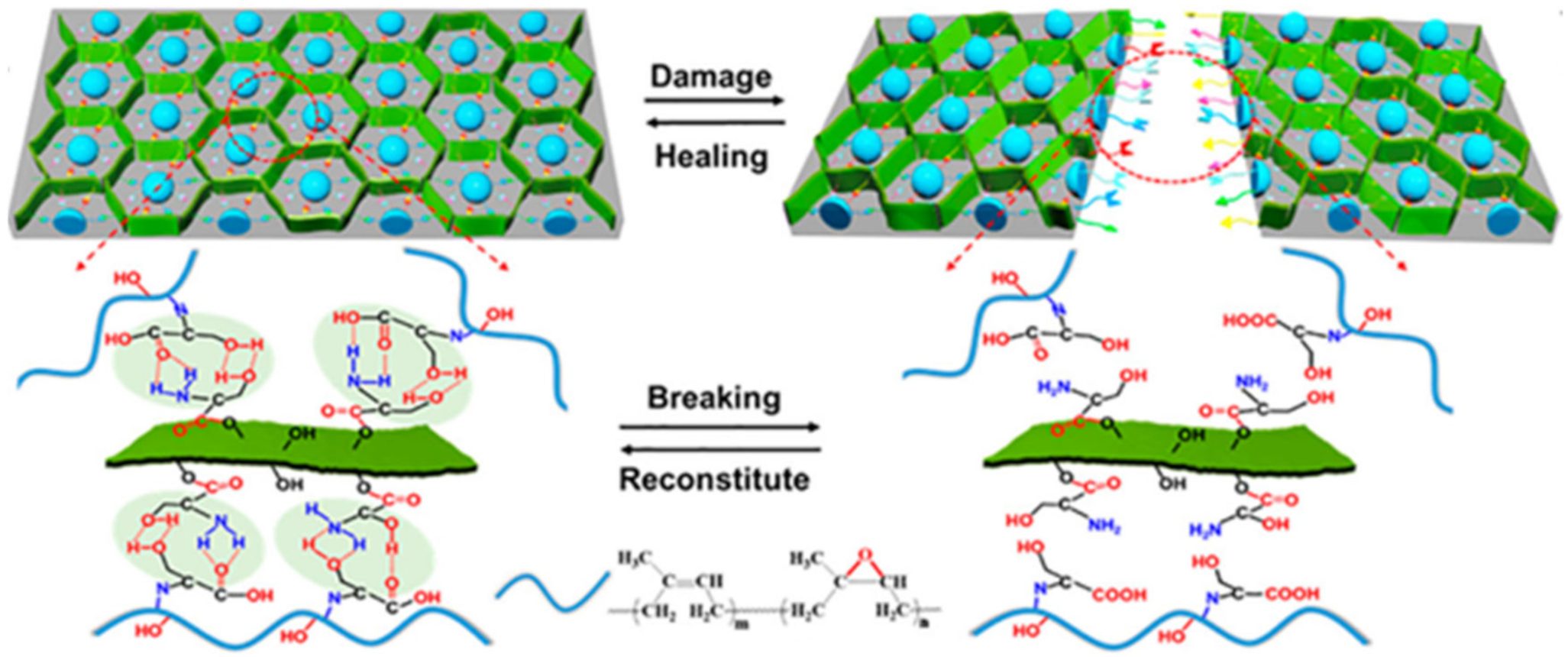{kind=link}
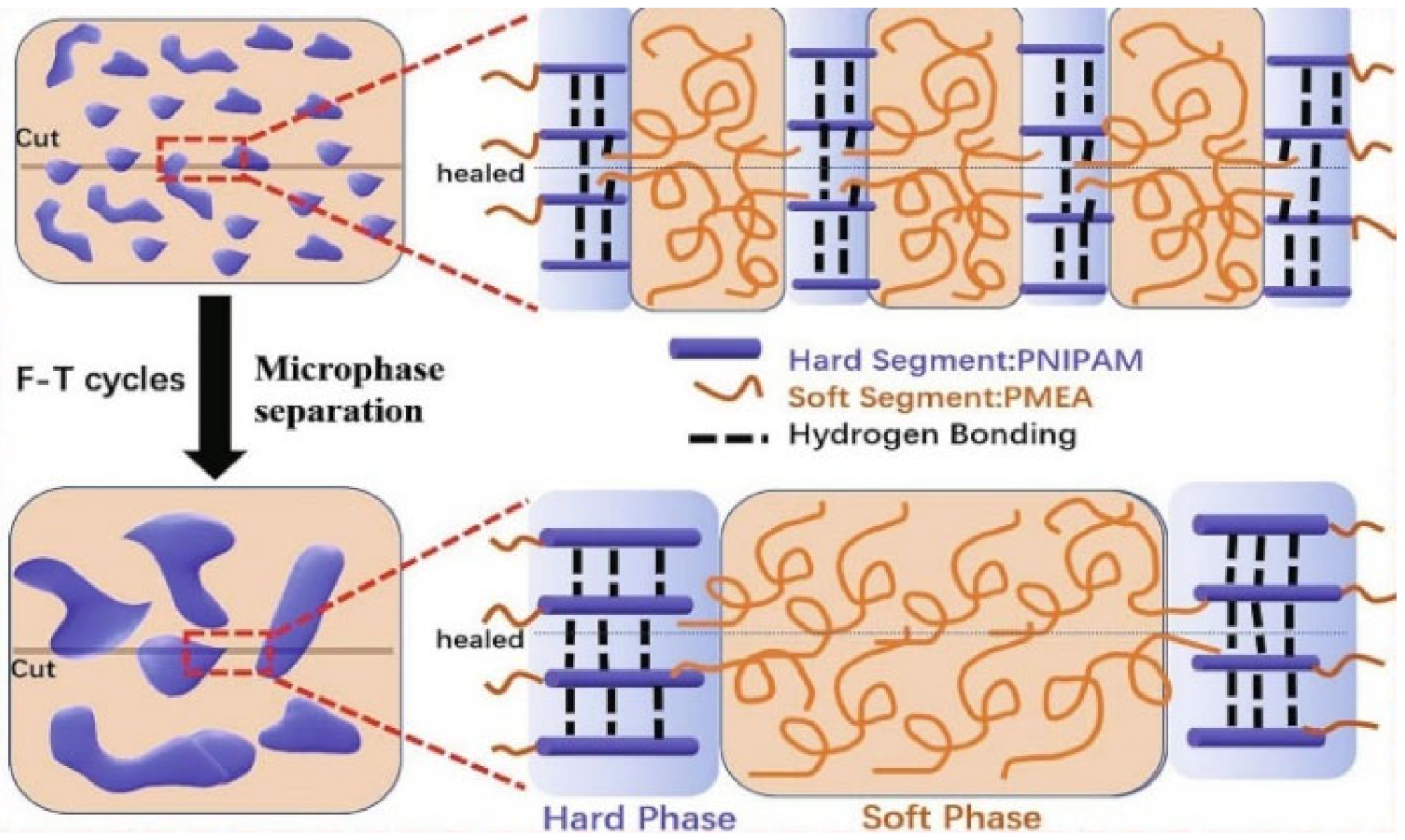{kind=link}
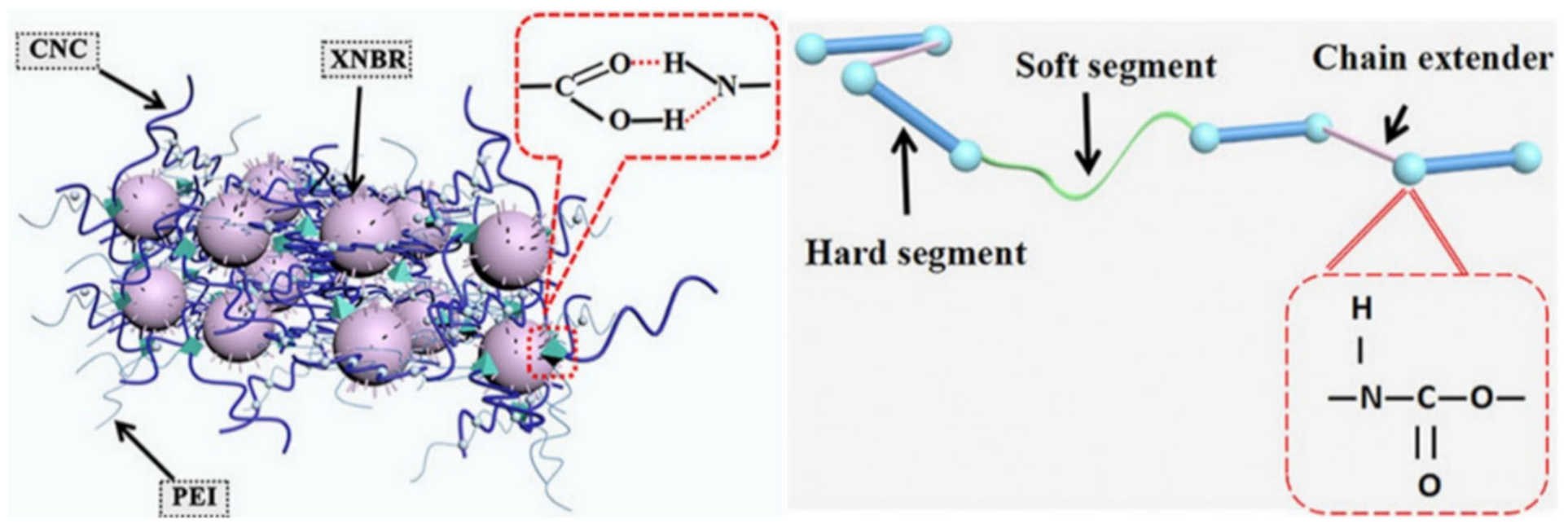{kind=link}
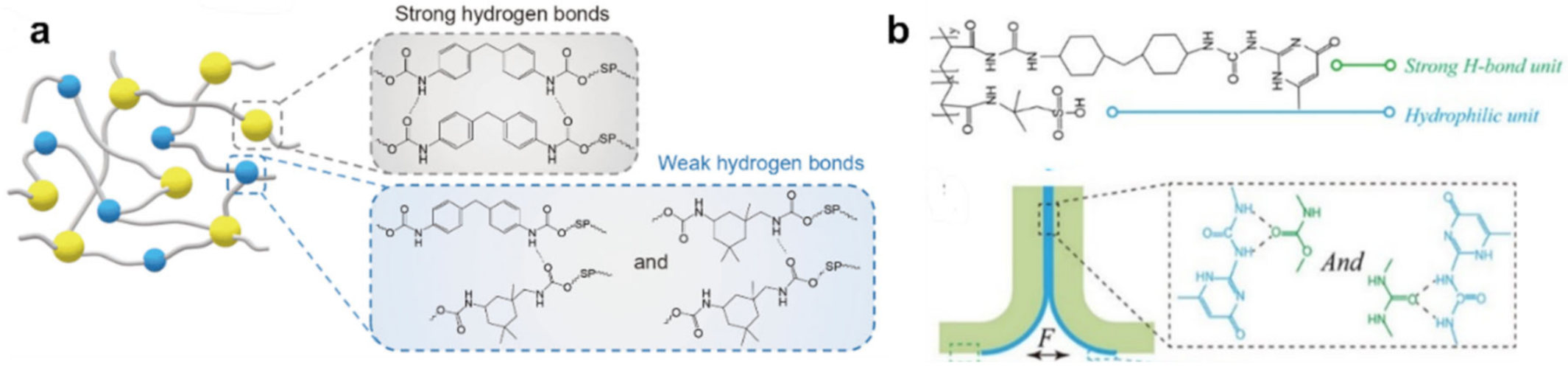{kind=link}
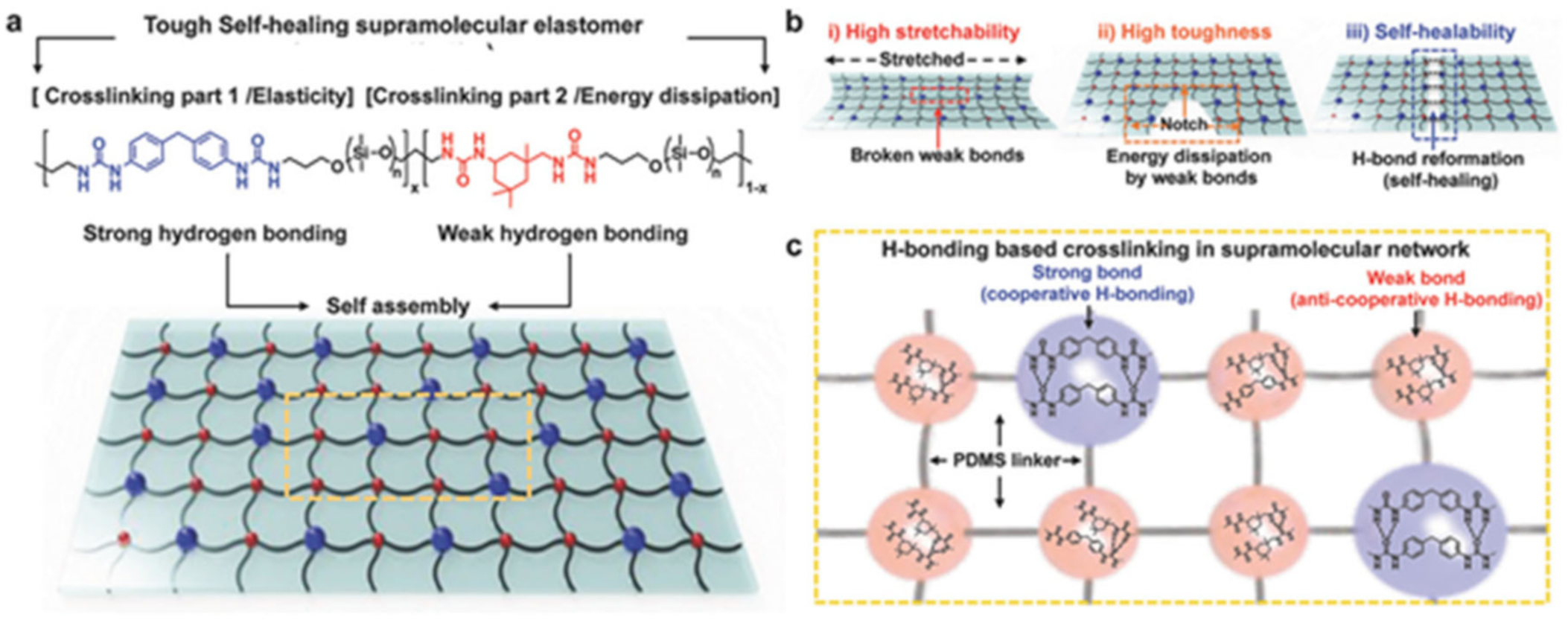{kind=link}
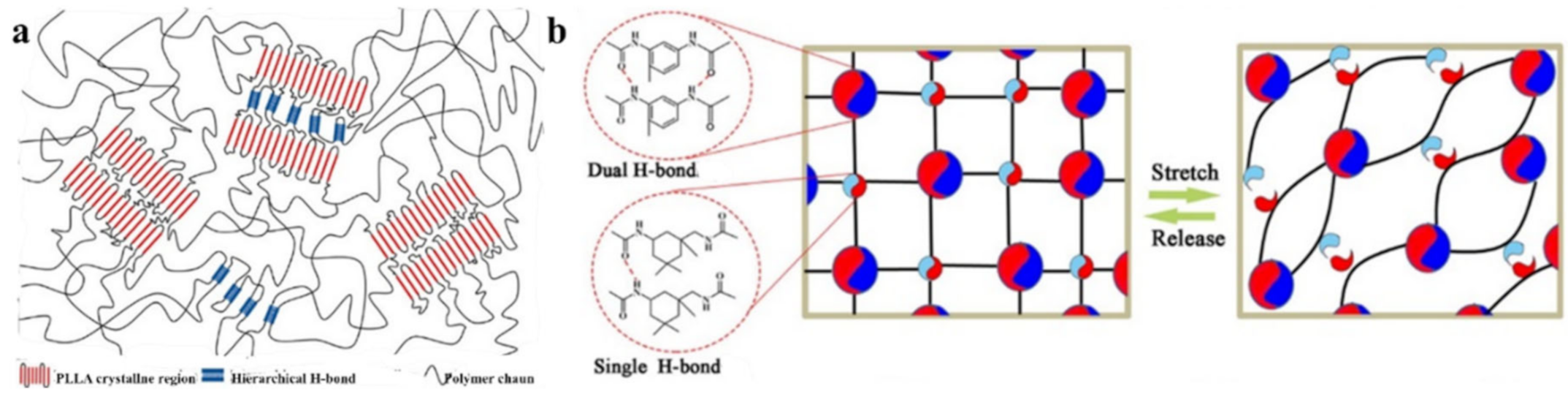{kind=link}
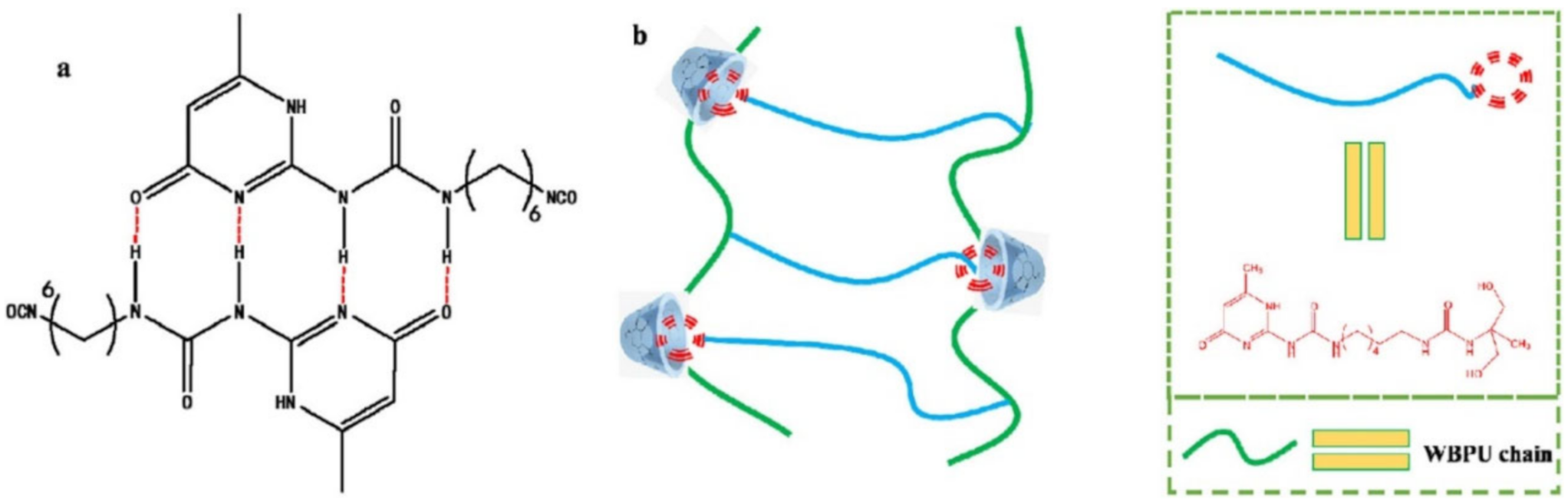{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
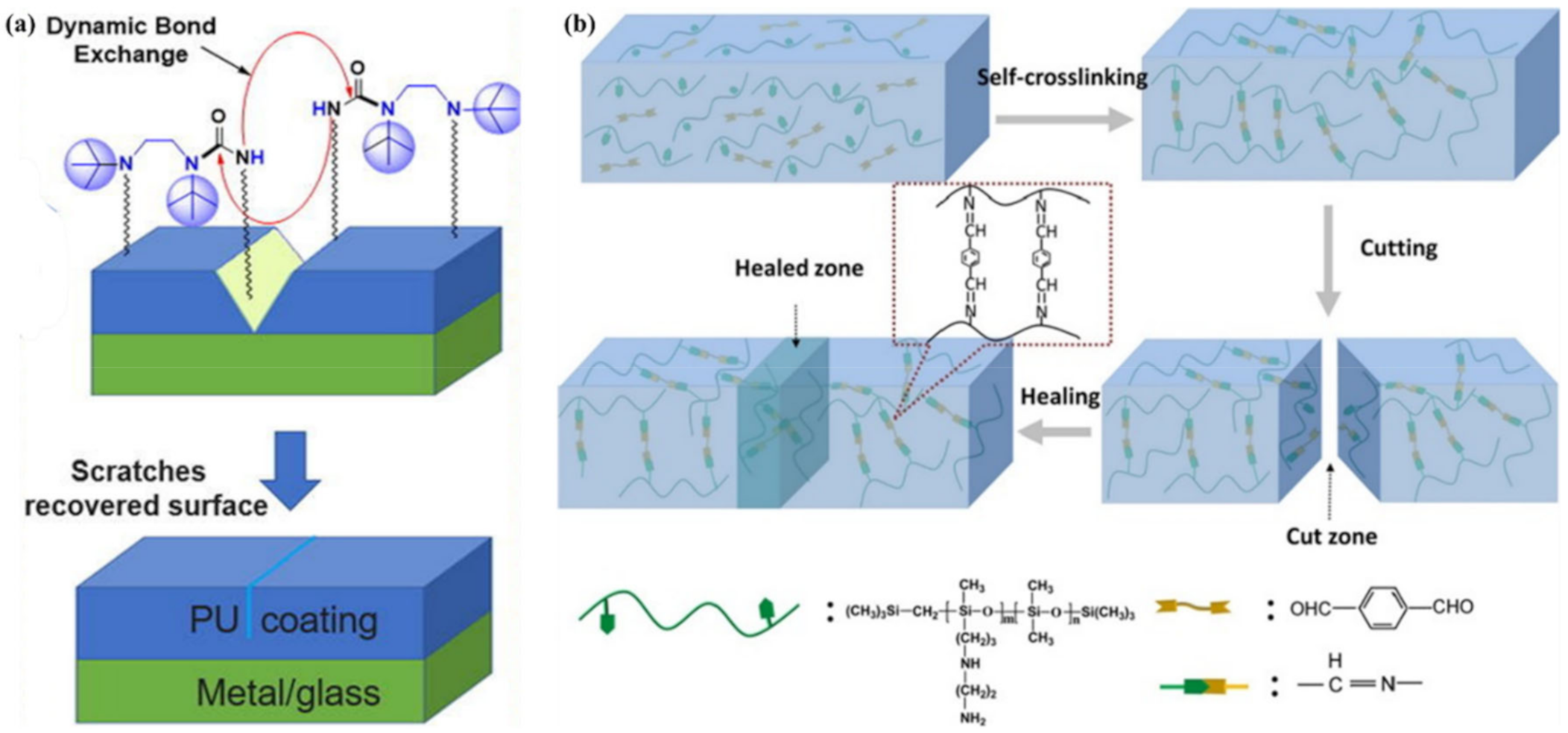{kind=link}
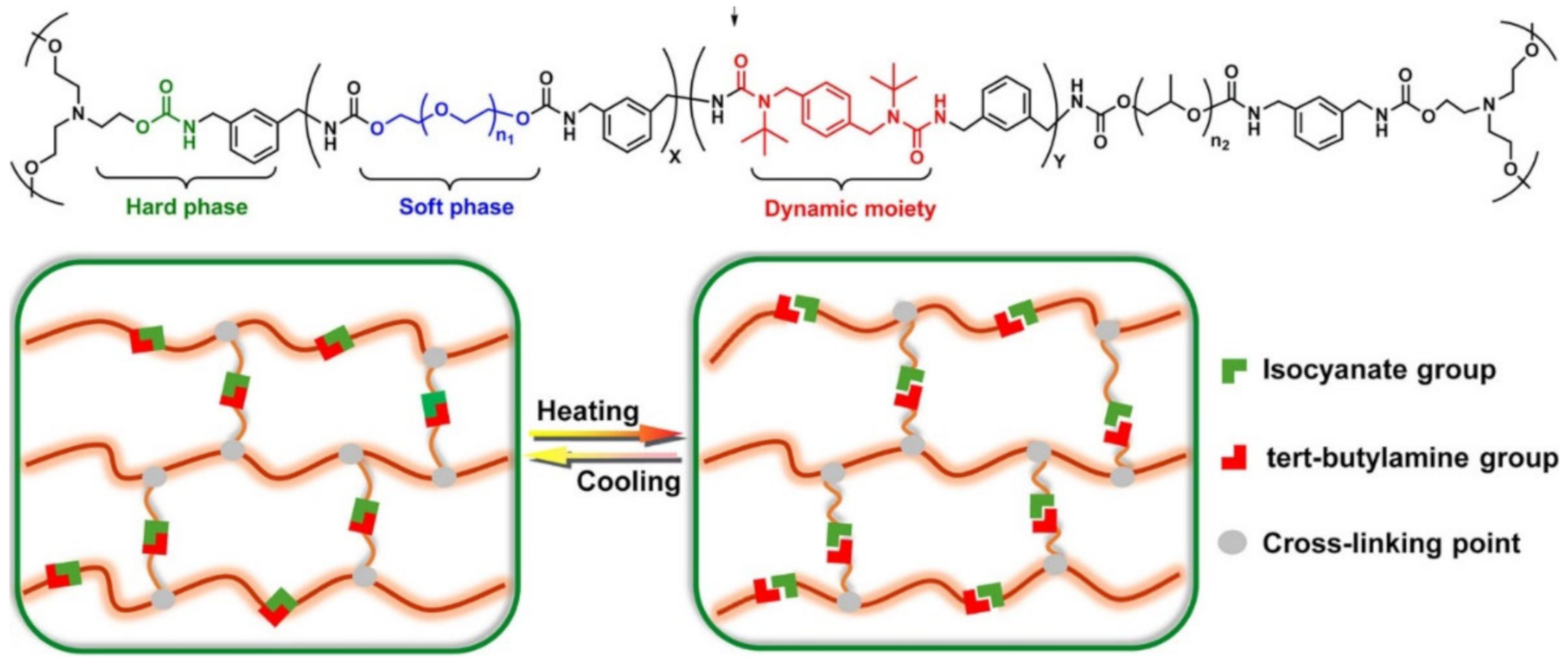{kind=link}
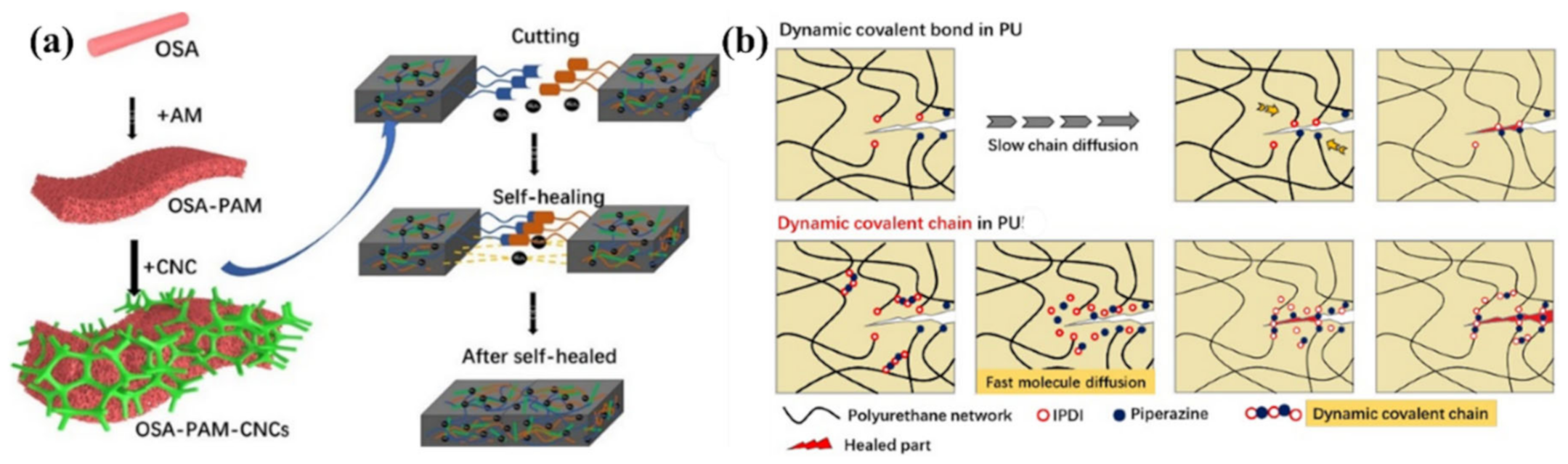{kind=link}
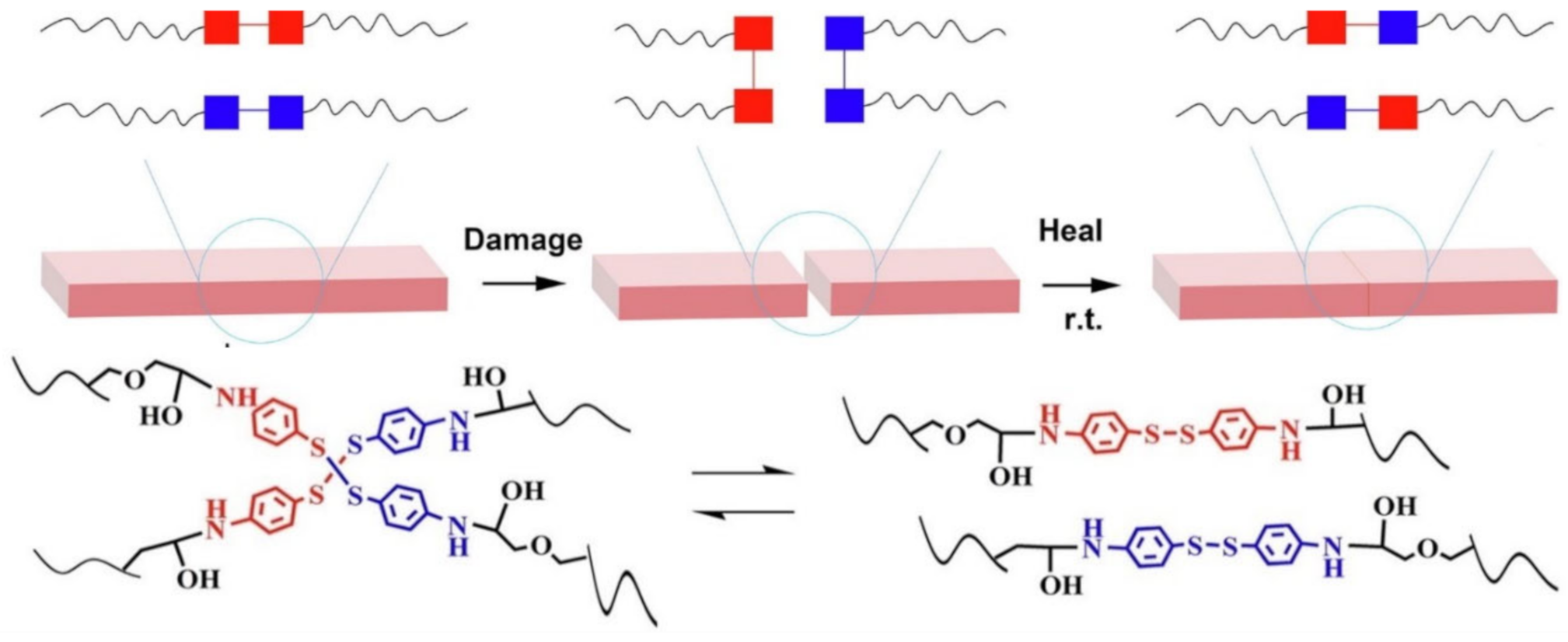{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
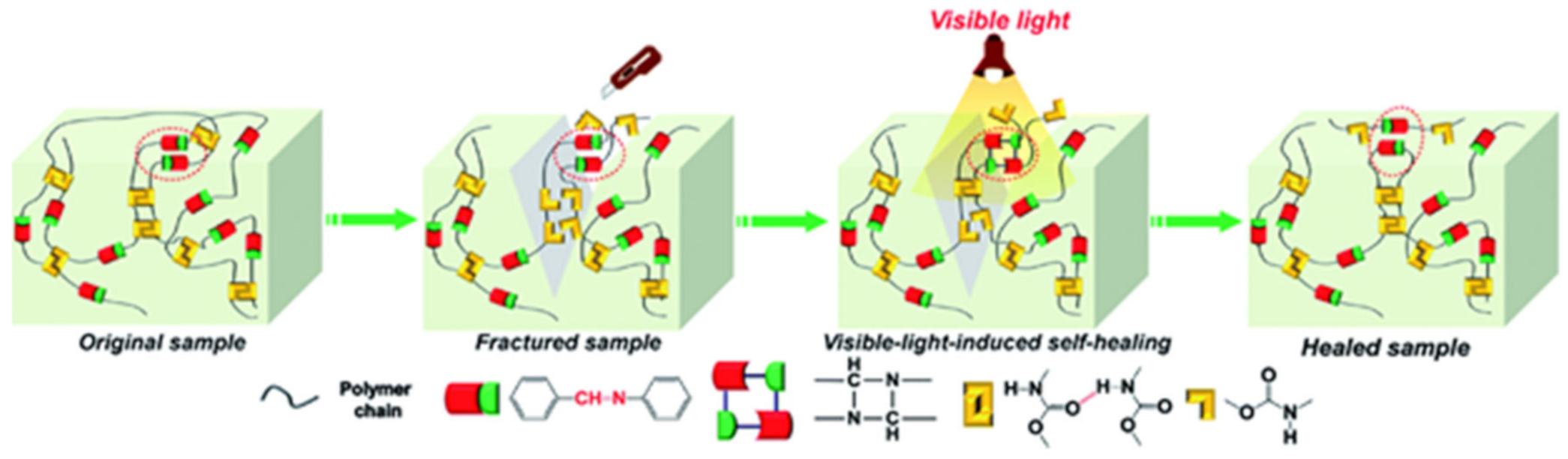{kind=link}
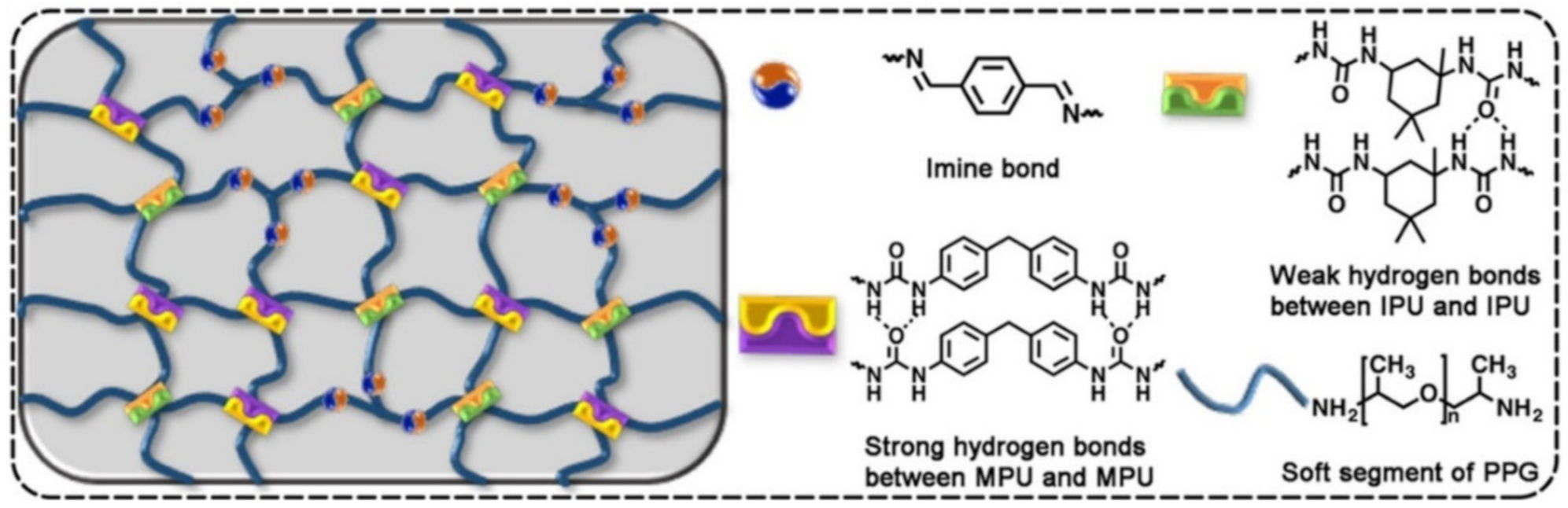{kind=link}
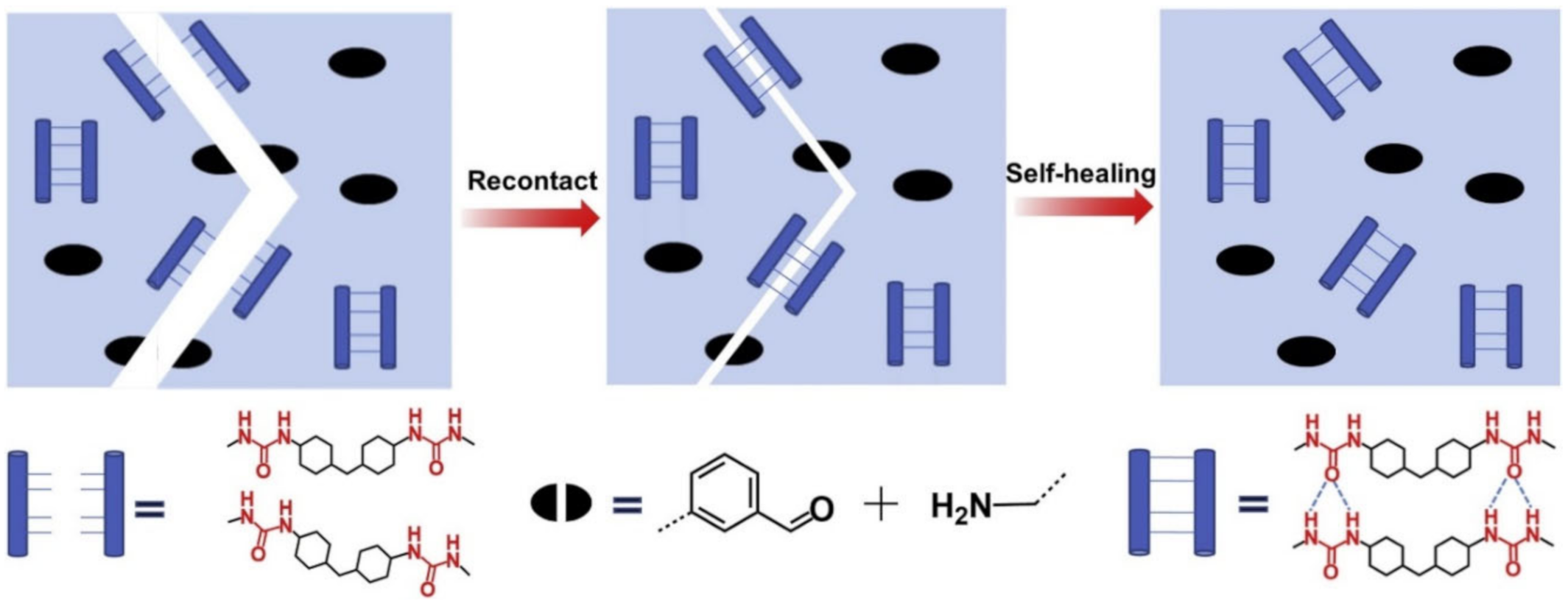{kind=link}
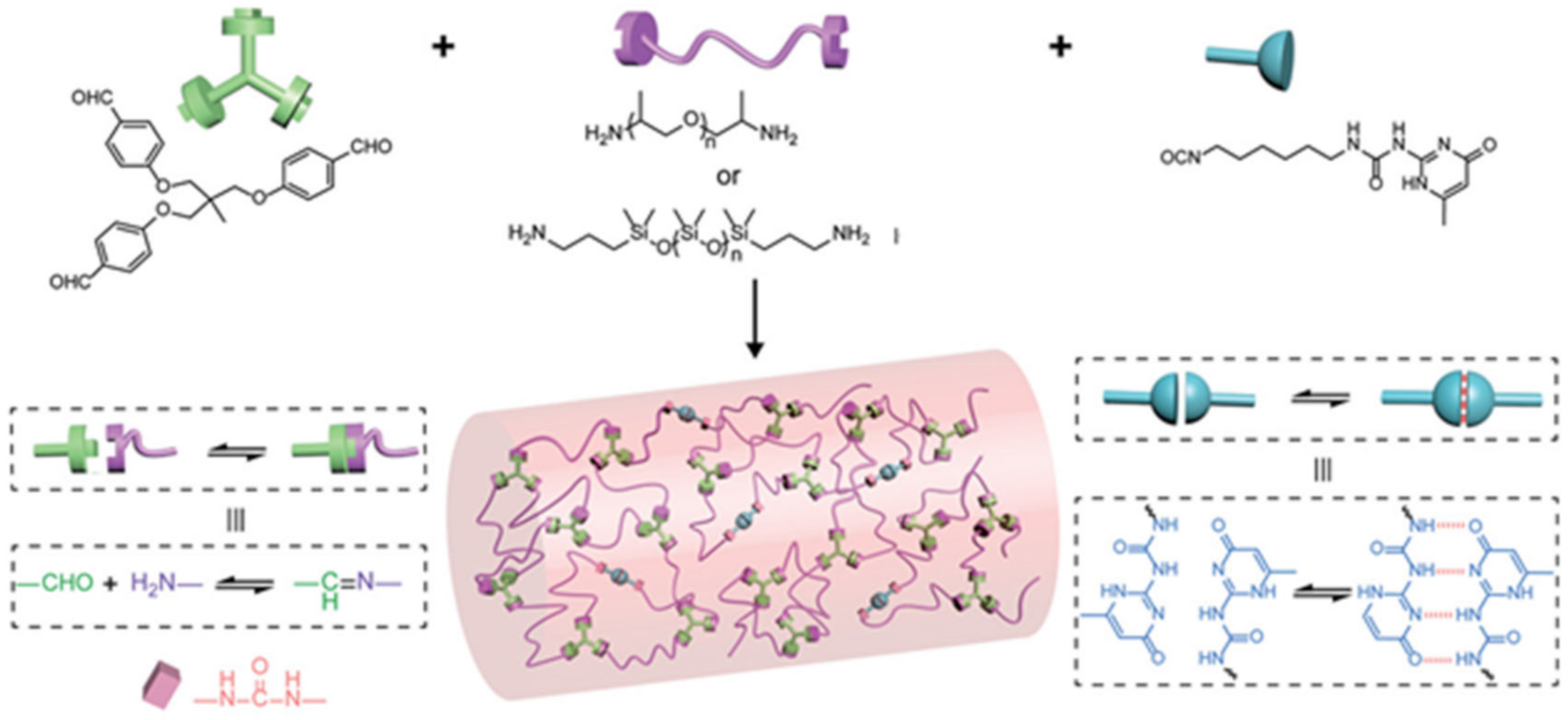{kind=link}
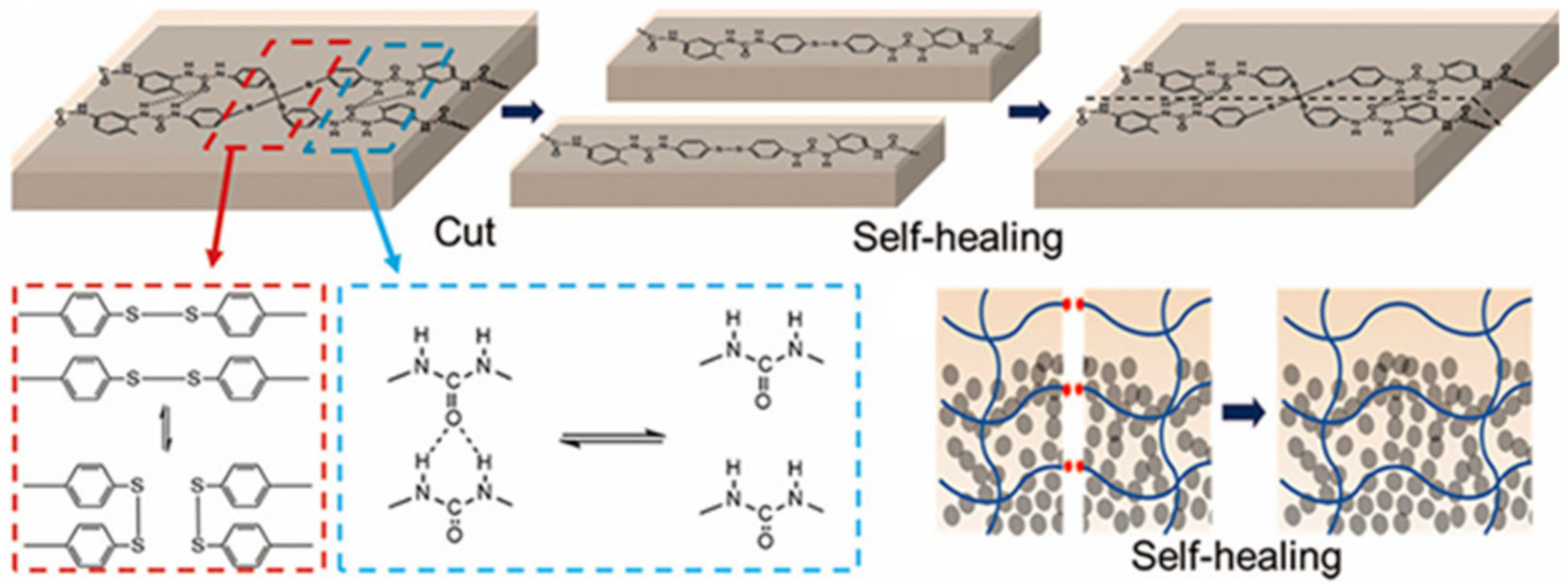{kind=link}
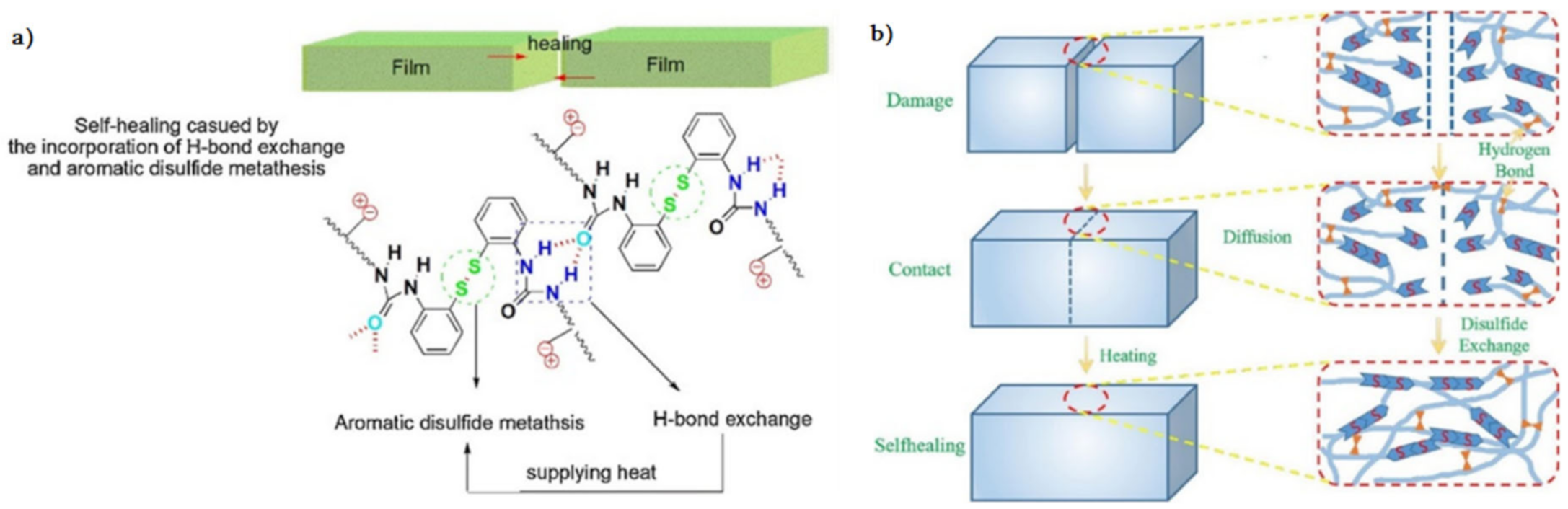{kind=link}
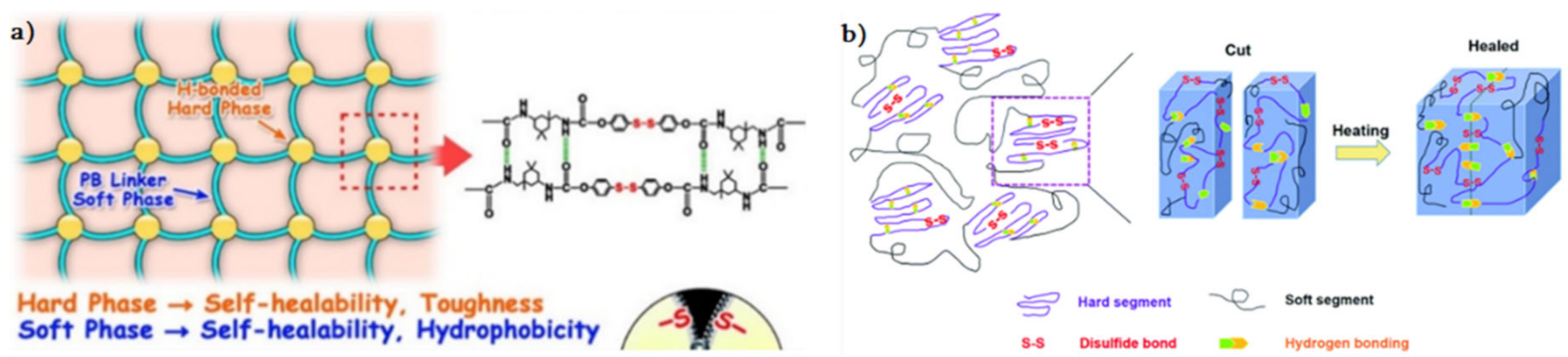{kind=link}
{kind=link}
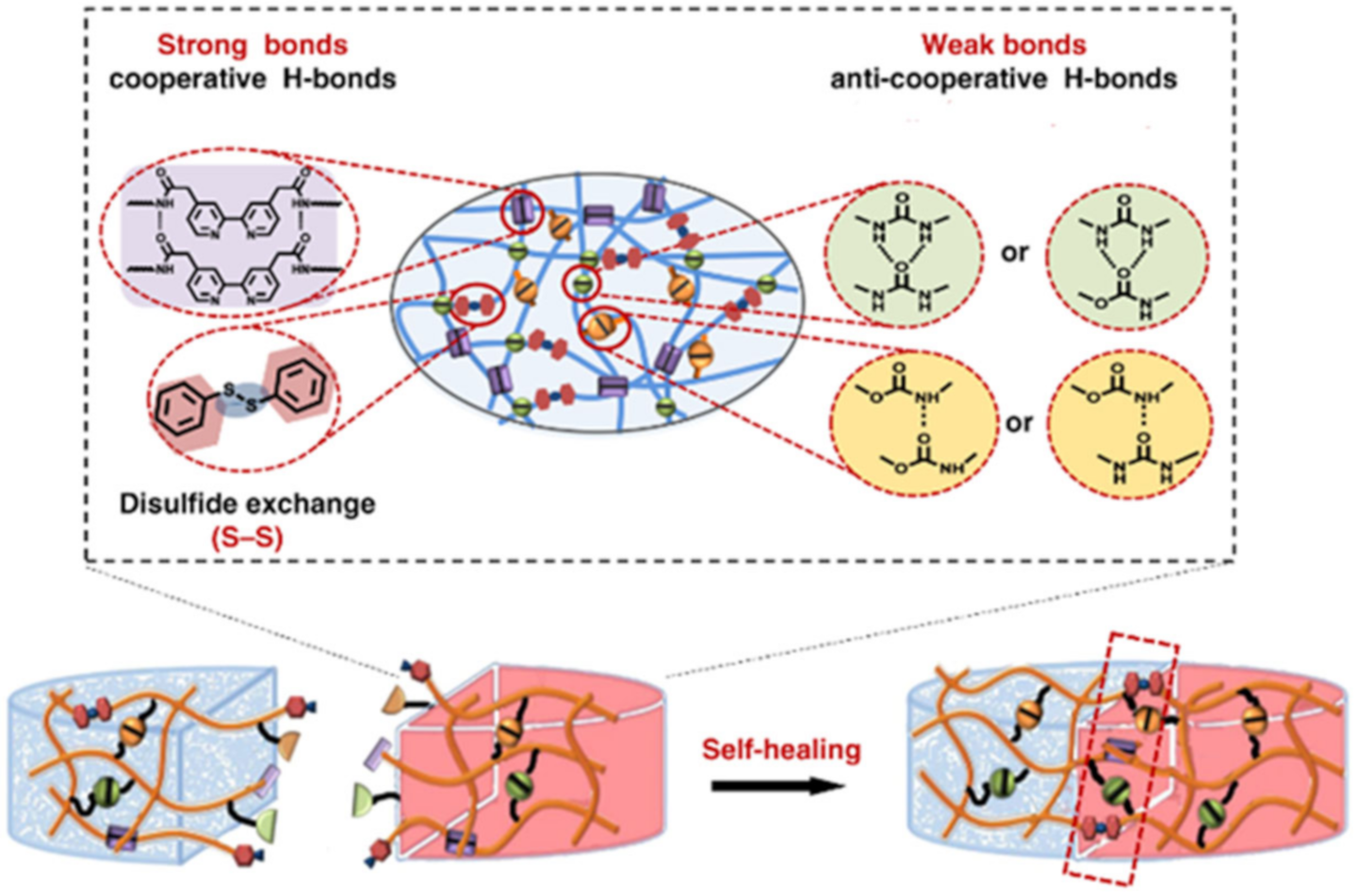{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
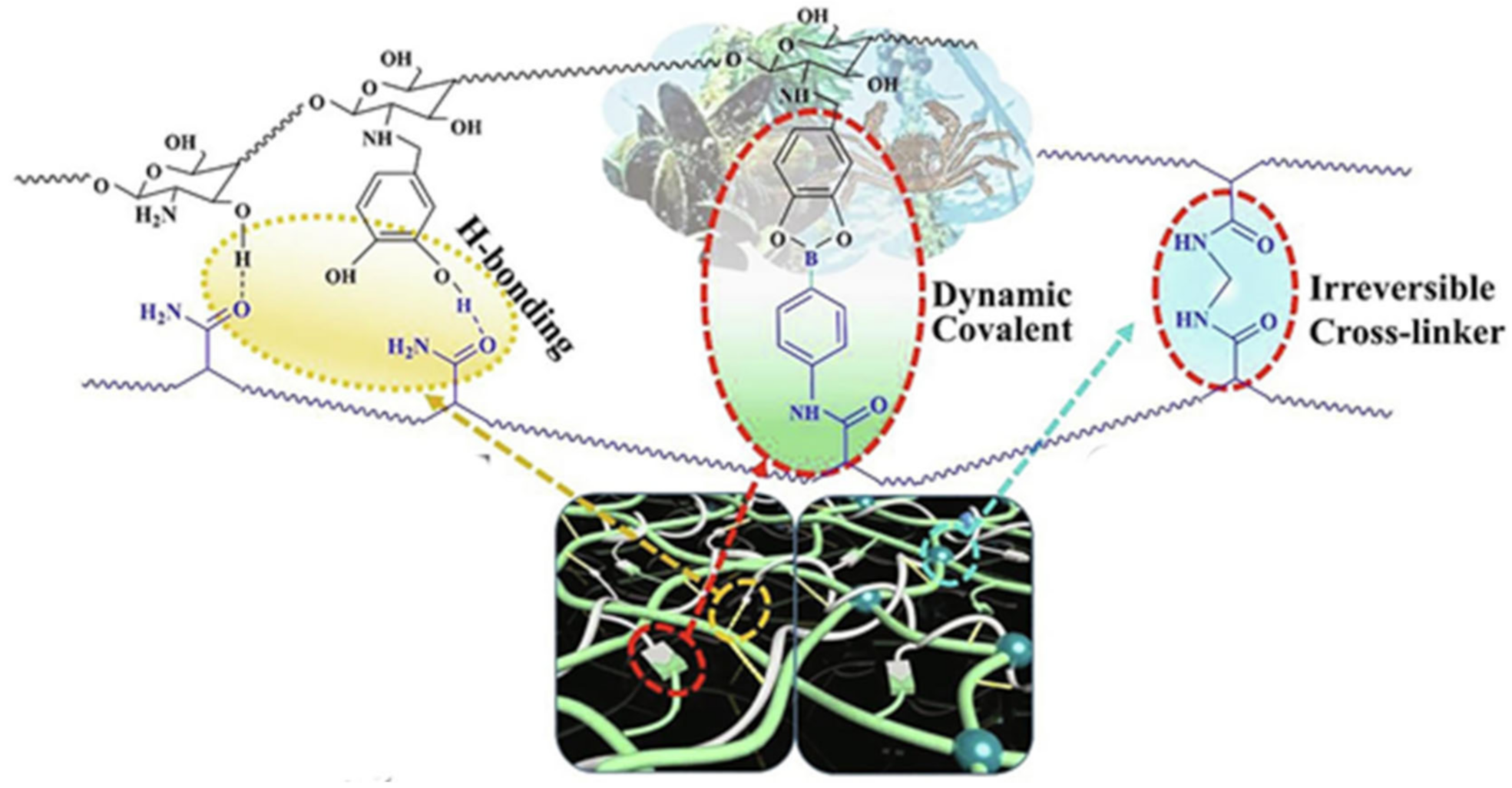{kind=link}
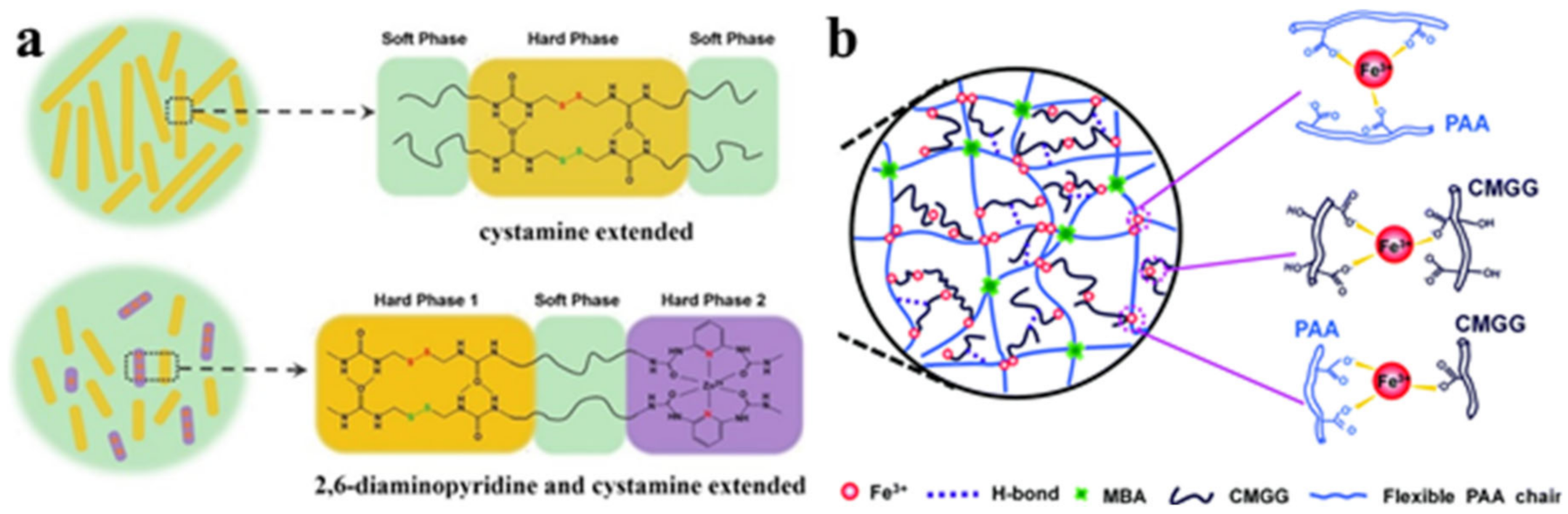{kind=link}
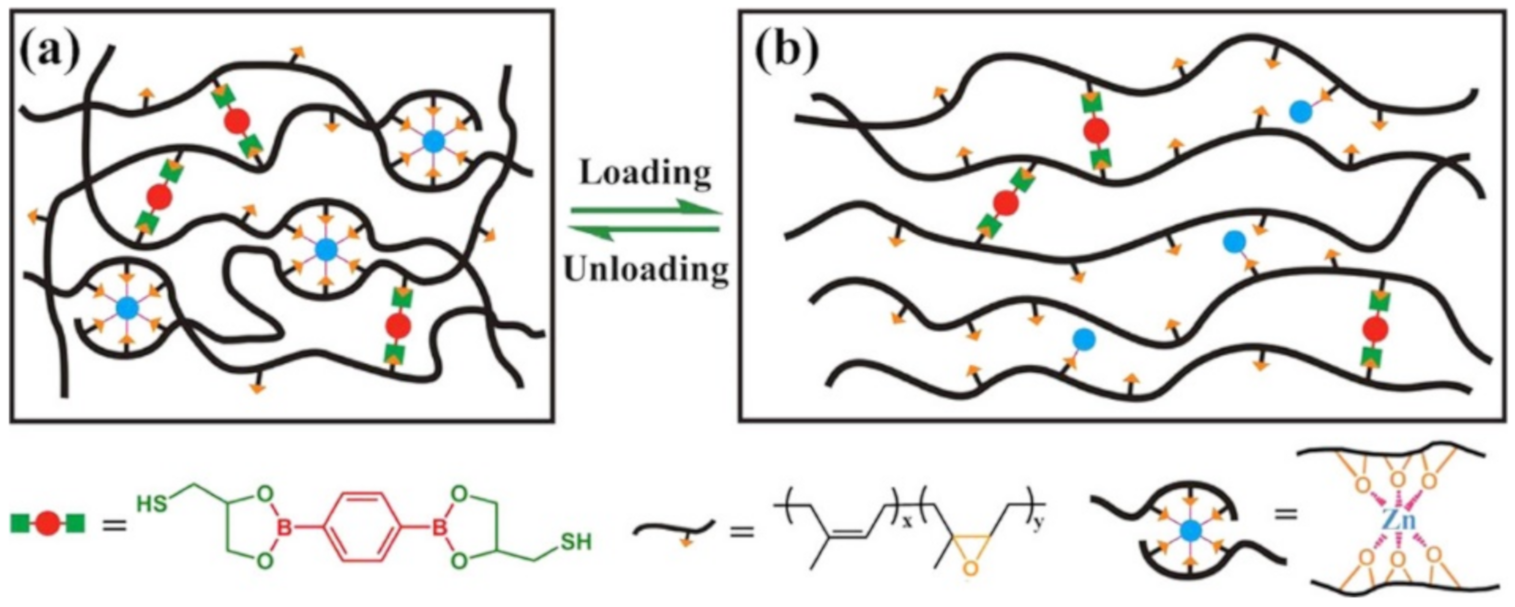{kind=link}
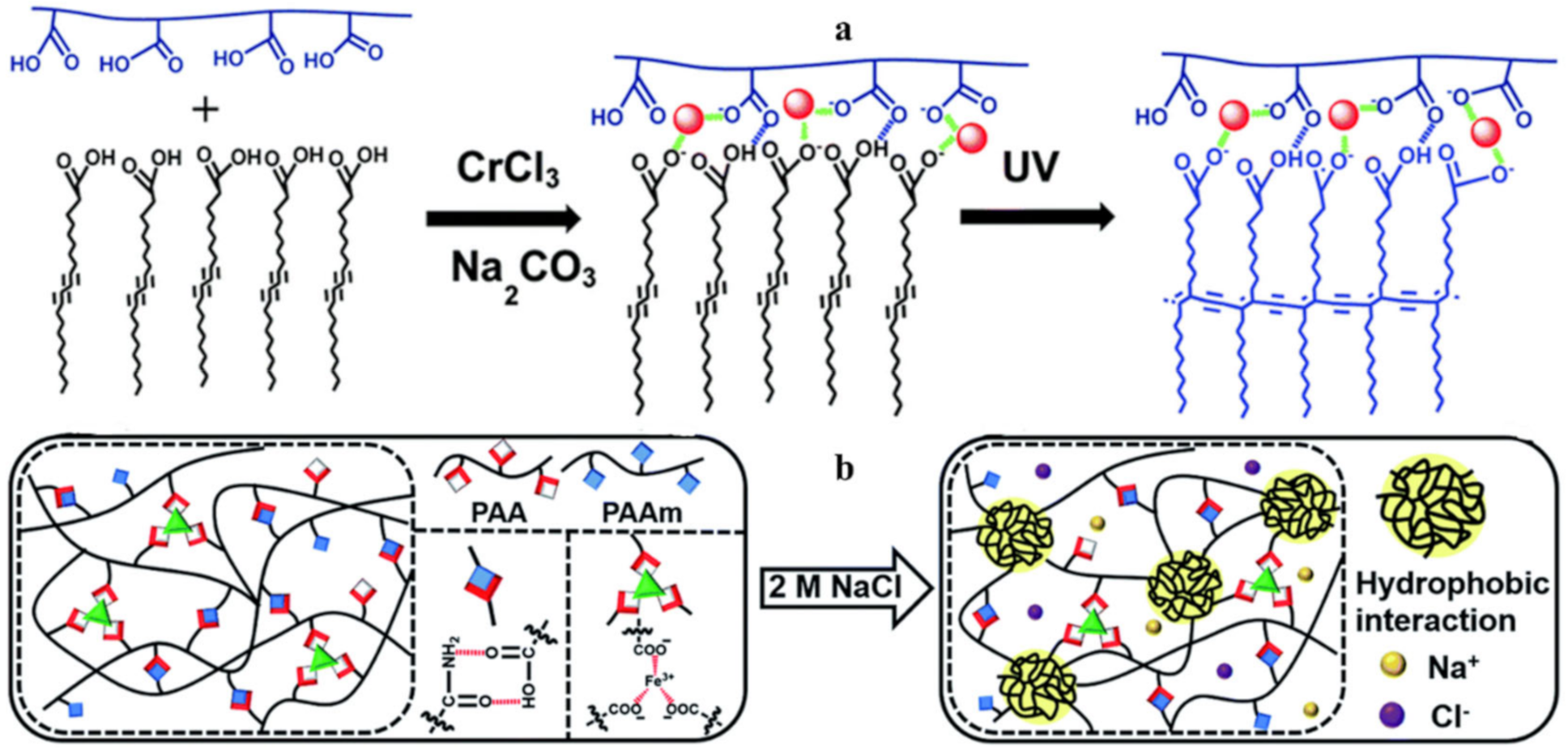{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
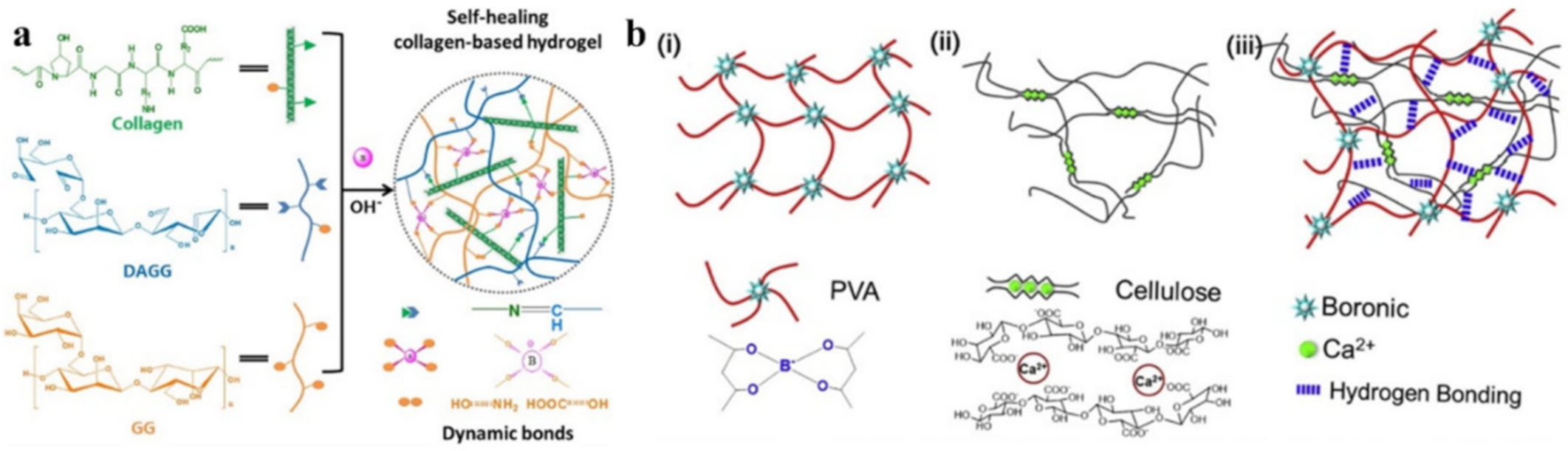{kind=link}
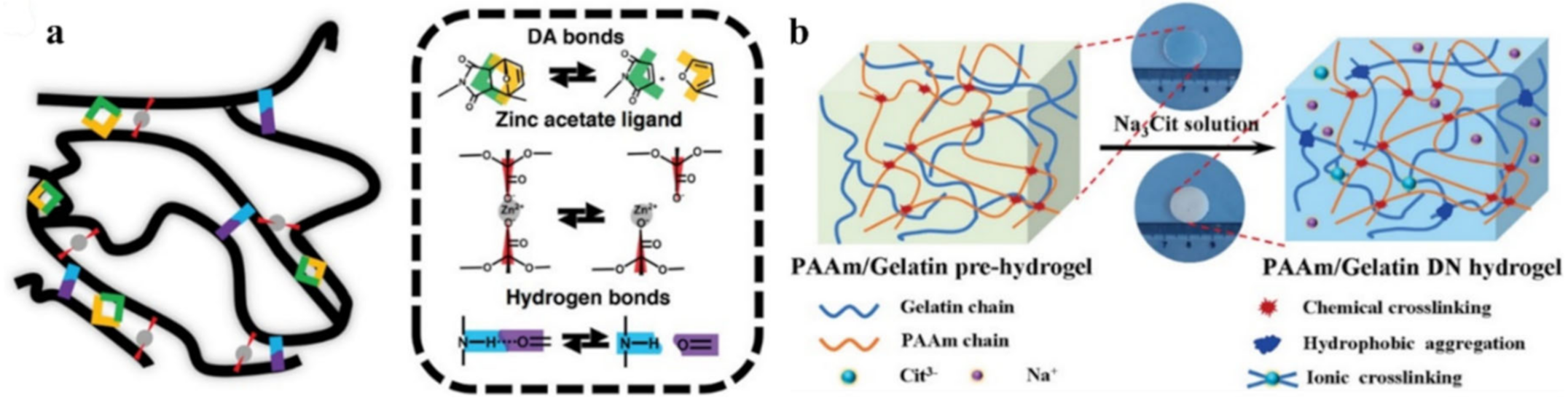{kind=link}
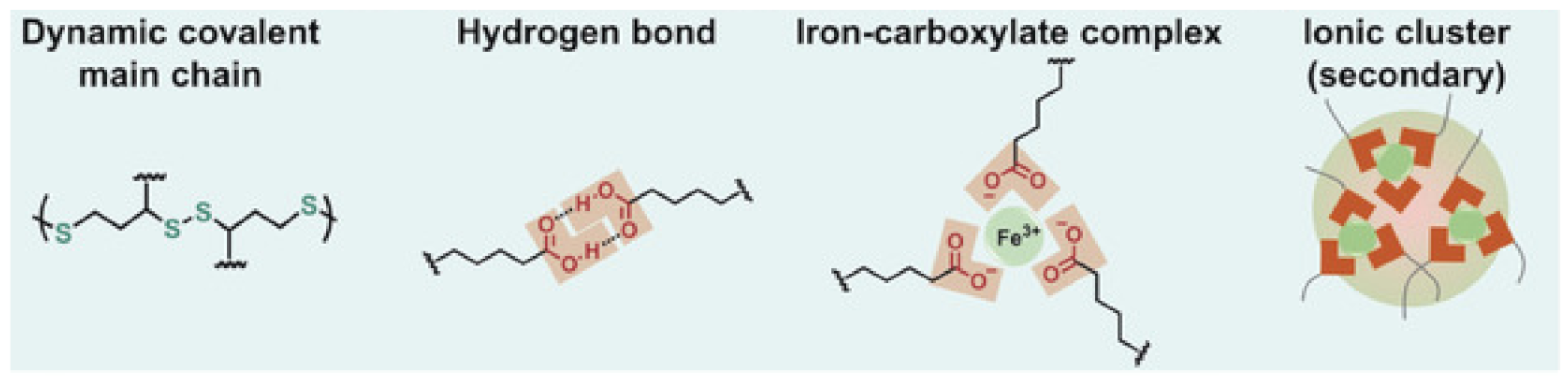{kind=link}
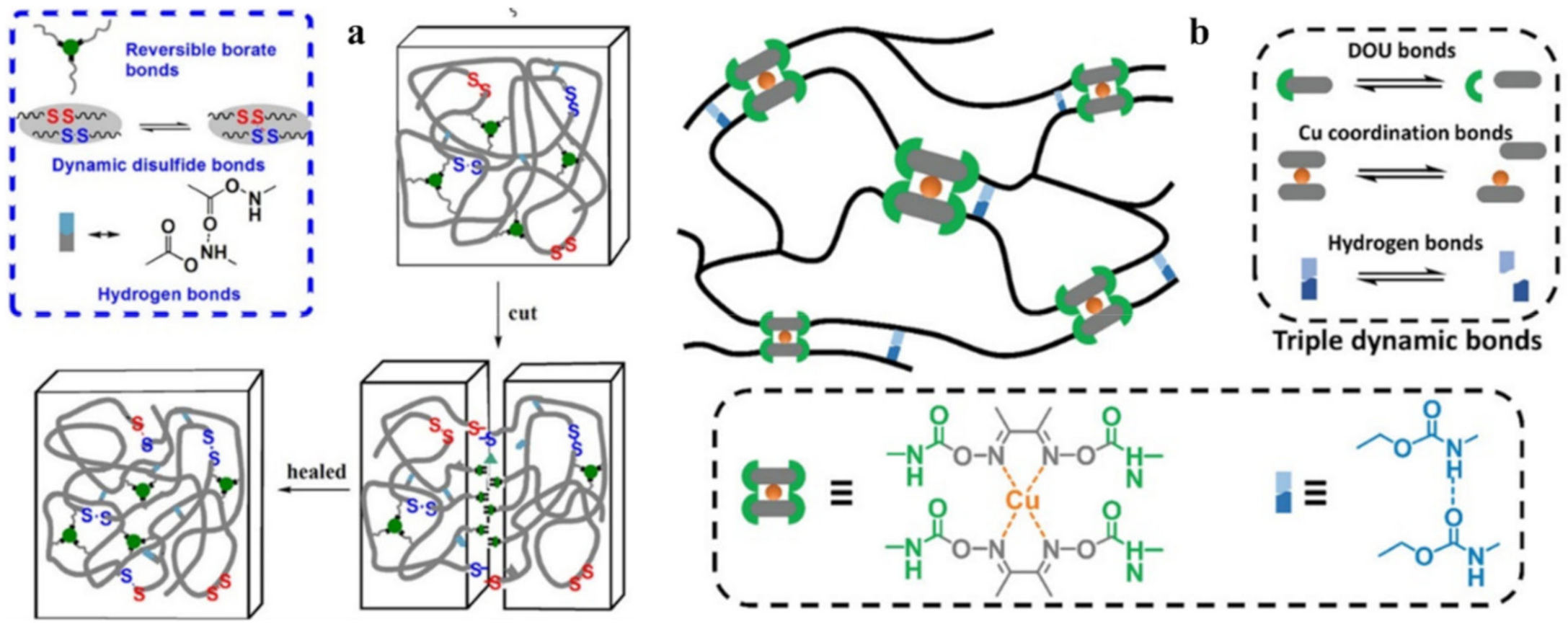{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
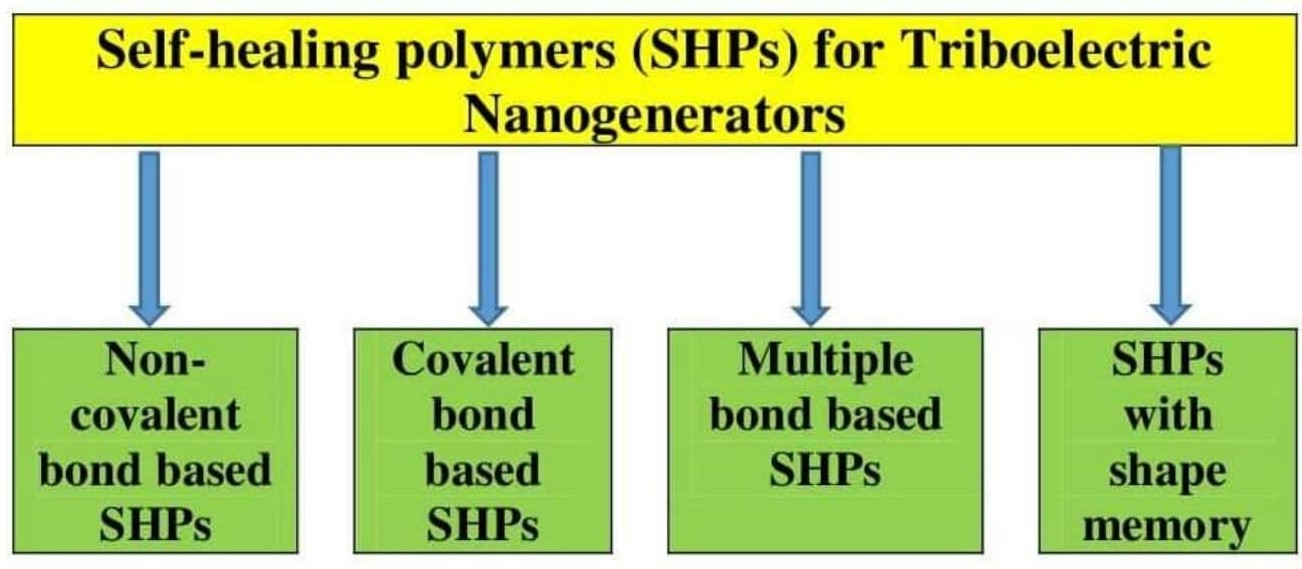
2. Non-Covalent Bond Based SHPs
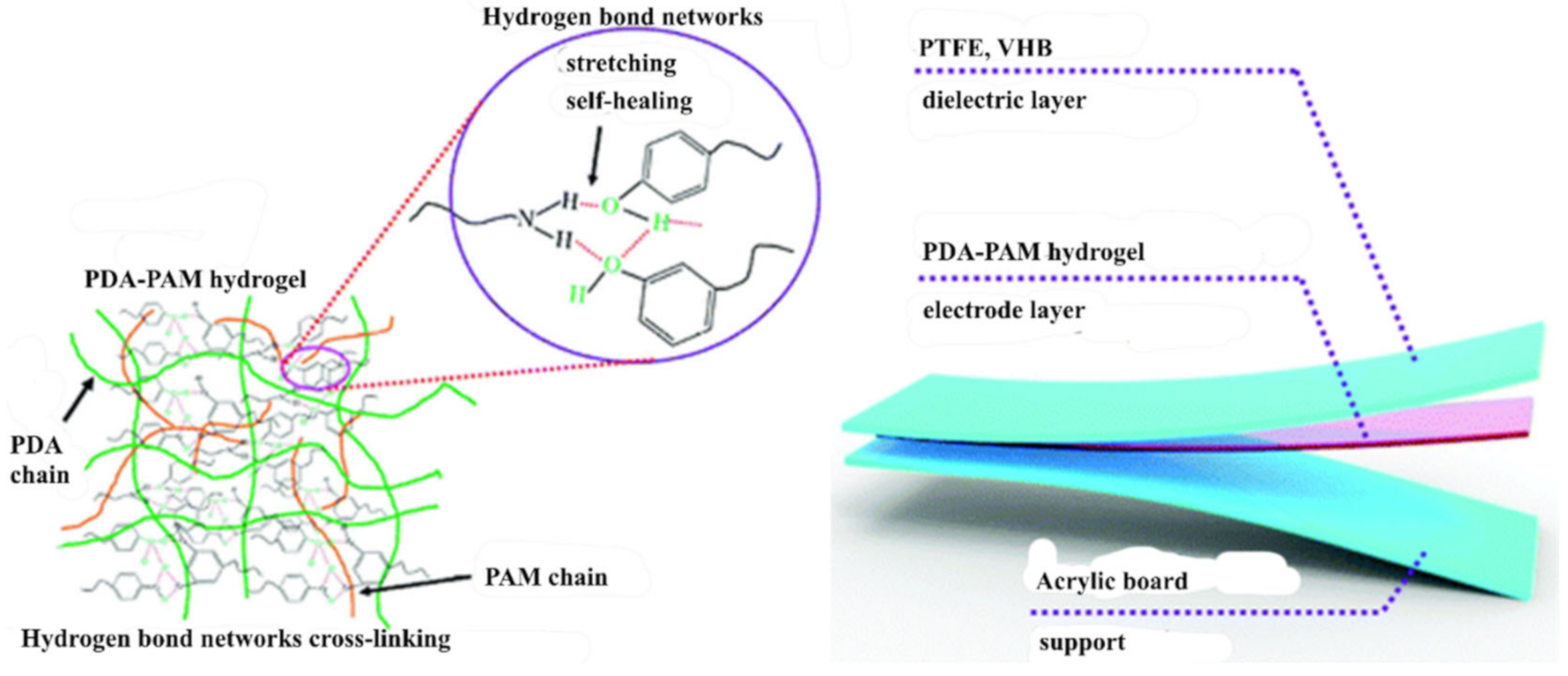
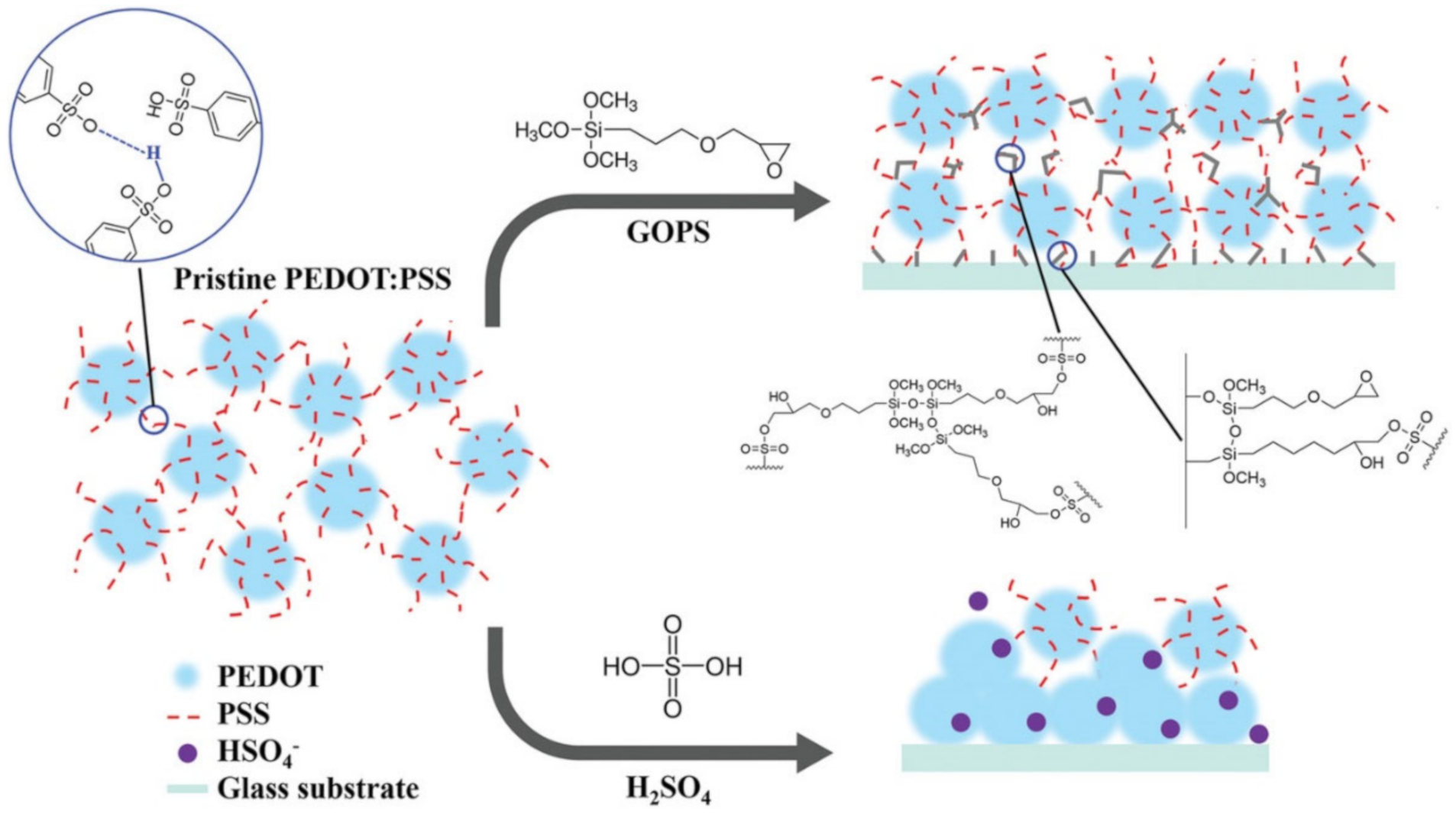
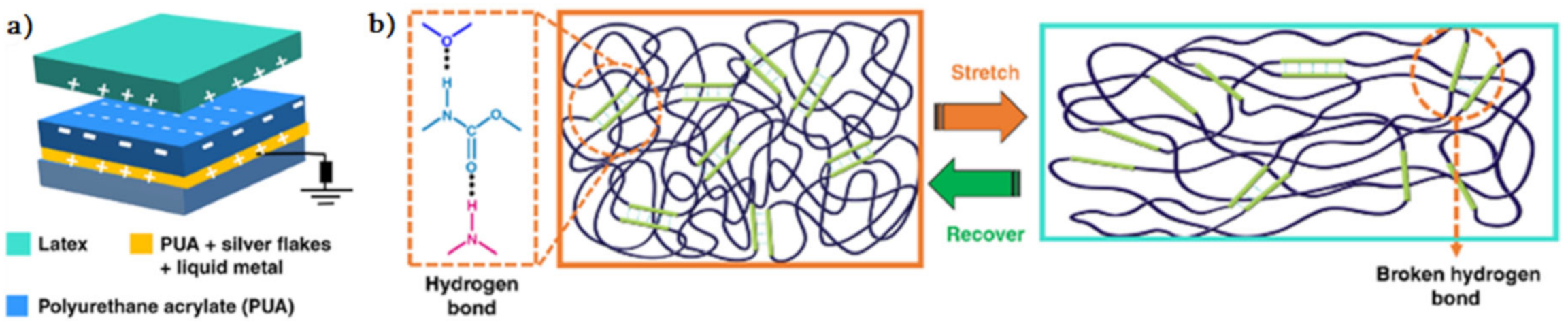
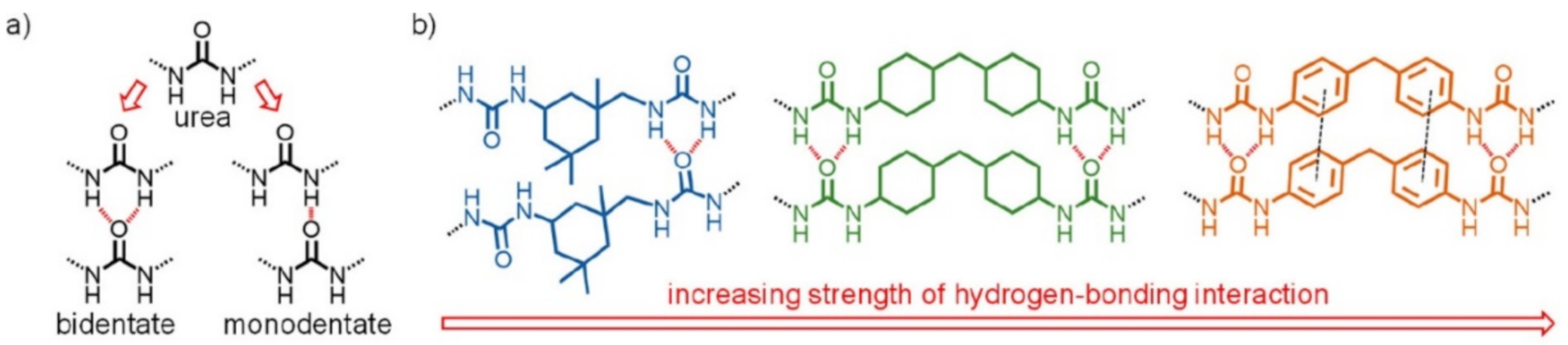
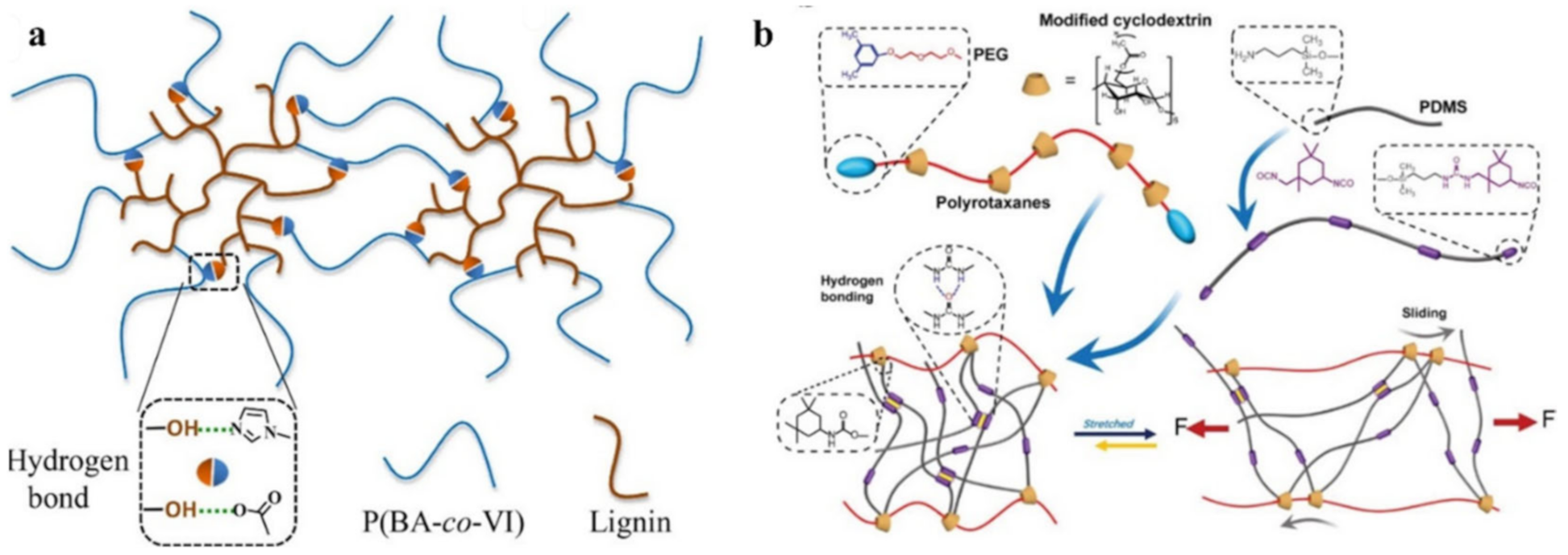
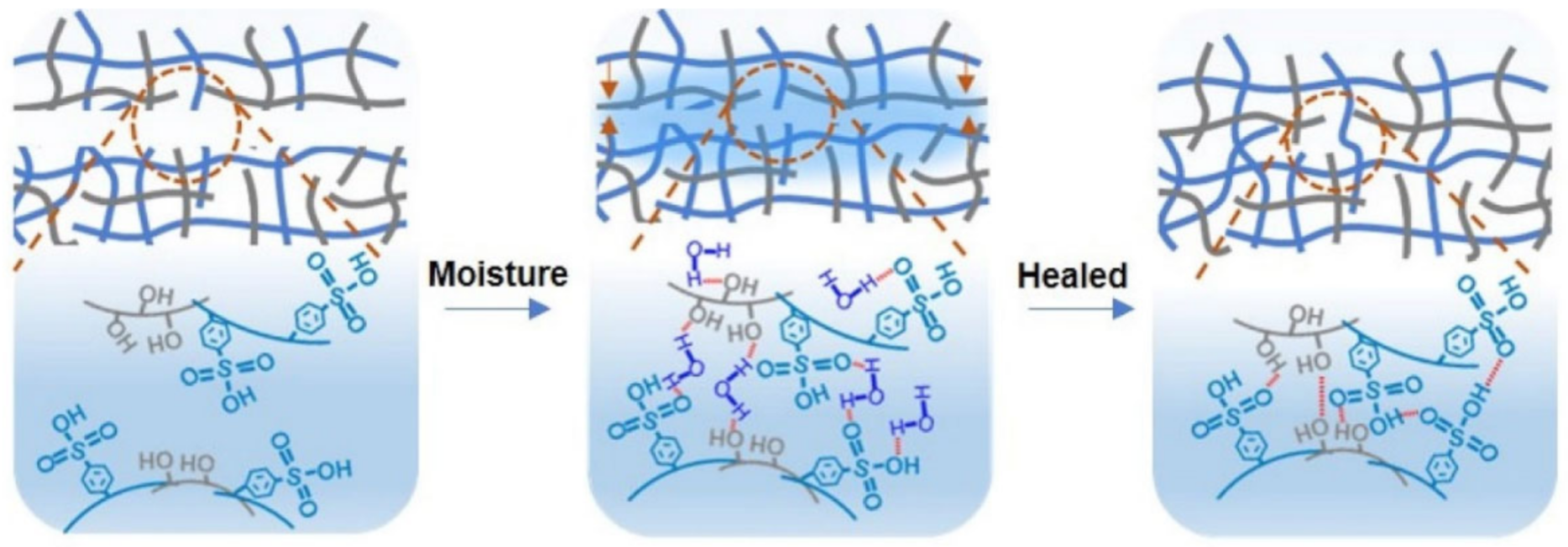
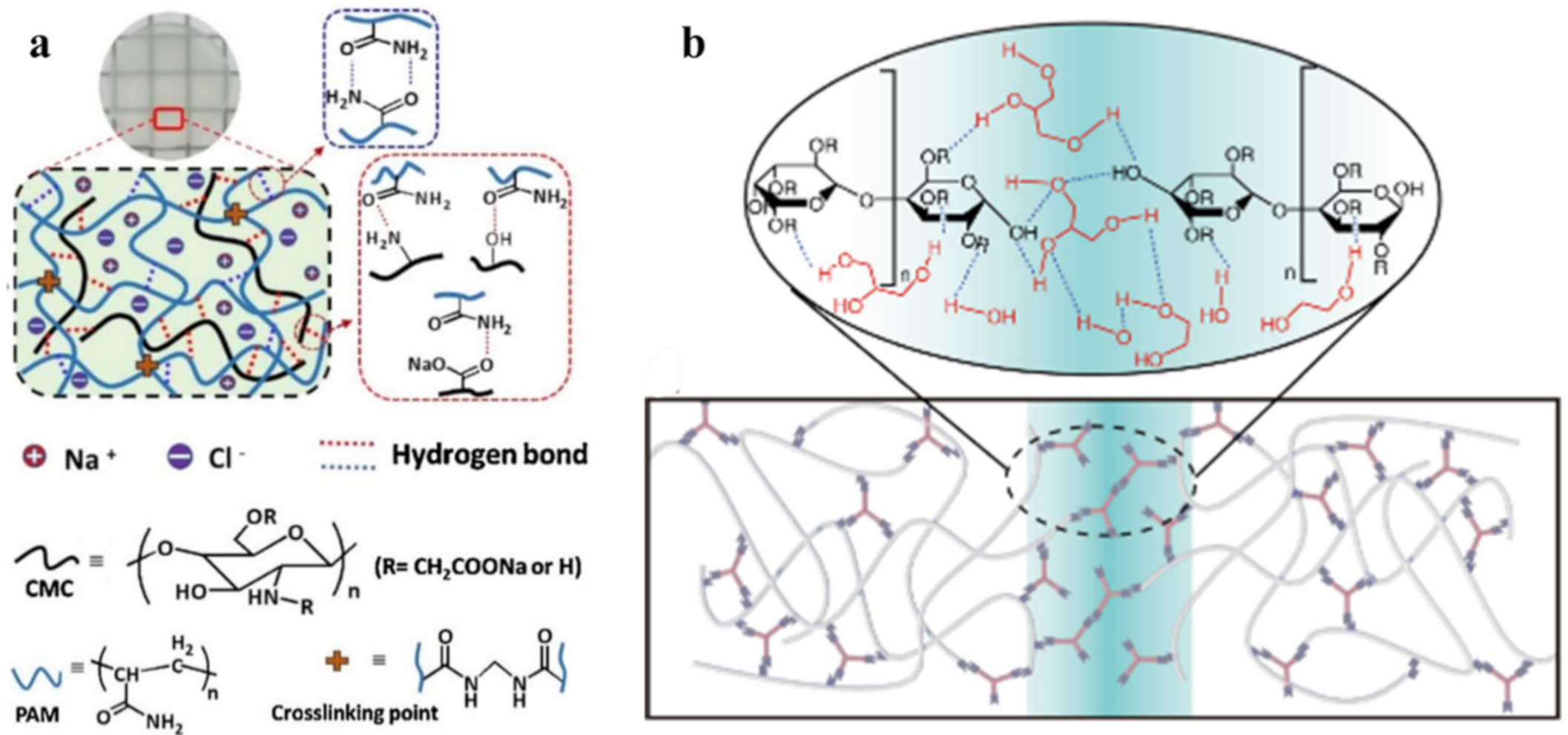
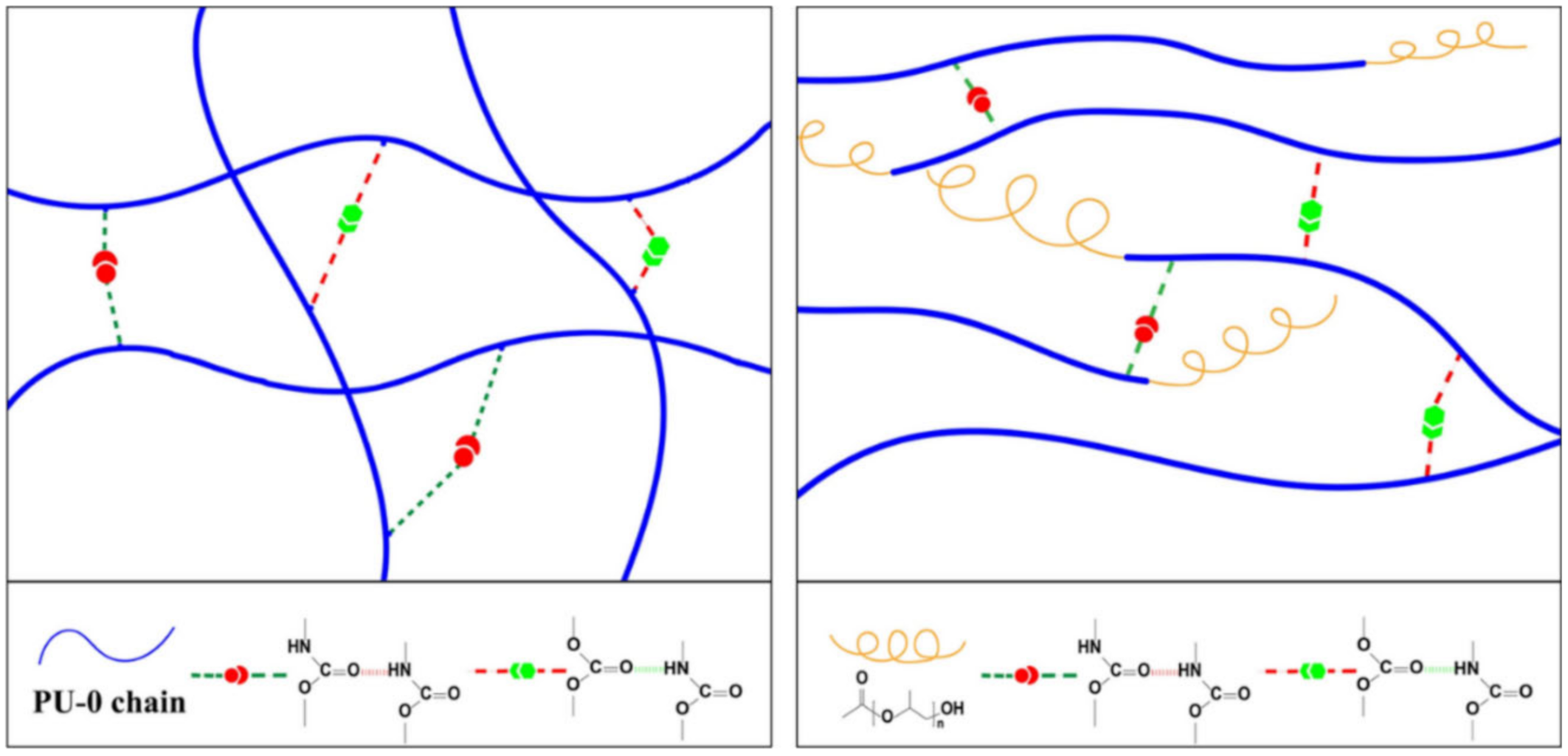
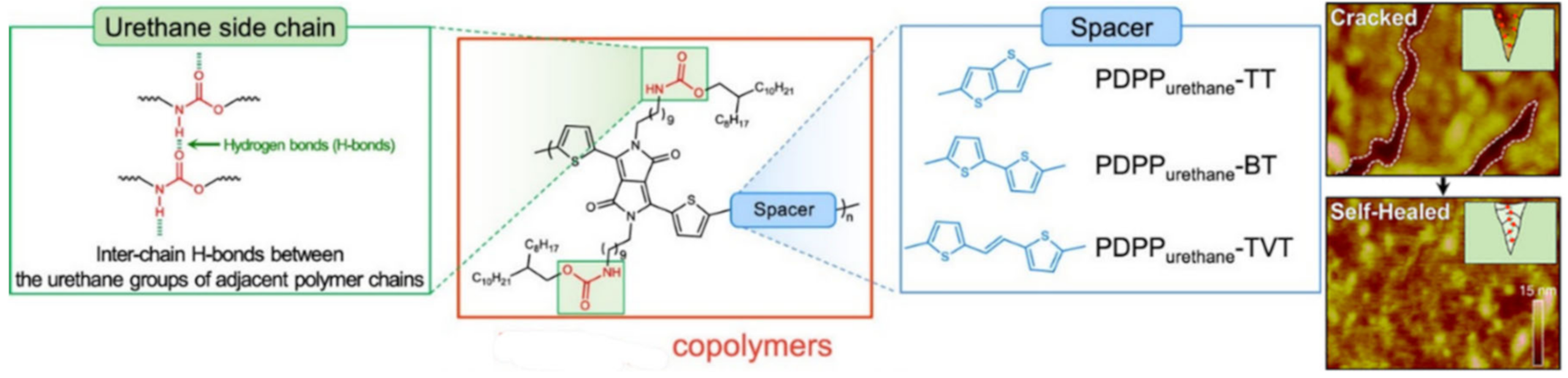
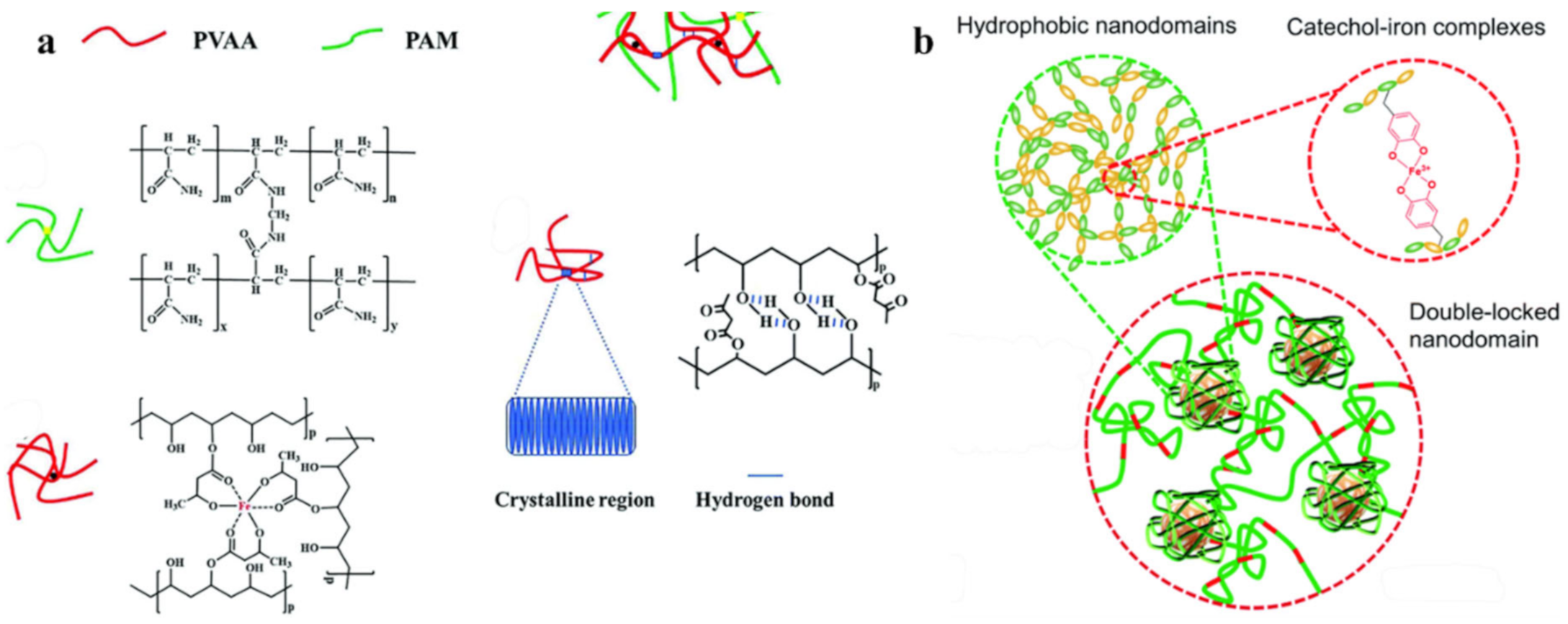
2.1. H-Bond Based SHPs
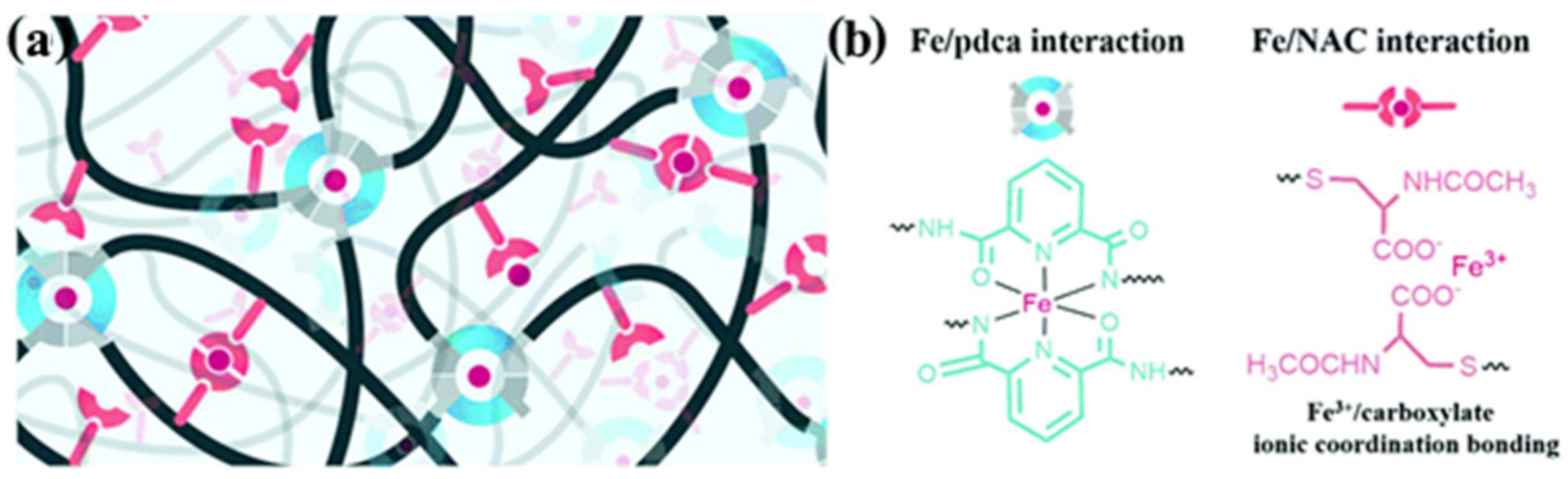
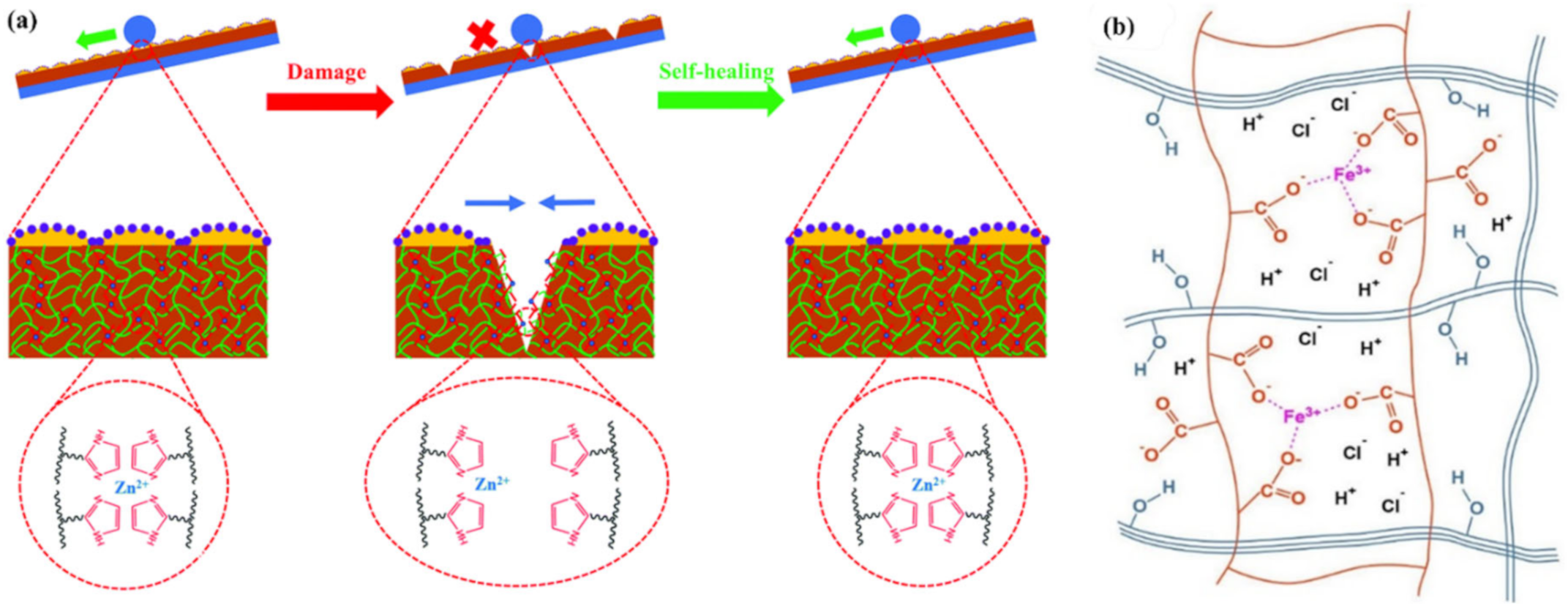
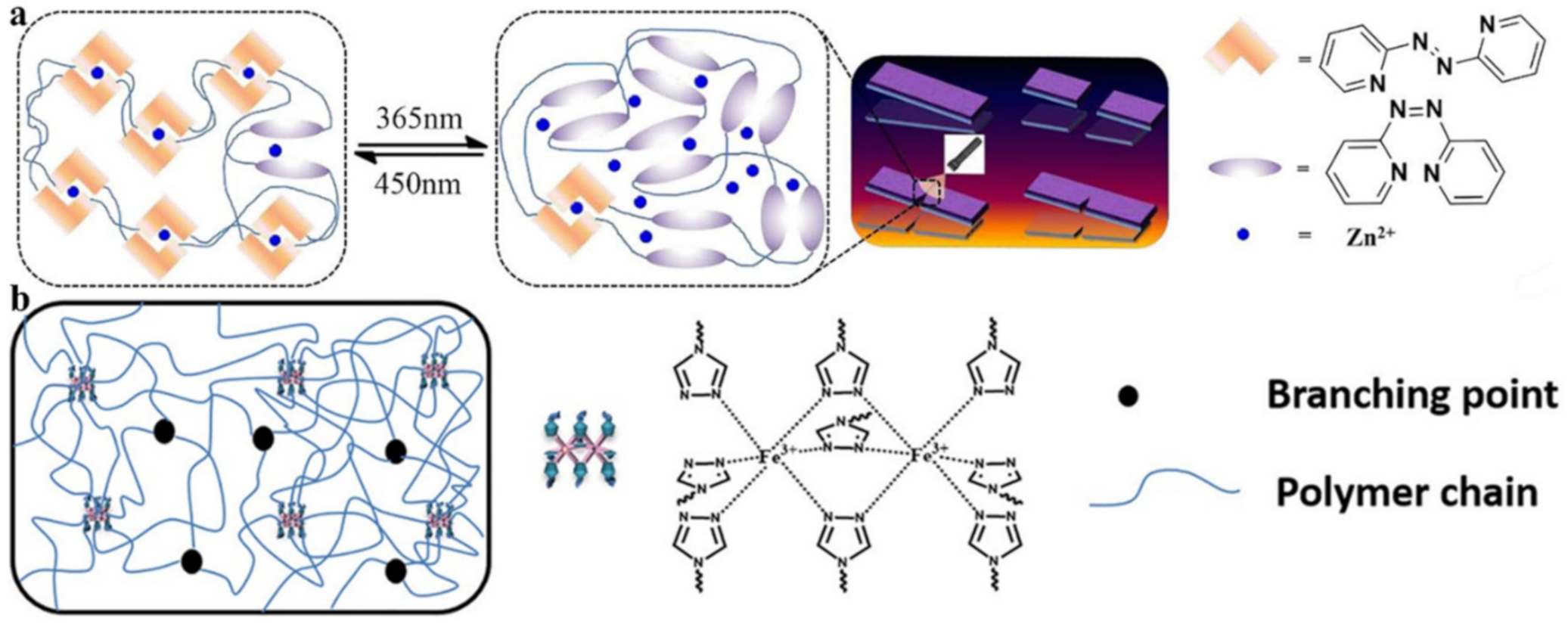
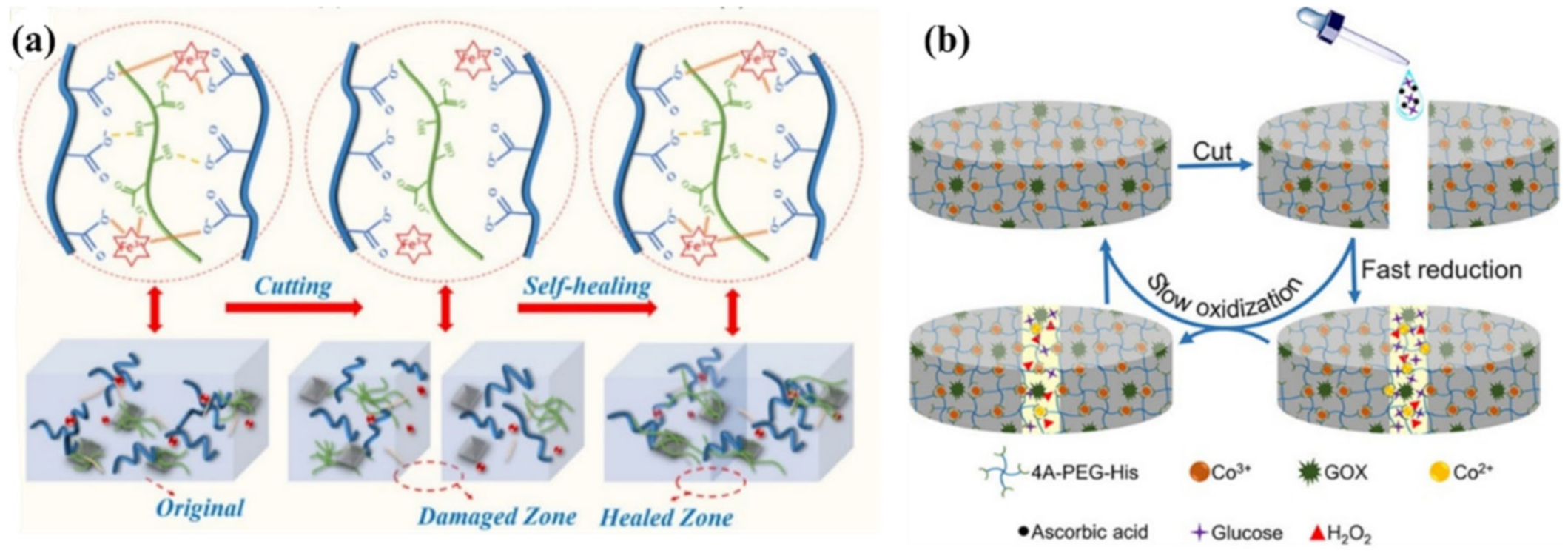
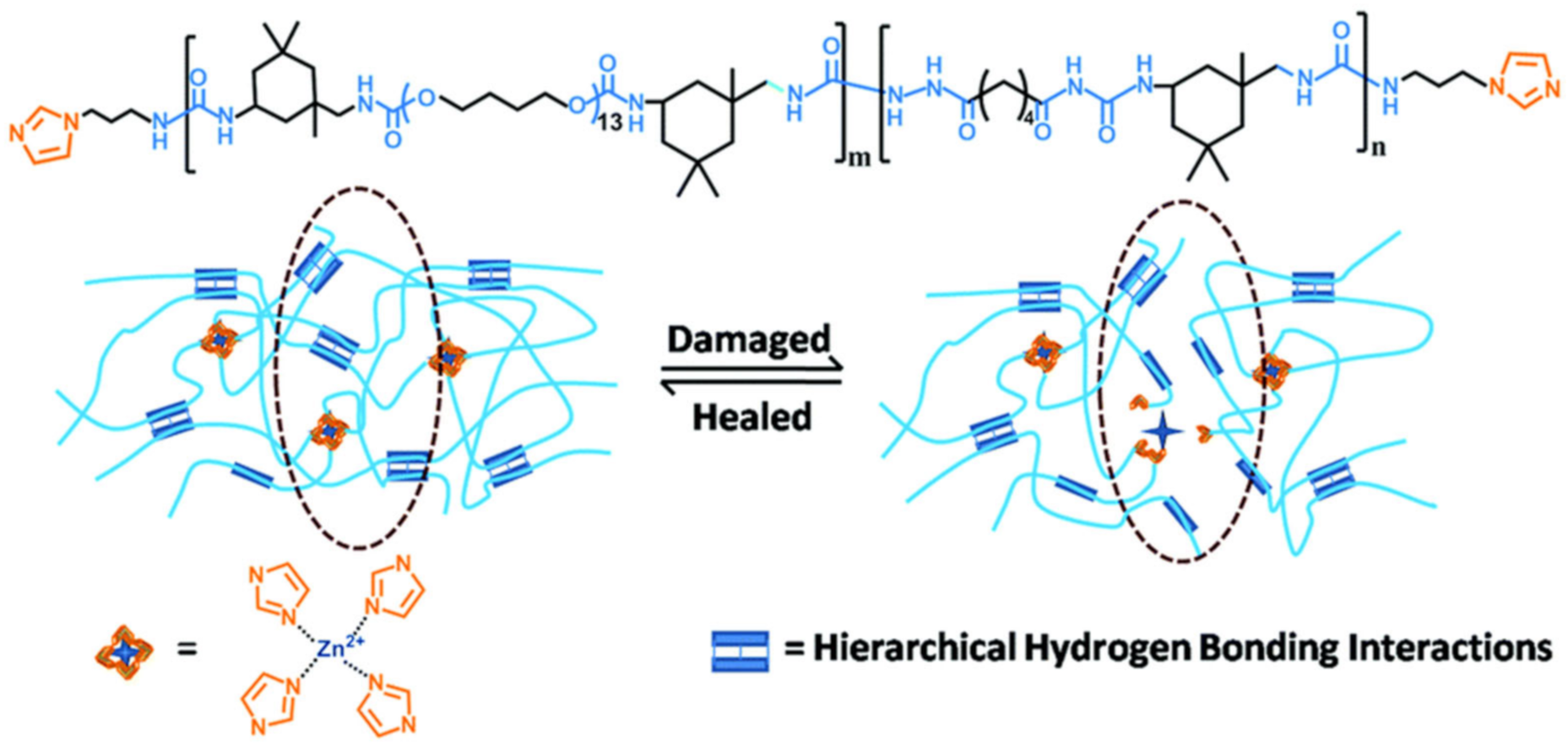
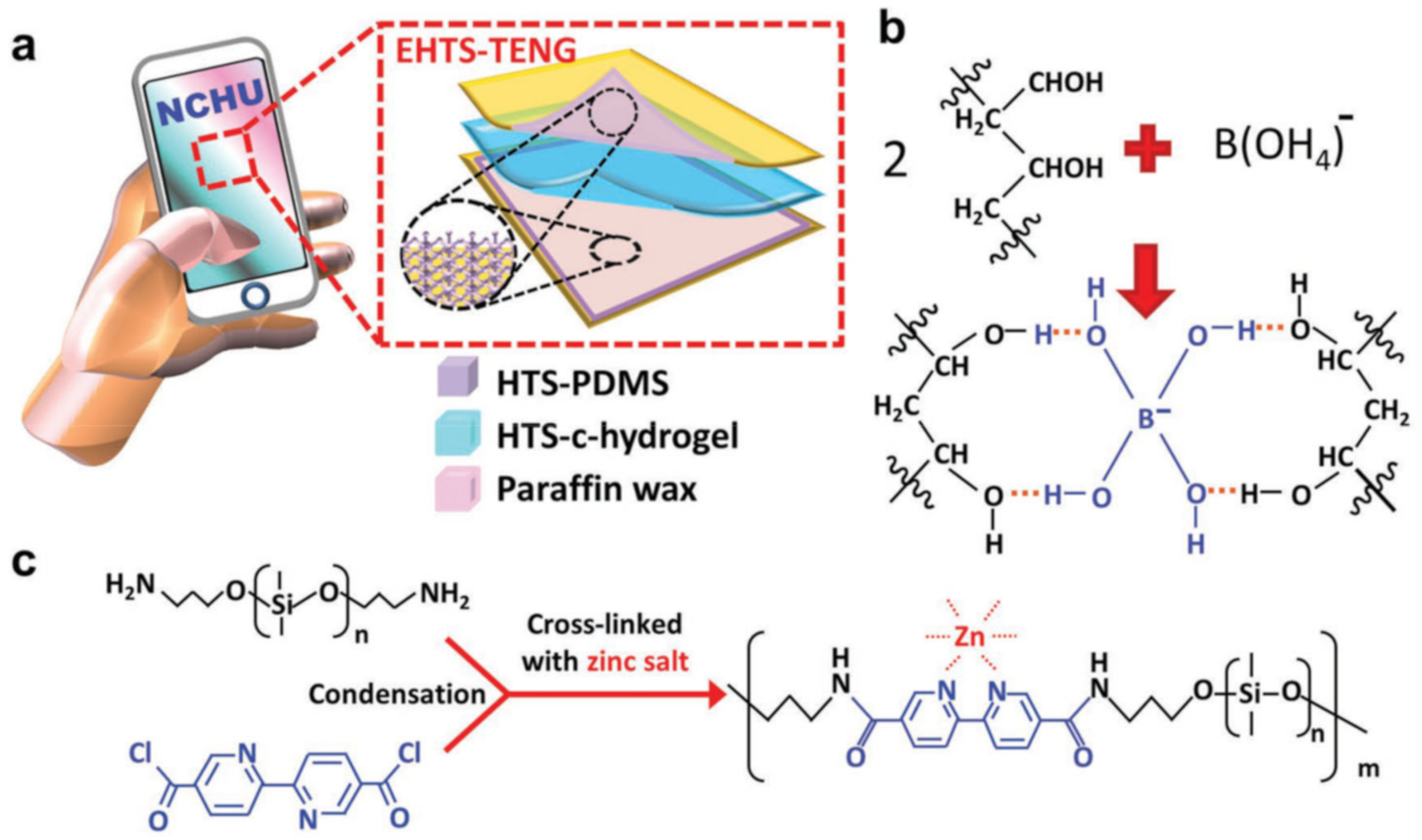
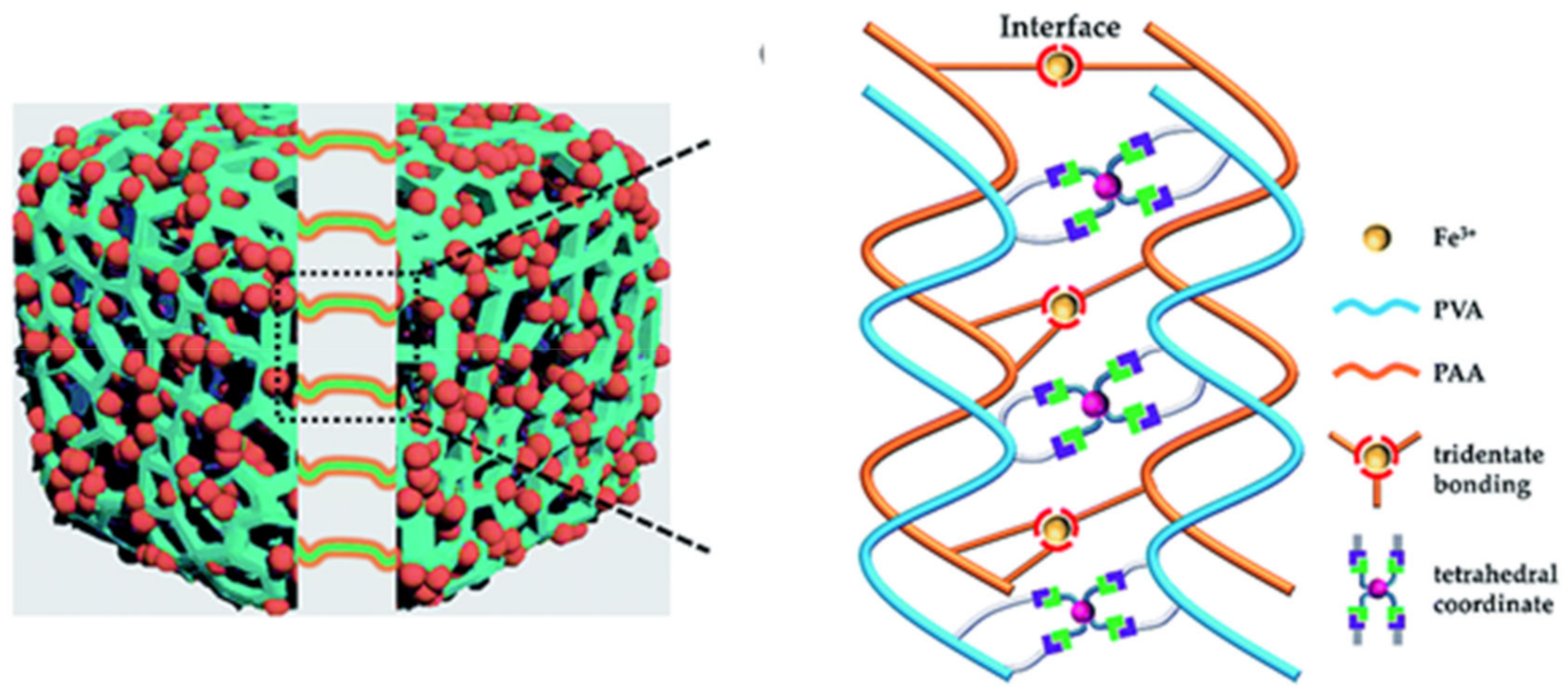
2.2. M–L Bond Based SHPs
3. Covalent Bond Based SHPs
3.1. Imine Bond Based SHPs
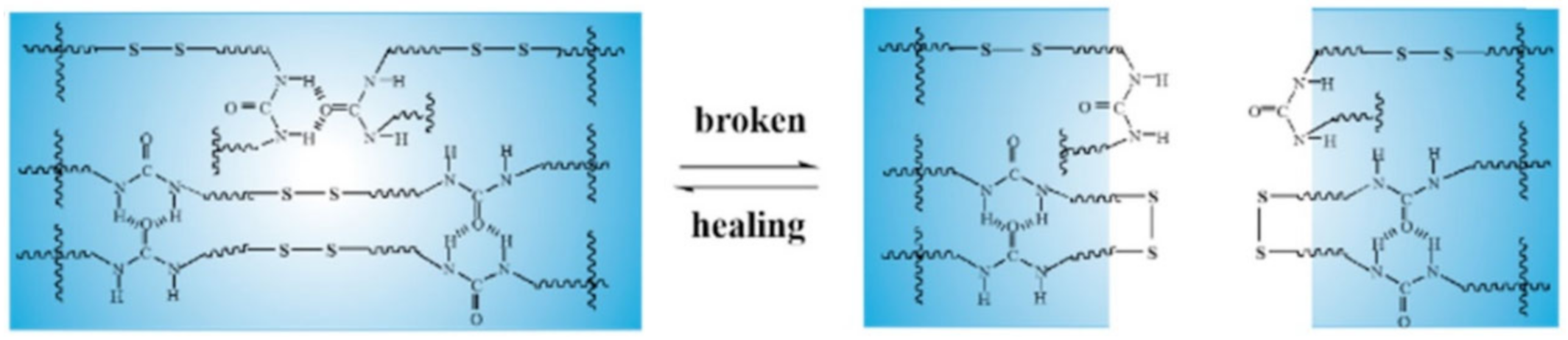
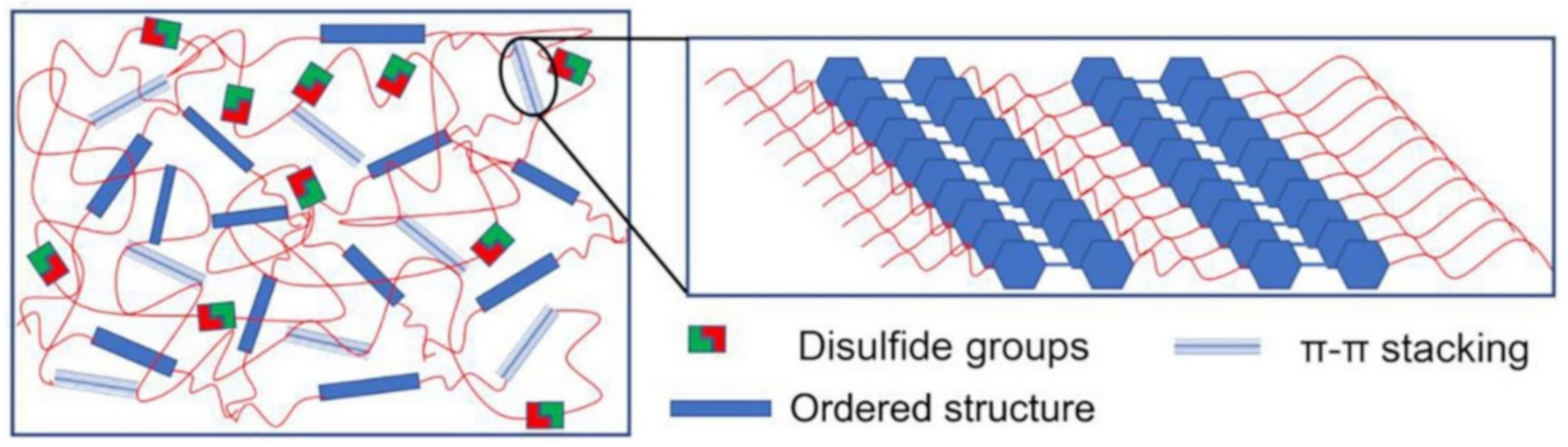
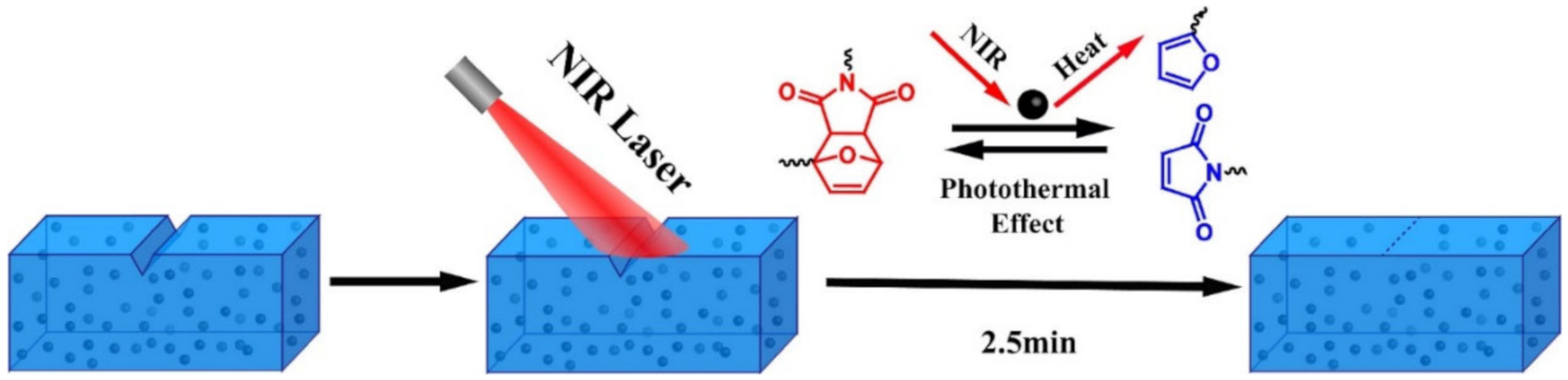
3.2. Disulfide Bond Based SHPs
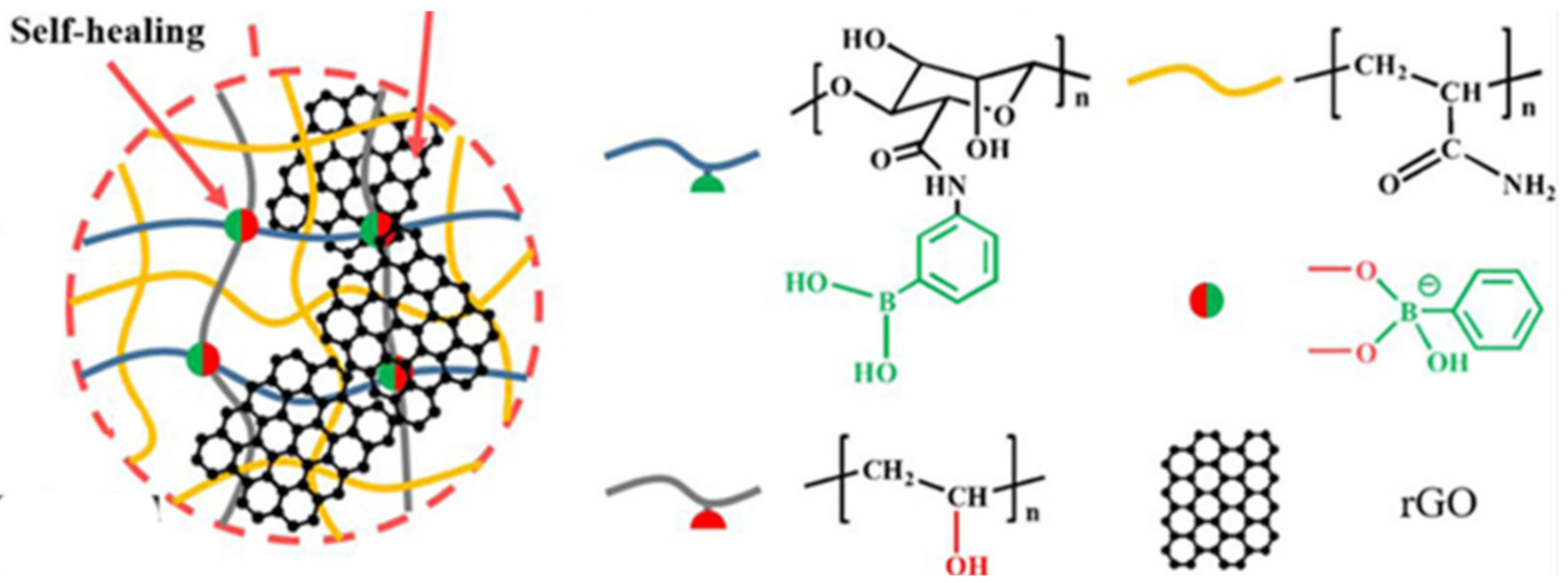
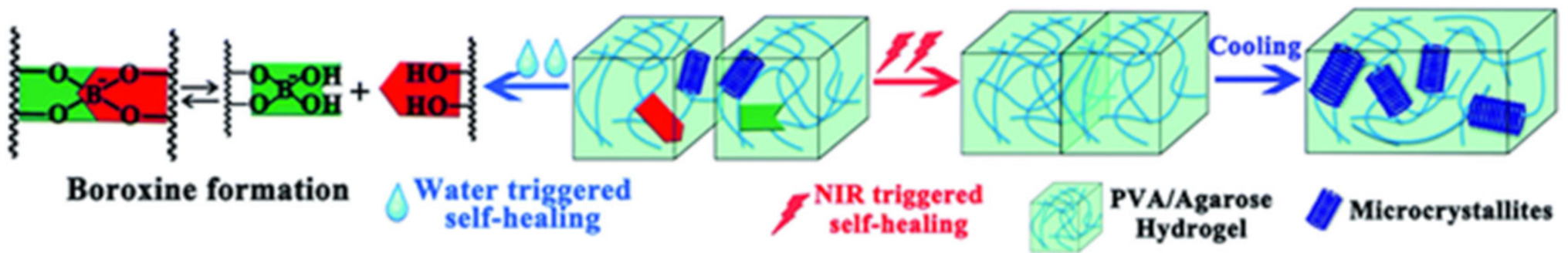
3.3. Borate Bond-Based SHPs
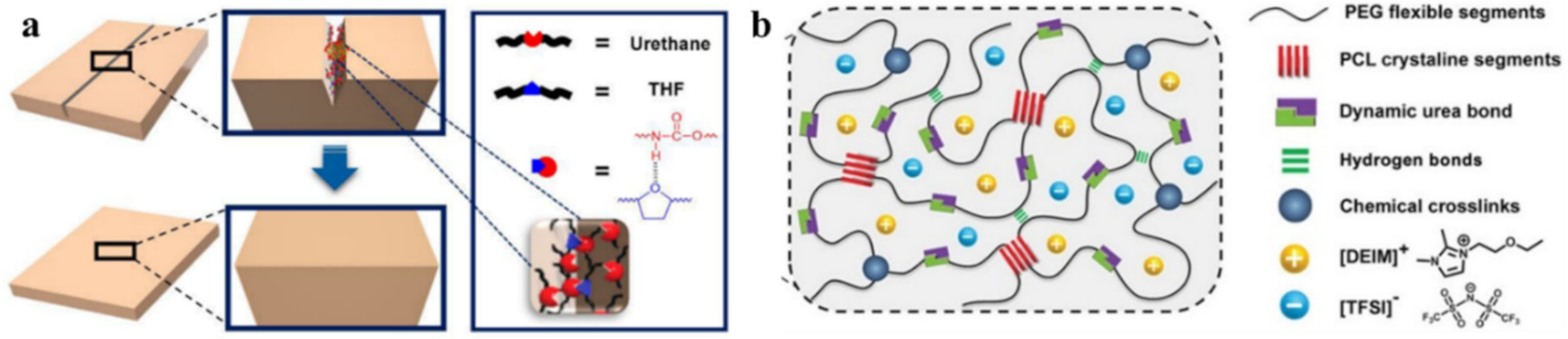
4. Multiple Bond-Based SHPs
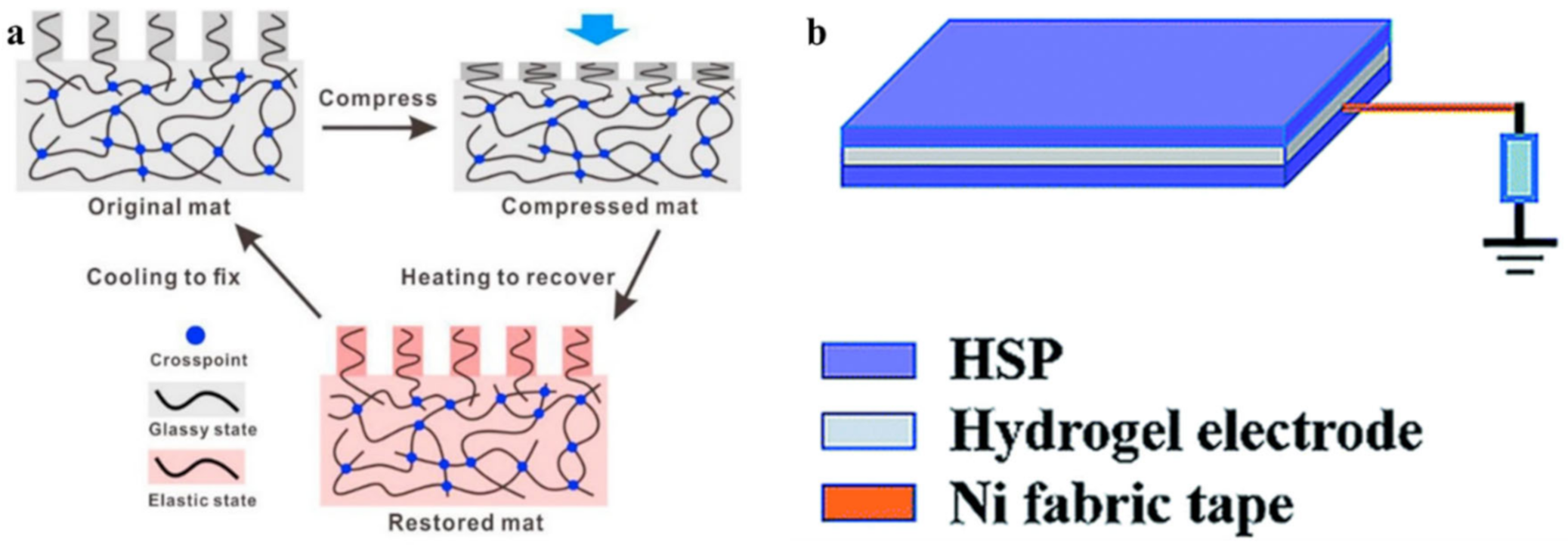
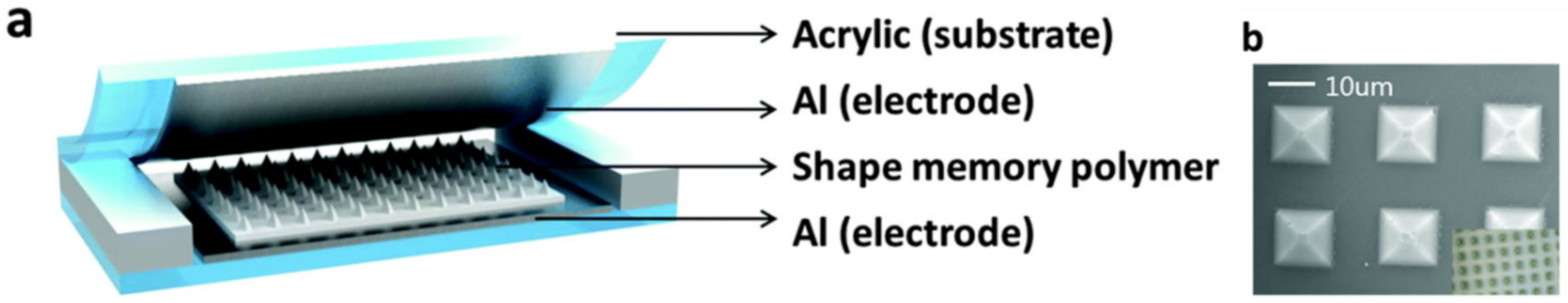
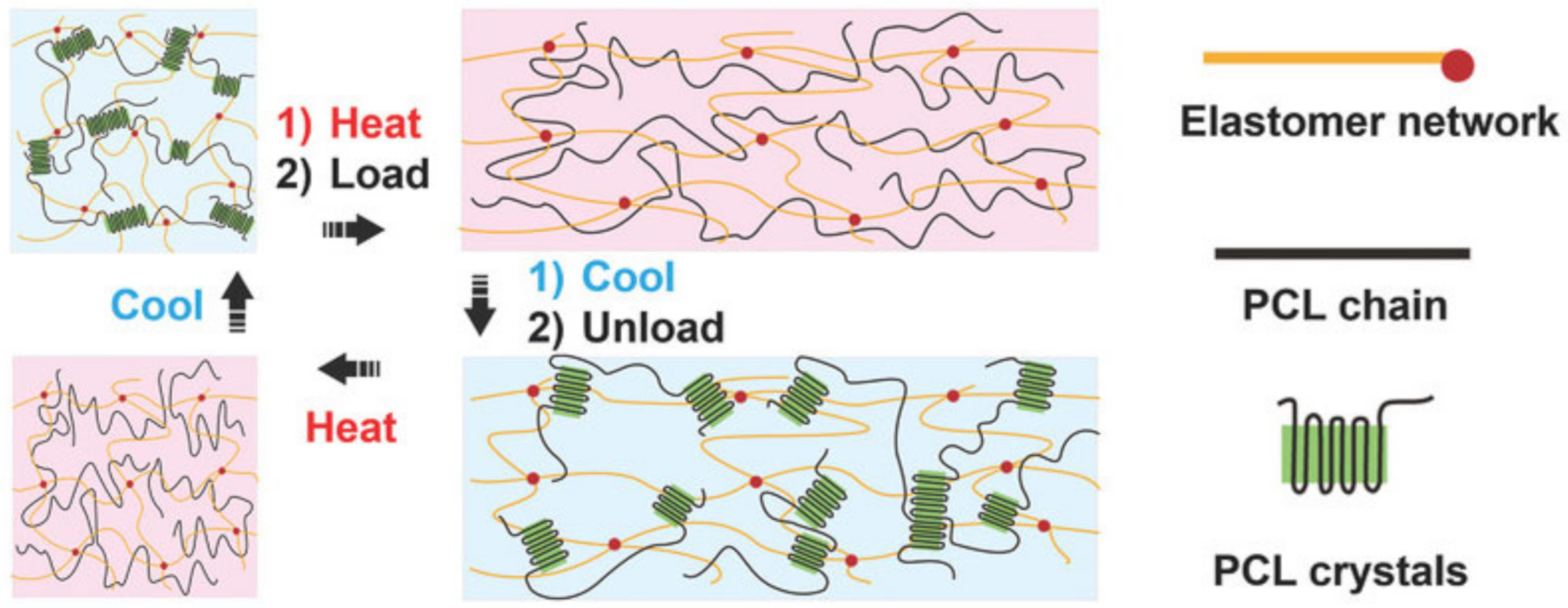
5. SHPs with Shape Memory
6. Summary and Future Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Encyclopedia of Renewable and Sustainable Materials; Hashmi, S., Choudhury, I.A., Eds.; Elsevier: Amsterdam, The Netherlands, 2020. [Google Scholar]
- Lee, S.; Shi, Q.; Lee, C. From flexible electronics technology in the era of IoT and artificial intelligence toward future implanted body sensor networks. APL Mater. 2019, 7, 031302. [Google Scholar] [CrossRef]
- Dong, K.; Peng, X.; Wang, Z.L. Fiber/Fabric-Based Piezoelectric and Triboelectric Nanogenerators for Flexible/Stretchable and Wearable Electronics and Artificial Intelligence. Adv. Mater. 2020, 32, 1902549. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Shi, Q.; Lee, C. A novel hybridized blue energy harvester aiming at all-weather IoT applications. Nano Energy 2020, 76, 105052. [Google Scholar] [CrossRef]
- Shi, Q.; Dong, B.; He, T.; Sun, Z.; Zhu, J.; Zhang, Z.; Lee, C. Progress in wearable electronics/photonics—Moving toward the era of artificial intelligence and internet of things. InfoMat 2020, 2, 1131–1162. [Google Scholar] [CrossRef]
- Fan, F.-R.; Tian, Z.-Q.; Wang, Z.L. Flexible triboelectric generator! Nano Energy 2012, 1, 328–334. [Google Scholar] [CrossRef]
- Zhou, L.; Liu, D.; Wang, J.; Wang, Z.L. Triboelectric nanogenerators: Fundamental physics and potential applications. Friction 2020, 8, 481–506. [Google Scholar] [CrossRef]
- Lin, H.; He, M.; Jing, Q.; Yang, W.; Wang, S.; Liu, Y.; Zhang, Y.; Li, J.; Li, N.; Ma, Y.; et al. Angle-shaped triboelectric nanogenerator for harvesting environmental wind energy. Nano Energy 2019, 56, 269–276. [Google Scholar] [CrossRef]
- Liu, G.; Guo, H.; Xu, S.; Hu, C.; Wang, Z.L. Oblate spheroidal triboelectric nanogenerator for all weather blue energy harvesting. Adv. Energy Mater. 2019, 9, 1900801. [Google Scholar] [CrossRef]
- Liu, S.; Li, Y.; Guo, W.; Huang, X.; Xu, L.; Lai, Y.-C.; Zhang, C.; Wu, H. Triboelectric nanogenerators enabled sensing and actuation for robotics. Nano Energy 2019, 65, 104005. [Google Scholar] [CrossRef]
- Tang, Q.; Pu, X.; Zeng, Q.; Yang, H.; Li, J.; Wu, Y.; Guo, H.; Huang, Z.; Hu, C. A strategy to promote efficiency and durability for sliding energy harvesting by designing alternating magnetic stripe arrays in triboelectric nanogenerator. Nano Energy 2019, 66, 104087. [Google Scholar] [CrossRef]
- Yang, H.; Deng, M.; Tang, Q.; He, W.; Hu, C.; Xi, Y.; Liu, R.; Wang, Z.L. A nonencapsulative pendulum-like paper–based hybrid nanogenerator for energy harvesting. Adv. Energy Mater. 2019, 9, 1901149. [Google Scholar] [CrossRef]
- Bai, Y.; Xu, L.; He, C.; Zhu, L.; Yang, X.; Jiang, T.; Nie, J.; Zhong, W.; Wang, Z.L. High-performance triboelectric nanogenerators for self-powered, in-situ and real-time water quality mapping. Nano Energy 2019, 66, 104117. [Google Scholar] [CrossRef]
- Wang, W.; Yu, A.; Liu, X.; Liu, Y.; Zhang, Y.; Zhu, Y.; Lei, Y.; Jia, M.; Zhai, J.; Wang, Z.L. Large-scale fabrication of robust textile triboelectric nanogenerators. Nano Energy 2020, 71, 104605. [Google Scholar] [CrossRef]
- Flexible and Stretchable Triboelectric Nanogenerator Devices: Toward Self-Powered Systems; Han, M., Zhang, X., Zhang, H., Eds.; Wiley: Weinheim, Germany, 2019. [Google Scholar]
- Seung, W.; Gupta, M.K.; Lee, K.Y.; Shin, K.-S.; Lee, J.-H.; Kim, T.Y.; Kim, S.; Lin, J.; Kim, J.H.; Kim, S.-W. Nanopatterned textile-based wearable triboelectric nanogenerator. ACS Nano 2015, 9, 3501–3509. [Google Scholar] [CrossRef]
- Li, H.Y.; Su, L.; Kuang, S.Y.; Pan, C.F.; Zhu, G.; Wang, Z.L. Significant enhancement of triboelectric charge density by fluorinated surface modification in nanoscale for converting mechanical energy. Adv. Funct. Mater. 2015, 25, 5691–5697. [Google Scholar] [CrossRef]
- Wang, S.; Zi, Y.; Zhou, Y.S.; Li, S.; Fan, F.; Lin, L.; Wang, Z.L. Molecular surface functionalization to enhance the power output of triboelectric nanogenerators. J. Mater. Chem. A 2016, 4, 3728–3734. [Google Scholar] [CrossRef]
- Feng, Y.; Zheng, Y.; Ma, S.; Wang, D.; Zhou, F.; Liu, W. High output polypropylene nanowire array triboelectric nanogenerator through surface structural control and chemical modification. Nano Energy 2016, 19, 48–57. [Google Scholar] [CrossRef]
- Long, L.; Wei, T.; Lin, W.Z. Inductively-coupled-plasma-induced electret enhancement for triboelectric nanogenerators. Nanotechnology 2017, 28, 035405. [Google Scholar]
- Dharmasena, R.D.I.G.; Deane, J.H.B.; Silva, S.R.P. Nature of power generation and output optimization criteria for triboelectric nanogenerators. Adv. Energy Mater. 2018, 8, 1802190. [Google Scholar] [CrossRef]
- Zhang, X.-S.; Han, M.; Kim, B.; Bao, J.-F.; Brugger, J.; Zhang, H. All-in-one self-powered flexible microsystems based on triboelectric Nanogenerators. Nano Energy 2018, 47, 410–426. [Google Scholar] [CrossRef]
- Wang, Z.L. Triboelectric nanogenerators as new energy technology and self-powered sensors–principles, problems and perspectives. Faraday Discuss. 2015, 176, 447–458. [Google Scholar] [CrossRef]
- Wang, Z.L.; Chen, J.; Lin, L. Progress in triboelectric nanogenerators as a new energy technology and self-powered sensors. Energy Environ. Sci. 2015, 8, 2250–2282. [Google Scholar] [CrossRef]
- Yun, Y.; La, M.; Cho, S.; Jang, S.; Choi, J.H.; Ra, Y.; Kam, D.; Park, S.J.; Choi, D. High Quality Electret Based Triboelectric Nanogenerator for Boosted and Reliable Electrical Output Performance. Int. J. Precis. Eng. Manuf.-Green Tech. 2020. [Google Scholar] [CrossRef]
- Jang, S.; La, M.; Cho, S.; Yun, Y.; Choi, J.H.; Ra, Y.; Park, S.J.; Choi, D. Monocharged electret based liquid-solid interacting triboelectric nanogenerator for its boosted electrical output performance. Nano Energy 2020, 70, 104541. [Google Scholar] [CrossRef]
- Ra, Y.; Choi, J.H.; Choi, S.-J.; La, M.; Park, S.J.; Kim, M.-J.; Choi, D. Cold rolled robust metal assisted triboelectric nanogenerator for extremely durable operation. Extrem. Mech. Lett. 2020, 40, 100910. [Google Scholar] [CrossRef]
- Cho, S.; Yun, Y.; Jang, S.; Ra, Y.; Choi, J.H.; Hwang, H.J.; Choi, D.; Choi, D. Universal biomechanical energy harvesting from joint movements using a direction-switchable triboelectric nanogenerator. Nano Energy 2020, 71, 104584. [Google Scholar] [CrossRef]
- Ra, Y.; Oh, S.; Lee, J.; Yun, Y.; Cho, S.; Choi, J.H.; Jang, S.; Hwang, H.J.; Choi, D.; Kim, J.-G.; et al. Triboelectric signal generation and its versatile utilization during gear-based ordinary power transmission. Nano Energy 2020, 73, 104745. [Google Scholar] [CrossRef]
- Choi, S.; Cho, S.; Yun, Y.; Jang, S.; Choi, J.H.; Ra, Y.; La, M.; Park, S.J.; Choi, D. Development of a High-Performance Handheld Triboelectric Nanogenerator with a Lightweight Power Transmission Unit. Adv. Mater. Technol. 2020, 5, 2000003. [Google Scholar] [CrossRef]
- Pu, X.; Liu, M.; Chen, X.; Sun, J.; Du, C.; Zhang, Y.; Zhai, J.; Hu, W.; Wang, Z.L. Ultrastretchable, transparent triboelectric nanogenerator as electronic skin for biomechanical energy harvesting and tactile sensing. Sci. Adv. 2017, 3, e1700015. [Google Scholar] [CrossRef]
- Kim, D.Y.; Kim, H.S.; Kong, D.S.; Choi, M.; Kim, H.B.; Lee, J.-H.; Murillo, G.; Lee, M.; Kim, S.S.; Jung, J.H. Floating buoy-based triboelectric nanogenerator for an effective vibrational energy harvesting from irregular and random water waves in wild sea. Nano Energy 2018, 45, 247–254. [Google Scholar] [CrossRef]
- Ning, C.; Tian, L.; Zhao, X.; Xiang, S.; Tang, Y.; Liang, E.; Mao, Y. Washable textile-structured single-electrode triboelectric nanogenerator for self-powered wearable electronics. J. Mater. Chem. A 2018, 6, 19143–19150. [Google Scholar] [CrossRef]
- Dong, K.; Wu, Z.; Deng, J.; Wang, A.C.; Zou, H.; Chen, C.; Hu, D.; Gu, B.; Sun, B.; Wang, Z.L. A stretchable yarn embedded triboelectric nanogenerator as electronic skin for biomechanical energy harvesting and multifunctional pressure sensing. Adv. Mater. 2018, 30, 1804944. [Google Scholar] [CrossRef]
- Yu, A.; Zhu, Y.; Wang, W.; Zhai, J. Progress in triboelectric materials: Toward high performance and widespread applications. Adv. Funct. Mater. 2019, 29, 1900098. [Google Scholar] [CrossRef]
- Xu, W.; Huang, L.B.; Wong, M.C.; Chen, L.; Bai, G.; Hao, J. Environmentally-friendly hydrogel-based triboelectric nanogenerators for versatile energy harvesting and self-powered sensors. Adv. Energy Mater. 2017, 7, 1601529. [Google Scholar] [CrossRef]
- Wang, X.; Niu, S.; Yi, F.; Yin, Y.; Hao, C.; Dai, K.; Zhang, Y.; You, Z.; Wang, Z.L. Harvesting Ambient Vibration Energy over a Wide Frequency Range for Self-Powered Electronics. ACS Nano 2017, 11, 1728–1735. [Google Scholar] [CrossRef]
- Chen, A.; Zhang, C.; Zhu, G.; Wang, Z.L. Polymer Materials for High-Performance Triboelectric Nanogenerators. Adv. Mater. 2020, 7, 2000186. [Google Scholar]
- Deng, J.; Kuang, X.; Liu, R.; Ding, W.; Wang, A.C.; Lai, Y.-C.; Dong, K.; Wen, Z.; Wang, Y.; Wang, L.; et al. Vitrimer Elastomer-Based Jigsaw Puzzle-Like Healable Triboelectric Nanogenerator for Self-Powered Wearable Electronics. Adv. Mater. 2018, 30, 1705918. [Google Scholar] [CrossRef]
- Lai, Y.-C.; Wu, H.-M.; Lin, H.-C.; Chang, C.-L.; Chou, H.-H.; Hsiao, Y.-C.; Wu, Y.-C. Entirely, Intrinsically, and Autonomously Self-Healable, Highly Transparent, and Superstretchable Triboelectric Nanogenerator for Personal Power Sources and Self-Powered Electronic Skins. Adv. Funct. Mater. 2019, 29, 1904626. [Google Scholar] [CrossRef]
- Huynh, T.-P.; Sonar, P.; Haick, H. Advanced Materials for Use in Soft Self-Healing Devices. Adv. Mater. 2017, 29, 1604973. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Song, Y.; Cheng, X.; Zhang, X. Self-powered electronic skin based on the triboelectric generator. Nano Energy 2019, 56, 252–268. [Google Scholar] [CrossRef]
- Guan, Q.; Dai, Y.; Yang, Y.; Bi, X.; Wen, Z.; Pan, Y. Near-infrared irradiation induced remote and efficient self-healable triboelectric nanogenerator for potential implantable electronics. Nano Energy 2018, 51, 333–339. [Google Scholar] [CrossRef]
- Sun, J.; Pu, X.; Liu, M.; Yu, A.; Du, C.; Zhai, J.; Hu, W.; Wang, Z.L. Self-Healable, Stretchable, Transparent Triboelectric Nanogenerators as Soft Power Sources. ACS Nano 2018, 12, 6147–6155. [Google Scholar] [CrossRef]
- Xu, W.; Huang, L.-B.; Hao, J. Fully self-healing and shape-tailorable triboelectric nanogenerators based on healable polymer and magnetic-assisted electrode. Nano Energy 2017, 40, 399–407. [Google Scholar] [CrossRef]
- Dzhardimalieva, G.I.; Yadav, B.C.; Singh, S.; Uflyand, I.E. Self-healing and shape memory metallopolymers: State-of-the-art and future perspectives. Dalton Trans. 2020, 49, 3042–3087. [Google Scholar] [CrossRef]
- Chang, T.; Panhwar, F.; Zhao, G. Flourishing Self-Healing Surface Materials: Recent Progresses and Challenges. Adv. Mater. Interfaces 2020, 7, 1901959. [Google Scholar] [CrossRef]
- Chen, D.; Wang, D.; Yang, Y.; Huang, Q.; Zhu, S.; Zheng, Z. Self-Healing Materials for Next-Generation Energy Harvesting and Storage Devices. Adv. Energy Mater. 2017, 7, 1700890. [Google Scholar] [CrossRef]
- An, T.; Cheng, W. Recent progress in stretchable supercapacitors. J. Mater. Chem. A 2018, 6, 15478–15494. [Google Scholar] [CrossRef]
- Ocheje, M.U.; Charron, B.P.; Nyayachavadi, A.; Rondeau-Gagné, S. Stretchable electronics: Recent progress in the preparation of stretchable and self-healing semiconducting conjugated polymers. Flex. Print. Electron. 2017, 2, 043002. [Google Scholar] [CrossRef]
- Su, C.C.; Chen, J.S. Self-Healing Polymeric Materials. Key Eng. Mater. 2017, 727, 482–489. [Google Scholar] [CrossRef]
- Urdl, K.; Kandelbauer, A.; Kern, W.; Müller, U.; Thebault, M.; Zikulnig-Rusch, E. Self-healing of densely crosslinked thermoset polymers—A critical review. Prog. Org. Coat. 2017, 104, 232–249. [Google Scholar] [CrossRef]
- Mai, W.; Yu, Q.; Han, C.; Kang, F.; Li, B. Self-Healing Materials for Energy-Storage Devices. Adv. Funct. Mater. 2020, 30, 1909912. [Google Scholar] [CrossRef]
- Chen, C.; Chen, S.; Guo, Z.; Hu, W.; Chen, Z.; Wang, J.; Hu, J.; Guo, J.; Yang, L. Highly efficient self-healing materials with excellent shape memory and unprecedented mechanical properties. J. Mater. Chem. A 2020, 8, 16203–16211. [Google Scholar] [CrossRef]
- Kang, J.; Tok, J.B.-H.; Bao, Z. Self-healing soft electronics. Nat. Electron. 2019, 2, 144–150. [Google Scholar] [CrossRef]
- Wang, S.; Urban, M.W. Self-healing polymers. Nat. Rev. Mater. 2020, 5, 562–583. [Google Scholar] [CrossRef]
- Wang, Z.; Scheres, L.; Xia, H.; Zuilhof, H. Developments and Challenges in Self-Healing Antifouling Materials. Adv. Funct. Mater. 2020, 30, 1908098. [Google Scholar] [CrossRef]
- Zhai, L.; Narkar, A.; Ahn, K. Self-healing polymers with nanomaterials and nanostructures. Nano Today 2020, 30, 100826. [Google Scholar] [CrossRef]
- Zhu, M.; Liu, J.; Gan, L.; Long, M. Research progress in bio-based self-healing materials. Eur. Polym. J. 2020, 129, 109651. [Google Scholar] [CrossRef]
- Neumann, L.N.; Weder, C.; Schrettl, S. Healing of Polymeric Solids by Supramolecular Means. Chimia 2019, 73, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Campanella, A.; Döhler, D.; Binder, W.H. Self-Healing in Supramolecular Polymers. Macromol. Rapid Commun. 2018, 39, 1700739. [Google Scholar] [CrossRef]
- Yang, Y.; Urban, M.W. Self-Healing of Polymers via Supramolecular Chemistry. Adv. Mater. Interfaces 2018, 5, 1800384. [Google Scholar] [CrossRef]
- Wang, Z.; Lu, X.; Sun, S.; Yu, C.; Xia, H. Preparation, characterization and properties of intrinsic self-healing elastomers. J. Mater. Chem. B 2019, 7, 4876–4926. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Carstensen, H.; Hölzl, K.; Lunzer, M.; Li, H.; Hilborn, J.; Ovsianikov, A.; Ossipov, D.A. Dynamic Coordination Chemistry Enables Free Directional Printing of Biopolymer Hydrogel. Chem. Mater. 2017, 29, 5816–5823. [Google Scholar] [CrossRef]
- Cui, J.; Ma, Z.; Pan, L.; An, C.-H.; Liu, J.; Zhou, Y.-F.; Li, Y.-S. Self-healable gradient copolymers. Mater. Chem. Front. 2019, 3, 464–471. [Google Scholar] [CrossRef]
- Dahlke, J.; Zechel, S.; Hager, M.D.; Schubert, U.S. How to Design a Self-Healing Polymer: General Concepts of Dynamic Covalent Bonds and Their Application for Intrinsic Healable Materials. Adv. Mater. Interfaces 2018, 5, 1800051. [Google Scholar] [CrossRef]
- Amabilino, D.B.; Smith, D.K.; Steed, J.W. Supramolecular materials. Chem. Soc. Rev. 2017, 46, 2404–2420. [Google Scholar] [CrossRef]
- Wurthner, F.; Saha-Moller, C.R.; Fimmel, B.; Ogi, S.; Leowanawat, P.; Schmidt, D. Perylene Bisimide Dye Assemblies as Archetype Functional Supramolecular Materials. Chem. Rev. 2016, 116, 962–1052. [Google Scholar] [CrossRef]
- Krieg, E.; Bastings, M.M.C.; Besenius, P.; Rybtchinski, B. Supramolecular Polymers in Aqueous Media. Chem. Rev. 2016, 116, 2414–2477. [Google Scholar] [CrossRef]
- Zhang, Z.P.; Rong, M.Z.; Zhang, M.Q. Polymer engineering based on reversible covalent chemistry: A promising innovative pathway towards new materials and new functionalities. Prog. Polym. Sci. 2018, 80, 39–93. [Google Scholar] [CrossRef]
- Mukherjee, S.; Brooks, W.L.A.; Dai, Y.Q.; Sumerlin, B.S. Doubly-dynamic-covalent polymers composed of oxime and oxanorbornene links. Polym. Chem. 2016, 7, 1971–1978. [Google Scholar] [CrossRef]
- Jiang, Z.; Bhaskaran, A.; Aitken, H.M.; Shackleford, I.C.G.; Connal, L.A. Using Synergistic Multiple Dynamic Bonds to Construct Polymers with Engineered Properties. Macromol. Rapid Commun. 2019, 40, 1900038. [Google Scholar] [CrossRef]
- Chakma, P.; Konkolewicz, D. Dynamic Covalent Bonds in Polymeric Materials. Angew. Chem. Int. Ed. 2019, 58, 9682–9695. [Google Scholar] [CrossRef]
- Nevejans, S.; Ballard, N.; Fernández, M.; Reck, B.; García, S.J.; Asua, J.M. The challenges of obtaining mechanical strength in self-healing polymers containing dynamic covalent bonds. Polymer 2019, 28, 121670. [Google Scholar] [CrossRef]
- Ling, L.; Li, J.; Zhang, G.; Sun, R.; Wong, C.-P. Self-Healing and Shape Memory Linear Polyurethane Based on Disulfide Linkages with Excellent Mechanical Property. Macromol. Res. 2018, 26, 365–373. [Google Scholar] [CrossRef]
- Taynton, P.; Ni, H.; Zhu, C.; Yu, K.; Loob, S.; Jin, Y.; Qi, H.J.; Zhang, W. Repairable Woven Carbon Fiber Composites with Full Recyclability Enabled by Malleable Polyimine Networks. Adv. Mater. 2016, 28, 2904–2909. [Google Scholar] [CrossRef]
- Zhang, G.; Zhao, Q.; Yang, L.; Zou, W.; Xi, X.; Xie, T. Exploring Dynamic Equilibrium of Diels–Alder Reaction for Solid State Plasticity in Remoldable Shape Memory Polymer Network. ACS Macro Lett. 2016, 5, 805–808. [Google Scholar] [CrossRef]
- Lee, H.Y.; Cha, S.H. Enhancement of self-healing property by introducing ethylene glycol group into thermally reversible Diels-Alder reaction based self-healable materials. Macromol. Res. 2017, 25, 640–647. [Google Scholar] [CrossRef]
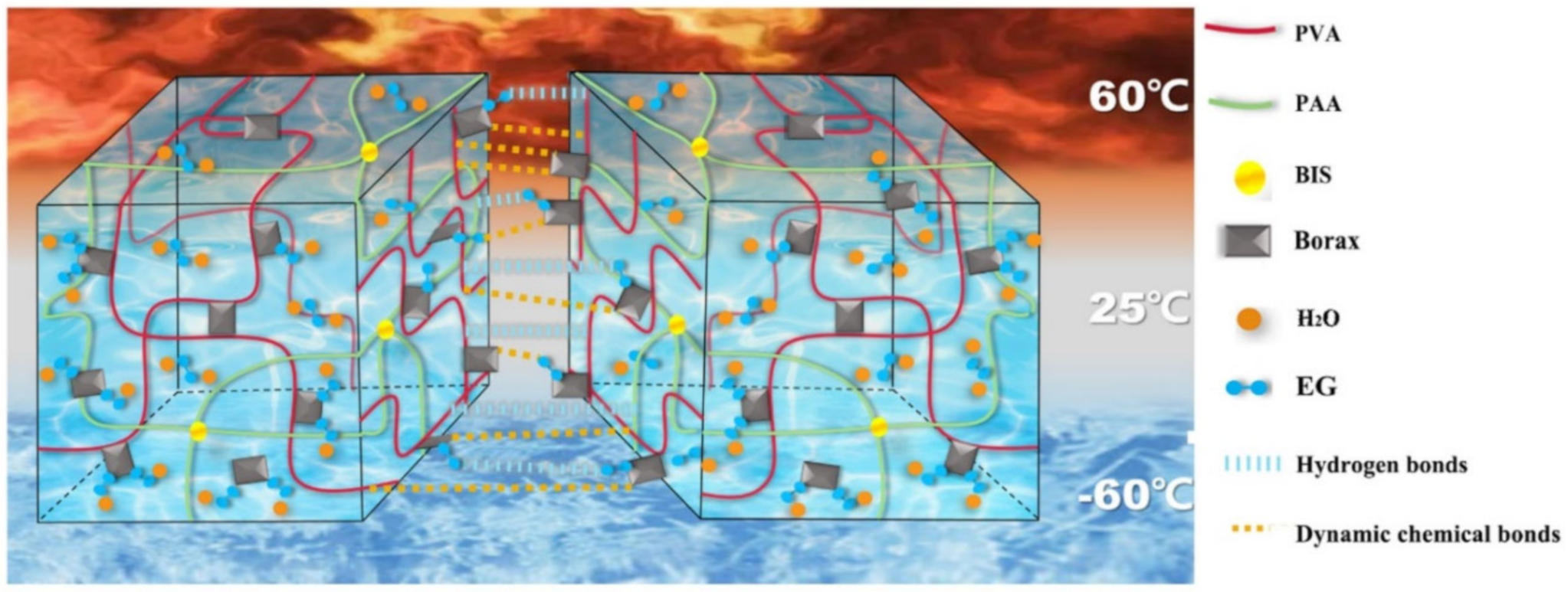
- Seidi, F.; Jin, Y.; Han, J.; Saeb, M.R.; Akbari, A.; Hosseini, S.H.; Shabanian, M.; Xiao, H. Self-healing Polyol/Borax Hydrogels: Fabrications, Properties and Applications. Chem. Rec. 2020, 20, 1142–1162. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, N.; Zhang, B.; Zhang, B.; Song, J. Multifunctional Self-Healing Ionogels from Supramolecular Assembly: Smart Conductive and Remarkable Lubricating Materials. ACS Appl. Mater. Interfaces 2018, 10, 44706–44715. [Google Scholar] [CrossRef]
- Gyarmati, B.; Szilágyi, B.Á.; Szilágyi, A. Reversible interactions in self-healing and shape memory hydrogels. Eur. Polym. J. 2017, 93, 642–669. [Google Scholar] [CrossRef]
- Liu, J.; Tan, C.S.; Yu, Z.; Lan, Y.; Abell, C.; Scherman, O.A. Biomimetic Supramolecular Polymer Networks Exhibiting both Toughness and Self-Recovery. Adv. Mater. 2017, 29, 1604951. [Google Scholar] [CrossRef]
- van der Kooij, H.M.; Susa, A.; Garcia, S.J.; van der Zwaag, S.; Sprakel, J. Imaging the Molecular Motions of Autonomous Repair in a Self-Healing Polymer. Adv. Mater. 2017, 29, 1701017. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, X.; Li, S.; Li, T.; Song, Y.; Li, Z.; Zhang, W.; Sun, J. Transparent, Healable Elastomers with High Mechanical Strength and Elasticity Derived from Hydrogen-Bonded Polymer Complexes. ACS Appl. Mater. Interfaces 2017, 9, 29120–29129. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.D.; Ye, Q.; Lu, X.M.; Lu, Q. Self-healing polymers with PEG oligomer side chains based on multiple H-bonding and adhesion properties. Polym. Chem. 2015, 6, 5086–5092. [Google Scholar] [CrossRef]
- Liu, L.; Pan, C.; Zhang, L.; Guo, B. A Novel and Non-Cytotoxic Self-Healing Supramolecular Elastomer Synthesized with Small Molecular Biological Acids. Macromol. Rapid Commun. 2016, 37, 1603–1610. [Google Scholar] [CrossRef]
- Mauro, M. Dynamic Metal–Ligand Bonds as Scaffolds for Autonomously Healing Multi-Responsive Materials. Eur. J. Inorg. Chem. 2018, 2018, 2090–2100. [Google Scholar] [CrossRef]
- Bentz, K.C.; Cohen, S.M. Supramolecular Metallopolymers: From Linear Materials to Infinite Networks. Angew. Chem. Int. Ed. 2018, 57, 14992–15001. [Google Scholar] [CrossRef] [PubMed]
- Levy, A.; Feinstein, R.; Diesendruck, C.E. Mechanical Unfolding and Thermal Refolding of Single-Chain Nanoparticles Using Ligand–Metal Bonds. J. Am. Chem. Soc. 2019, 141, 7256–7260. [Google Scholar] [CrossRef]
- Li, C.-H.; Zuo, J.-L. Self-Healing Polymers Based on Coordination Bonds. Adv. Mater. 2020, 32, 1903762. [Google Scholar] [CrossRef]
- Uflyand, I.E.; Dzhardimalieva, G.I. Molecular design of supramolecular polymers with chelated units and their application as functional materials. J. Coord. Chem. 2018, 71, 1272–1356. [Google Scholar] [CrossRef]
- Zhang, X.; Vidavsky, Y.; Aharonovich, S.; Yang, S.J.; Buche, M.R.; Diesendruck, C.E.; Silberstein, M.N. Bridging experiments and theory: Isolating the effects of metal–ligand interactions on viscoelasticity of reversible polymer networks. Soft Matter 2020, 16, 8591–8601. [Google Scholar] [CrossRef]
- Zhu, Y.X.; Xuan, H.Y.; Ren, J.Y.; Ge, L. Self-healing multilayer polyelectrolyte composite film with chitosan and poly(acrylic acid). Soft Matter 2015, 11, 8452–8459. [Google Scholar] [CrossRef]
- Ren, Y.; Lou, R.Y.; Liu, X.C.; Gao, M.; Zheng, H.; Yang, T.; Xie, H.; Yu, W.; Ma, X. A self-healing hydrogel formation strategy via exploiting endothermic interactions between polyelectrolytes. Chem. Commun. 2016, 52, 6273–6276. [Google Scholar] [CrossRef]
- Miwa, Y.; Kurachi, J.; Sugino, Y.; Udagawa, T.; Kutsumizu, S. Toward strong self-healing polyisoprene elastomers with dynamic ionic crosslinks. Soft Matter 2020, 16, 3384–3394. [Google Scholar] [CrossRef] [PubMed]
- Takashima, Y.; Yonekura, K.; Koyanagi, K.; Iwaso, K.; Nakahata, M.; Yamaguchi, H.; Harada, A. Multifunctional Stimuli-Responsive Supramolecular Materials with Stretching, Coloring, and Self-Healing Properties Functionalized via Host–Guest Interactions. Macromolecules 2017, 50, 4144–4150. [Google Scholar] [CrossRef]
- Guo, K.; Zhang, D.L.; Zhang, X.M.; Zhang, J.; Ding, L.-S.; Li, B.-J.; Zhang, S. Conductive Elastomers with Autonomic Self-Healing Properties. Angew. Chem. Int. Ed. 2015, 54, 12127–12133. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Wei, K.; Zhang, K.; Yang, B.; Tian, F.; Wang, G.; Bian, L. One-pot solvent exchange preparation of non-swellable, thermoplastic, stretchable and adhesive supramolecular hydrogels based on dual synergistic physical crosslinking. NPG Asia Mater. 2018, 10, e455. [Google Scholar] [CrossRef]
- Yang, Y.; Urban, M.W. Self-healing of glucose-modified polyurethane networks facilitated by damage-induced primary amines. Polym. Chem. 2017, 8, 303–309. [Google Scholar] [CrossRef]
- Zeng, G.; Qiu, L.; Wen, T. Recent advances in crystallization and self-assembly of polypeptoid polymers. Polym. Cryst. 2019, 2, e10065. [Google Scholar] [CrossRef]
- Deng, Z.; Wang, H.; Ma, P.X.; Guo, B. Self-healing conductive hydrogels: Preparation, properties and applications. Nanoscale 2020, 12, 1224–1246. [Google Scholar] [CrossRef]
- Ko, J.; Surendran, A.; Febriansyah, B.; Leong, W.L. Self-healable electrochromic ion gels for low power and robust displays. Org. Electron. 2019, 71, 199–205. [Google Scholar] [CrossRef]
- Tamate, R.; Watanabe, M. Recent progress in self-healable ion gels. Sci. Technol. Adv. Mater. 2020, 21, 388. [Google Scholar] [CrossRef]
- Long, Y.; Chen, Y.; Liu, Y.; Chen, G.; Guo, W.; Kang, X.; Pu, X.; Hu, W.; Wang, Z.L. A flexible triboelectric nanogenerator based on a super-stretchable and self-healable hydrogel as the electrode. Nanoscale 2020, 12, 12753. [Google Scholar] [CrossRef]
- Zhao, J.; Li, Y.; Wang, M. Fabrication of robust transparent hydrogel with stretchable, self-healing, easily recyclable and adhesive properties and its application. Mater. Res. Bull. 2019, 112, 292–296. [Google Scholar] [CrossRef]
- Chen, C.; Duan, N.; Chen, S.; Guo, Z.; Hu, J.; Guo, J.; Chen, Z.; Yang, L. Synthesis, mechanical properties and self-healing behavior of aliphatic polycarbonate hydrogels based on cooperation hydrogen bonds. J. Mol. Liq. 2020, 319, 114134. [Google Scholar] [CrossRef]
- Zhao, J.; Gong, J.; Wang, G.; Zhu, K.; Ye, K.; Yan, J.; Cao, D. A self-healing hydrogel electrolyte for flexible solid-state supercapacitors. Chem. Eng. J. 2020, 401, 125456. [Google Scholar] [CrossRef]
- Wang, H.; Guo, M.; Wu, Y.; Zhang, J.; Xue, S.; Yang, X.; Li, Z. Tough, highly stretchable and self-healing poly(acrylic acid) hydrogels reinforced by functionalized basalt fibers. Mater. Res. Express 2020, 7, 065307. [Google Scholar] [CrossRef]
- Wei, P.; Chen, T.; Chen, G.; Liu, H.; Mugaanire, I.T.; Hou, K.; Zhu, M. Conductive Self-Healing Nanocomposite Hydrogel Skin Sensors with Antifreezing and Thermoresponsive Properties. ACS Appl. Mater. Interfaces 2020, 12, 3068–3079. [Google Scholar] [CrossRef]
- Luo, J.; Yang, J.; Zheng, X.; Ke, X.; Chen, Y.; Tan, H.; Li, J. A Highly Stretchable, Real-Time Self-Healable Hydrogel Adhesive Matrix for Tissue Patches and Flexible Electronics. Adv. Health Mater. 2020, 9, 1901423. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, S.; Hamad, N.; Kim, K.; Liu, L.; Lerond, M.; Cicoira, F. Tailoring the Self-Healing Properties of Conducting Polymer Films. Macromol. Biosci. 2020. [Google Scholar] [CrossRef]
- Li, Y.; Li, X.; Zhang, S.; Liu, L.; Hamad, N.; Bobbara, S.R.; Pasini, D.; Cicoira, F. Autonomic Self-Healing of PEDOT:PSS Achieved Via Polyethylene Glycol Addition. Adv. Funct. Mater. 2020, 30, 2002853. [Google Scholar]
- Parida, K.; Thangavel, G.; Cai, G.; Zhou, X.; Park, S.; Xiong, J.; Lee, P.S. Extremely stretchable and self-healing conductor based on thermoplastic elastomer for all-three-dimensional printed triboelectric nanogenerator. Nat. Commun. 2019, 10, 2158. [Google Scholar] [CrossRef]
- Xu, C.; Ma, B.; Yuan, S.; Zhao, C.; Liu, H. High-Resolution Patterning of Liquid Metal on Hydrogel for Flexible, Stretchable, and Self-Healing Electronics. Adv. Electron. Mater. 2019, 6, 1900721. [Google Scholar] [CrossRef]
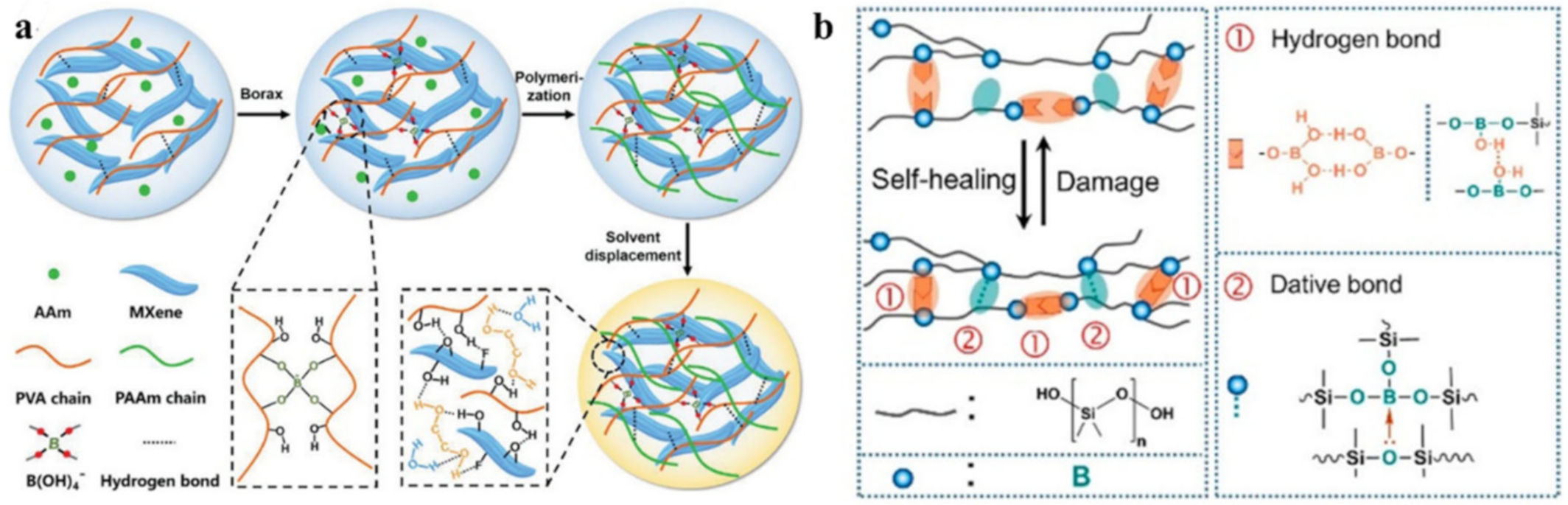
- Guo, Q.; Zhang, X.; Zhao, F.; Song, Q.; Su, G.; Tan, Y.; Tao, Q.; Zhou, T.; Yu, Y.; Zhou, Z.; et al. Protein Inspired Self-Healable Ti3C2 MXenes/Rubber-Based Supramolecular Elastomer for Intelligent Sensing. ACS Nano 2020, 14, 2788–2797. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhang, C.; Yang, L.; Bai, L.; Liang, Y.; Wang, W.; Yang, H.; Wei, D.; Chen, H. Tailoring LaB6 nanoparticle-based self-healing film for heat-shielding window. Bull. Mater. Sci. 2020, 43, 62. [Google Scholar] [CrossRef]
- Niu, H.; Du, X.; Zhao, S.; Yuan, Z.; Zhang, X.; Cao, R.; Yin, Y.; Zhang, C.; Zhou, T.; Li, C. Polymer nanocomposite-enabled high-performance triboelectric nanogenerator with self-healing capability. RSC Adv. 2018, 8, 30661–30668. [Google Scholar] [CrossRef]
- Jeong, S.H.; Zhang, S.; Hjort, K.; Hilborn, J.; Wu, Z. PDMS-Based Elastomer Tuned Soft, Stretchable, and Sticky for Epidermal Electronics. Adv. Mater. 2016, 28, 5830–5836. [Google Scholar] [CrossRef]
- Liu, L.; Liang, S.; Huang, Y.; Hu, C.; Yang, J. A stretchable polysiloxane elastomer with self-healing capacity at room temperature and solvatochromic properties. Chem. Commun. 2017, 53, 12088–12091. [Google Scholar] [CrossRef]
- Yan, S.; Zhang, G.; Jiang, H.; Li, F.; Zhang, L.; Xia, Y.; Wang, Z.; Wu, Y.; Li, H. Highly stretchable room-temperature self-healing conductors based on wrinkled graphene films for flexible electronics. ACS Appl. Mater. Interfaces 2019, 11, 10736–10744. [Google Scholar] [CrossRef]
- Yeasmin, R.; Duy, L.T.; Han, S.; Seo, H. Intrinsically Stretchable and Self-Healing Electroconductive Composites Based on Supramolecular Organic Polymer Embedded with Copper Microparticles. Adv. Electron. Mater. 2020, 128, 2000527. [Google Scholar] [CrossRef]
- Zhao, H.; Yan, S.; Jin, X.; Niu, P.; Zhang, G.; Wu, Y.; Li, H. Tough, Self-Healable and Conductive Elastomers Based on Freezing-Thawing Strategy. Chem. Eng. J. 2020, 402, 125421. [Google Scholar] [CrossRef]
- Gao, Z.; Lou, Z.; Han, W.; Shen, G. A self-healable bifunctional electronic skin. ACS Appl. Mater. Interfaces 2020, 12, 24339–24347. [Google Scholar] [CrossRef]
- Wei, D.; Wang, H.; Zhu, J.; Luo, L.; Huang, H.; Li, L.; Yu, X. Highly Stretchable, Fast Self-Healing, Responsive Conductive Hydrogels for Supercapacitor Electrode and Motion Sensor. Macromol. Mater. Eng. 2020, 305, 2000018. [Google Scholar] [CrossRef]
- Zhao, K.; Lv, C.; Zheng, J. A robust mechanochromic self-healing poly(dimethylsiloxane) elastomer. Sci. China Technol. Sci. 2020, 63, 740–747. [Google Scholar] [CrossRef]
- Chen, H.; Koh, J.J.; Liu, M.; Li, P.; Fan, X.; Liu, S.; Yeo, J.C.C.; Tan, Y.; Tee, B.C.K.; He, C. Super Tough and Self-Healable Poly(dimethylsiloxane) Elastomer via Hydrogen Bonding Association and Its Applications as Triboelectric Nanogenerators. ACS Appl. Mater. Interfaces 2020, 12, 31975–31983. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Son, D.; Wang, G.N.; Liu, Y.; Lopez, J.; Kim, Y.; Oh, J.Y.; Katsumata, T.; Mun, J.; Lee, Y.; et al. Tough and Water-Insensitive Self-Healing Elastomer for Robust Electronic Skin. Adv. Mater. 2018, 30, 1706846. [Google Scholar] [CrossRef]
- Wan, D.; Jiang, Q.; Song, Y.; Pan, J.; Qi, T.; Li, G.L. Biomimetic Tough Self-Healing Polymers Enhanced by Crystallization Nanostructures. ACS Appl. Polym. Mater. 2020, 2, 879–886. [Google Scholar] [CrossRef]
- Wu, X.; Wang, J.; Huang, J.; Yang, S. Room temperature readily self-healing polymer via rationally designing molecular chain and crosslinking bond for flexible electrical sensor. J. Colloid Interface Sci. 2020, 559, 152–161. [Google Scholar] [CrossRef]
- Xie, H.; Sheng, D.; Zhou, Y.; Xu, S.; Wu, H.; Tian, X.; Sun, Y.; Liu, X.; Yan, Y. Thermally healable polyurethane with tailored mechanical performance using dynamic crosslinking motifs. New, J. Chem. 2020, 44, 13584. [Google Scholar] [CrossRef]
- Xiao, L.; Shi, J.; Wu, K.; Lu, M. Self-healing supramolecular waterborne polyurethane based on host–guest interactions and multiple hydrogen bonds. React. Funct. Polym. 2020, 148, 104482. [Google Scholar] [CrossRef]
- Yang, Y.; Ye, Z.; Liu, X.; Su, J. A healable waterborne polyurethane synergistically cross-linked by hydrogen bonds and covalent bonds for composite conductors. J. Mater. Chem. C 2020, 8, 5280–5292. [Google Scholar] [CrossRef]
- Chen, L.; Peng, H.; Wei, Y.; Wang, X.; Jin, Y.; Liu, H.; Jiang, Y. Self-Healing Properties of PDMS Elastomers via Guanine and Cytosine Base Pairs. Macromol. Chem. Phys. 2019, 220, 1900280. [Google Scholar] [CrossRef]
- Wu, P.; Cheng, H.; Wang, Y.; Shi, R.; Wu, Z.; Arai, M.; Zhao, F. New Kind of Thermoplastic Polyurea Elastomers Synthesized from CO2 and with Self-Healing Properties. ACS Sustain. Chem. Eng. 2020, 8, 12677–12685. [Google Scholar] [CrossRef]
- Döhler, D.; Kang, J.; Cooper, C.B.; Tok, J.B.-H.; Rupp, H.; Binder, W.H.; Bao, Z. Tuning the Self-Healing Response of Poly(dimethylsiloxane)-Based Elastomers. ACS Appl. Polym. Mater. 2020, 2, 4127–4139. [Google Scholar] [CrossRef]
- Wang, W.; Wang, F.; Zhang, C.; Tang, J.; Zeng, X.; Wan, X. Versatile Value-added Application of Hyperbranched Lignin Derivatives: Water-resistance Adhesive, UV Protection Coating, Self-healing and Skin-adhesive Sensing. Chem. Eng. J. 2020, 404, 126358. [Google Scholar] [CrossRef]
- Du, R.; Xu, Z.; Zhu, C.; Jiang, Y.; Yan, H.; Wu, H.-C.; Vardoulis, O.; Cai, Y.; Zhu, X.; Bao, Z.; et al. A Highly Stretchable and Self-Healing Supramolecular Elastomer Based on Sliding Crosslinks and Hydrogen Bonds. Adv. Funct. Mater. 2019, 30, 1907139. [Google Scholar] [CrossRef]
- Niu, W.; Zhu, Y.-L.; Wang, R.; Lu, Z.; Liu, X.; Sun, J. Remalleable, Healable, Highly Sustainable Supramolecular Polymeric Materials Combining Super-High Strength and Ultra-High Toughness. ACS Appl. Mater. Interfaces 2020, 12, 30805–30814. [Google Scholar] [CrossRef]
- Chen, Z.; Ma, H.; Li, Y.; Meng, J.; Yao, Y.; Yao, C. Biomass Polyamide Elastomers Based on Hydrogen Bonds with Rapid Self-healing Properties. Eur. Polym. J. 2020, 133, 109802. [Google Scholar] [CrossRef]
- Wang, H.; Cheng, H.; Huang, Y.; Yang, C.; Wang, D.; Li, C.; Qu, L. Transparent, self-healing, arbitrary tailorable moist-electric film generator. Nano Energy 2020, 67, 104238. [Google Scholar] [CrossRef]
- Zhao, W.; Liu, Y.; Zhang, Z.; Feng, X.; Xu, H.; Xu, J.; Hu, J.; Wang, S.; Wu, Y.; Yan, S. High-Strength, Fast Self-Healing, Aging-Insensitive Elastomers with Shape Memory Effect. ACS Appl. Mater. Interfaces 2020, 12, 35445–35452. [Google Scholar] [CrossRef]
- Muradyan, H.; Mozhdehi, D.; Guan, Z. Self-healing magnetic nanocomposites with robust mechanical properties and high magnetic actuation potential prepared from commodity monomers via graft-from approach. Polym. Chem. 2020, 11, 1292–1297. [Google Scholar] [CrossRef]
- Berkem, A.S.; Capoglu, A.; Nugay, T.; Sancaktar, E.; Anac, I. Self-Healable Supramolecular Vanadium Pentoxide Reinforced Polydimethylsiloxane-Graft-Polyurethane Composites. Polymers 2019, 11, 41. [Google Scholar] [CrossRef]
- Ding, H.; Liang, X.; Wang, Q.; Wang, M.; Li, Z.; Sun, G. A semi-interpenetrating network ionic composite hydrogel with low modulus, fast self-recoverability and high conductivity as flexible sensor. Carbohydr. Polym. 2020, 248, 116797. [Google Scholar] [CrossRef]
- Hao, M.; Li, L.; Wang, S.; Sun, F.; Bai, Y.; Cao, Z.; Qu, C.; Zhang, T. Stretchable, self-healing, transient macromolecular elastomeric gel for wearable electronics. Microsyst. Nanoeng. 2019, 5, 9. [Google Scholar] [CrossRef]
- Kim, J.; Hong, P.H.; Choi, K.; Moon, G.; Kang, J.; Lee, S.; Lee, S.; Jung, H.W.; Ko, M.J.; Hong, S.W. A Heterocyclic Polyurethane with Enhanced Self-Healing Efficiency and Outstanding Recovery of Mechanical Properties. Polymers 2020, 12, 968. [Google Scholar]
- Li, T.; Wang, Y.; Li, S.; Liu, X.; Sun, J. Mechanically Robust, Elastic, and Healable Ionogels for Highly Sensitive Ultra-Durable Ionic Skins. Adv. Mater. 2020, 32, 2002706. [Google Scholar]
- Liang, Z.; Huang, D.; Zhao, L.; Nie, Y.; Zhou, Z.; Hao, T.; Li, S. Self-healing Polyurethane Elastomer Based on Molecular Design: Combination of Reversible Hydrogen Bonds and High Segment Mobility. J. Inorg. Organomet. Polym. Mater. 2020. [Google Scholar] [CrossRef]
- Lee, M.Y.; Dharmapurikar, S.; Lee, S.J.; Cho, Y.; Yang, C.; Oh, J.H. Regular H-bonding-containing Polymers with Stretchability up to 100% External Strain for Self-healable Plastic Transistors. Chem. Mater. 2020, 32, 1914–1924. [Google Scholar] [CrossRef]
- Tian, M.; Zuo, H.; Wang, J.; Ning, N.; Yu, B.; Zhang, L. A silicone elastomer with optimized and tunable mechanical strength and self-healing ability based on strong and weak coordination bonds. Polym. Chem. 2020, 11, 4047–4057. [Google Scholar] [CrossRef]
- Liu, Y.; Tang, Z.; Wang, D.; Wu, S.; Guo, B. Biomimetic design of elastomeric vitrimers with unparalleled mechanical properties, improved creep resistance and retained malleability by metal–ligand coordination. J. Mater. Chem. A. 2019, 7, 26867–26876. [Google Scholar] [CrossRef]
- Shan, Y.; Zhou, Z.; Bai, H.; Wang, T.; Liu, L.; Zhao, X.; Huang, Y. Recovery of the self-cleaning property of silicon elastomers utilizing the concept of reversible coordination bonds. Soft Matter 2020, 16, 8473–8481. [Google Scholar] [CrossRef]
- Shin, S.-H.; Lee, W.; Kim, S.-M.; Lee, M.; Koo, J.M.; Hwang, S.Y.; Oh, D.X.; Park, J. Ion-conductive self-healing hydrogels based on an interpenetrating polymer network for a multimodal sensor. Chem. Eng. J. 2019, 371, 452–460. [Google Scholar] [CrossRef]
- Peng, B.; Li, H.; Li, Y.; Lv, Z.; Wu, M.; Zhao, C. A photoresponsive azopyridine-based supramolecular elastomer for self-healing strain sensors. Chem. Eng. J. 2020, 395, 125079. [Google Scholar] [CrossRef]
- Xu, S.; Sheng, D.; Zhou, Y.; Wu, H.; Xie, H.; Liu, X.; Yang, Y. A robust and healable polyurethane based on coordination bonds. Polym. Int. 2020, 69, 876–882. [Google Scholar] [CrossRef]
- Zheng, C.; Lu, K.; Lu, Y.; Zhu, S.; Yue, Y.; Xu, X.; Mei, C.; Xiao, H.; Wu, Q.; Han, J. A stretchable, self-healing conductive hydrogels based on nanocellulose supported graphene towards wearable monitoring of human motion. Carbohydr. Polym. 2020, 250, 116905. [Google Scholar] [CrossRef]
- Li, P.; Xia, Y.; Hao, J.; Wang, X. Transient Healability of Metallosupramolecular Polymer Networks Mediated by Kinetic Control of Competing Chemical Reactions. Macromolecules 2020, 53, 2856–2863. [Google Scholar] [CrossRef]
- Liang, Y.; Ye, L.; Sun, X.; Lv, Q.; Liang, H. Tough and Stretchable Dual Ionically Cross-Linked Hydrogel with High Conductivity and Fast Recovery Property for High-Performance Flexible Sensors. ACS Appl. Mater. Interfaces 2020, 12, 1577–1587. [Google Scholar] [CrossRef]
- Zhou, H.; Li, S.; Liu, H.; Zheng, B.; Jin, X.; Ma, A.; Chen, W. High-Performance Flexible Sensors of Self-Healing, Reversibly Adhesive, and Stretchable Hydrogels for Monitoring Large and Subtle Strains. Macromol. Mater. Eng. 2020, 305, 1900621. [Google Scholar] [CrossRef]
- Heidarian, P.; Kouzani, A.Z.; Kaynak, A.; Zolfagharian, A.; Yousefi, H. Dynamic Mussel-Inspired Chitin Nanocomposite Hydrogels for Wearable Strain Sensors. Polymers 2020, 12, 1416. [Google Scholar] [CrossRef]
- Khan, A.; Huang, K.; Sarwar, M.G.; Cheng, K.; Li, Z.; Tuhin, M.O.; Rabnawaz, M. Self-healing and Self-cleaning Clear Coating. J. Colloid Interface Sci. 2020, 577, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Yang, L.; Dai, B.; Yang, Z.; Guo, S.; Gao, G.; Xu, L.; Sun, M.; Yao, K.; Zhu, J. A self-healing transparent polydimethylsiloxane elastomer based on imine bonds. Eur. Polym. J. 2020, 123, 109382. [Google Scholar] [CrossRef]
- Tan, Y.J.; Wu, J.; Li, H.; Tee, B.C.K. Self-Healing Electronic Materials for a Smart and Sustainable Future. ACS Appl. Mater. Interfaces 2018, 10, 15331–15345. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.; Pu, W.; Wang, Z.; Lu, X.; Sun, S.; Yu, C.; Xia, H. A facile dynamic crosslinked healable poly(oxime-urethane) elastomer with high elastic recovery and recyclability. J. Mater. Chem. A 2018, 6, 18154–18164. [Google Scholar] [CrossRef]
- Wang, S.; Yang, Y.; Ying, H.; Jing, X.; Wang, B.; Zhang, Y.; Cheng, J. Recyclable, Self-Healable, and Highly Malleable Poly(urethane-urea)s with Improved Thermal and Mechanical Performances. ACS Appl. Mater. Interfaces 2020, 12, 35403–35414. [Google Scholar] [CrossRef]
- Hu, J.; Mo, R.; Sheng, X.; Zhang, X. A self-healing polyurethane elastomer with excellent mechanical properties based on phase-locked dynamic imine bonds. Polym. Chem. 2020, 11, 2585–2594. [Google Scholar] [CrossRef]
- Wu, H.; Sheng, D.; Liu, X.; Zhou, Y.; Dong, L.; Ji, F.; Xu, S.; Yang, Y. NIR induced self-healing polyurethane/polypyrrole nanocomposites. Polymer 2020, 189, 122181. [Google Scholar] [CrossRef]
- Zhu, M.; Jin, H.; Shao, T.; Li, Y.; Liu, J.; Gan, L.; Long, M. Polysaccharide-based fast self-healing ion gel based on acylhydrazone and metal coordination bonds. Mater. Des. 2020, 192, 108723. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, H.; Dai, Z.; Zhao, Z.; Fu, F.; Liu, X. The dynamic chain effect on healing performance and thermo-mechanical properties of a polyurethane network. React. Funct. Polym. 2020, 146, 104444. [Google Scholar] [CrossRef]
- Liu, J.; Song, H.; Wang, Z.; Zhang, J.; Zhang, J.; Ba, X. Stretchable, self-healable, and reprocessable chemical cross-linked ionogels electrolytes based on gelatin for flexible supercapacitors. J. Mater. Sci. 2020, 55, 3991–4004. [Google Scholar] [CrossRef]
- Jia, H.; Gu, S.-Y. Remote and efficient infrared induced self-healable stretchable substrate for wearable electronics. Eur. Polym. J. 2020, 126, 109542. [Google Scholar] [CrossRef]
- Li, D.; Yuan, L.; Liang, G.; Gu, A. Self-healable and remoldable transparent polyurethane film with high dielectric constant from the synergistic effect between lithium salt and ionic liquid. Ind. Eng. Chem. Res. 2020, 59, 6600–6608. [Google Scholar] [CrossRef]
- Zhao, L.; Yin, Y.; Jiang, B.; Guo, Z.; Qu, C.; Huang, Y. Fast Room-Temperature Self-Healing Siloxane Elastomer for Healable Stretchable Electronics. J. Colloid Interface Sci. 2020, 573, 105–114. [Google Scholar] [CrossRef]
- Zhang, Q.; Chen, G.; Wu, K.; Shi, J.; Liang, L.; Lu, M. Self-healable and reprocessible liquid crystalline elastomer and its highly thermal conductive composites by incorporating graphene via in-situ polymerization. J. Appl. Polym. Sci. 2020, 138, 49748. [Google Scholar]
- Lai, Y.; Kuang, X.; Zhu, P.; Huang, M.; Dong, X.; Wang, D. Colorless, Transparent, Robust, and Fast Scratch-Self-Healing Elastomers via a Phase-Locked Dynamic Bonds Design. Adv. Mater. 2018, 30, 1802556. [Google Scholar] [CrossRef]
- Li, D.; Zhang, Y.; Yuan, L.; Liang, G.; Gu, A. Simultaneously achieving high strength, thermal resistance and high self-healing efficiency for polyacrylate coating by constructing a Diels–Alder reversible covalent structure with multi-maleimide terminated hyperbranched polysiloxane. Polym. Int. 2019, 69, 110–120. [Google Scholar] [CrossRef]
- Liu, Z.; Hong, P.; Huang, Z.; Zhang, T.; Xu, R.; Chen, L.; Xiang, H.; Liu, X. Self-healing, reprocessing and 3D printing of transparent and hydrolysis-resistant silicone elastomers. Chem. Eng. J. 2020, 387, 124142. [Google Scholar] [CrossRef]
- Bapat, A.P.; Sumerlin, B.S.; Sutti, A. Bulk network polymers with dynamic B–O bonds: Healable and reprocessable materials. Mater. Horiz. 2020, 7, 694–714. [Google Scholar] [CrossRef]
- Niu, W.; O’Sullivan, C.; Rambo, B.M.; Smith, M.D.; Lavigne, J.J. Self-repairing polymers: Poly(dioxaborolane)s containing trigonal planar boron. Chem. Commun. 2005, 4342–4344. [Google Scholar] [CrossRef] [PubMed]
- Cash, J.J.; Kubo, T.; Bapat, A.P.; Sumerlin, B.S. Room-Temperature Self-Healing Polymers Based on Dynamic-Covalent Boronic Esters. Macromolecules 2015, 48, 2098–2106. [Google Scholar] [CrossRef]
- Stukalin, E.B.; Cai, L.-H.; Kumar, N.A.; Leibler, L.; Rubinstein, M. Self-Healing of Unentangled Polymer Networks with Reversible Bonds. Macromolecules 2013, 46, 7525–7541. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Wu, X.; Wang, Y.; Wan, P.; Zhang, L. Wearable, Antifreezing, and Healable Epidermal Sensor Assembled from Long-Lasting Moist Conductive Nanocomposite Organohydrogel. ACS Appl. Mater. Interfaces 2019, 11, 41701–41709. [Google Scholar] [CrossRef]
- Guan, Q.; Lin, G.; Gong, Y.; Wang, J.; Tan, W.; Bao, D.; Liu, Y.; You, Z.; Sun, X.; Wen, Z.; et al. Highly efficient self-healable and dual responsive hydrogel-based deformable triboelectric nanogenerators for wearable electronics. J. Mater. Chem. A 2019, 7, 13948–13955. [Google Scholar] [CrossRef]
- Tsai, M.-S.; Shen, T.L.; Wu, H.-M.; Liao, Y.-M.; Liao, Y.-K.; Lee, W.-Y.; Kuo, H.-C.; Lai, Y.-C.; Chen, Y.-F. Self-powered, Self-healed and Shape-adaptive Ultraviolet Photodetectors. ACS Appl. Mater. Interfaces 2020, 12, 9755–9765. [Google Scholar] [CrossRef]
- Fan, W.; Jin, Y.; Shi, L.; Zhou, R.; Du, W. Developing visible-light-induced dynamic aromatic Schiff base bonds for room-temperature self-healable and reprocessable waterborne polyurethanes with high mechanical properties. J. Mater. Chem. A 2020, 8, 6757–6767. [Google Scholar] [CrossRef]
- Shi, Z.; Kang, J.; Zhang, L. Water-Enabled Room-Temperature Self-Healing and Recyclable Polyurea Materials with Super-Strong Strength, Toughness, and Large Stretchability. ACS Appl. Mater. Interfaces 2020, 12, 23484–23493. [Google Scholar] [CrossRef]
- Yang, Z.; Li, H.; Zhang, L.; Lai, X.; Zeng, X. Highly stretchable, transparent and room-temperature self-healable polydimethylsiloxane elastomer for bending sensor. J. Colloid Interface Sci. 2020, 570, 1–10. [Google Scholar] [CrossRef]
- Chen, G.; Sun, Z.; Wang, Y.; Zheng, J.; Wen, S.; Zhang, J.; Wang, L.; Hou, J.; Lin, C.; Yue, Z. Designed preparation of silicone protective materials with controlled self-healing and toughness properties. Prog. Org. Coat. 2020, 140, 105483. [Google Scholar] [CrossRef]
- Dai, X.; Huang, L.-B.; Du, Y.; Han, J.; Zheng, Q.; Kong, J.; Hao, J. Self-Healing, Flexible, and Tailorable Triboelectric Nanogenerators for Self-Powered Sensors based on Thermal Effect of Infrared Radiation. Adv. Funct. Mater. 2020, 30, 1910723. [Google Scholar] [CrossRef]
- Xun, X.; Zhang, Z.; Zhao, X.; Zhao, B.; Gao, F.; Kang, Z.; Liao, Q.; Zhang, Y. Highly Robust and Self-Powered Electronic Skin Based on Tough Conductive Self-Healing Elastomer. ACS Nano 2020, 14, 9066–9072. [Google Scholar] [CrossRef]
- Huang, H.; Zhou, W.; Zhong, Z.; Peng, S.; Peng, X. Self-antiglare waterborne coating with superior mechanical robustness and highly efficient room-temperature self-healing capability. Prog. Org. Coat. 2020, 146, 105717. [Google Scholar]
- Liu, M.; Zhong, J.; Li, Z.; Rong, J.; Yang, K.; Zhou, J.; Shen, L.; Gao, F.; Huang, X.; He, H. A high stiffness and self-healable polyurethane based on disulfide bonds and hydrogen bonding. Eur. Polym. J. 2020, 124, 109475. [Google Scholar] [CrossRef]
- Liu, Q.; Liu, Y.; Zheng, H.; Li, C.; Zhang, Y.; Zhang, Q. Design and development of self-repairable and recyclable crosslinked poly(thiourethane-urethane) via enhanced aliphatic disulfide chemistry. J. Polym. Sci. 2020, 58, 1092–1104. [Google Scholar] [CrossRef]
- Wen, J.; Jia, Z.; Zhang, X.; Pan, M.; Yuan, J.; Zhu, L. Tough, thermo-Responsive, biodegradable and fast self-healing polyurethane hydrogel based on microdomain-closed dynamic bonds design. Mater. Today Commun. 2020, 25, 101569. [Google Scholar]
- Ying, W.B.; Yu, Z.; Kim, D.H.; Lee, K.J.; Hu, H.; Liu, Y.; Kong, Z.; Wang, K.; Shang, J.; Zhang, R.; et al. Water Proof, Highly Tough and Fast Self-healing Polyurethane for Durable Electronic Skin. ACS Appl. Mater. Interfaces 2020, 12, 11072–11083. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, H.; Yang, B.; Wang, L.; Sun, H. A colorless, transparent and self-healing polyurethane elastomer modulated by dynamic disulfide and hydrogen bonds. New J. Chem. 2020, 44, 5746–5754. [Google Scholar] [CrossRef]
- Jo, Y.H.; Li, S.; Zuo, C.; Zhang, Y.; Gan, H.; Li, S.; Yu, L.; He, D.; Xie, X.; Xue, Z. Self-Healing Solid Polymer Electrolyte Facilitated by a Dynamic Cross-Linked Polymer Matrix for Lithium-Ion Batteries. Macromolecules 2020, 53, 1024–1032. [Google Scholar] [CrossRef]
- Khatib, M.; Zohar, O.; Saliba, W.; Srebnik, S.; Haick, H. Highly Efficient and Water-Insensitive Self-Healing Elastomer for Wet and Underwater Electronics. Adv. Funct. Mater. 2020, 30, 1910196. [Google Scholar] [CrossRef]
- Guo, H.; Han, Y.; Zhao, W.; Yang, J.; Zhang, L. Universally autonomous self-healing elastomer with high stretchability. Nat. Commun. 2020, 11, 2037. [Google Scholar] [CrossRef]
- Bai, L.; Lei, Y.L.; Huang, H.; Xiang, Y. Neuron-Inspired Self-Healing Composites via Dynamic Construction of Polypyrrole-Decorated Carbon Nanotubes for Smart Physiochemical Sensing. ACS Appl. Mater. Interfaces 2020, 12, 33139–33151. [Google Scholar] [CrossRef]
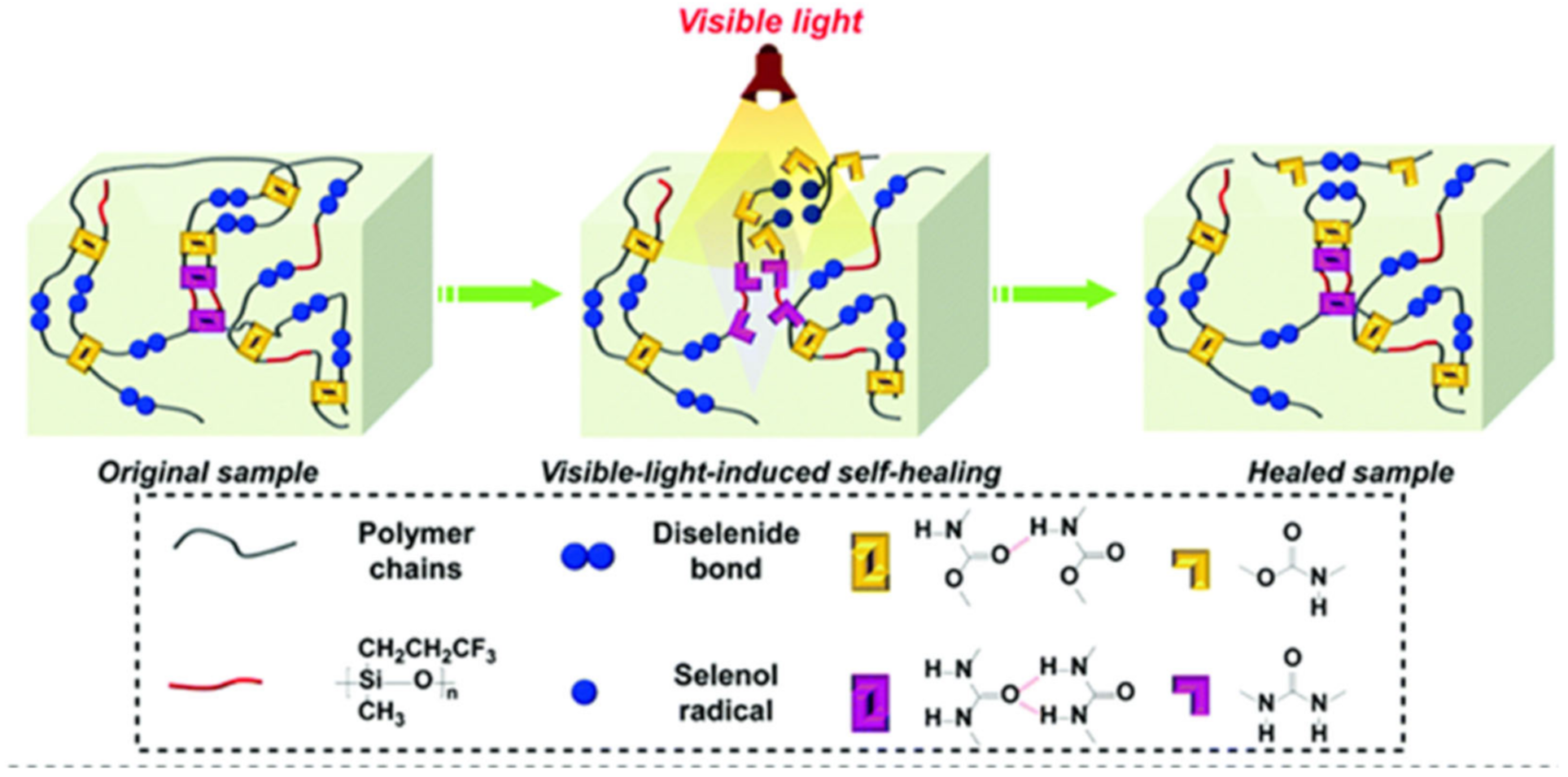
- Fan, W.; Jin, Y.; Shi, L. Mechanically robust and tough waterborne polyurethane films based on diselenide bonds and dual H-bonding interactions with fast visible-light-triggered room-temperature self-healability. Polym. Chem. 2020, 11, 5463–5474. [Google Scholar] [CrossRef]
- Fan, W.; Jin, Y.; Shi, L.; Du, W.; Zhou, R. Transparent, eco-friendly, super-tough “living” supramolecular polymers with fast room-temperature self-healability and reprocessability under visible light. Polymer 2020, 190, 122199. [Google Scholar] [CrossRef]
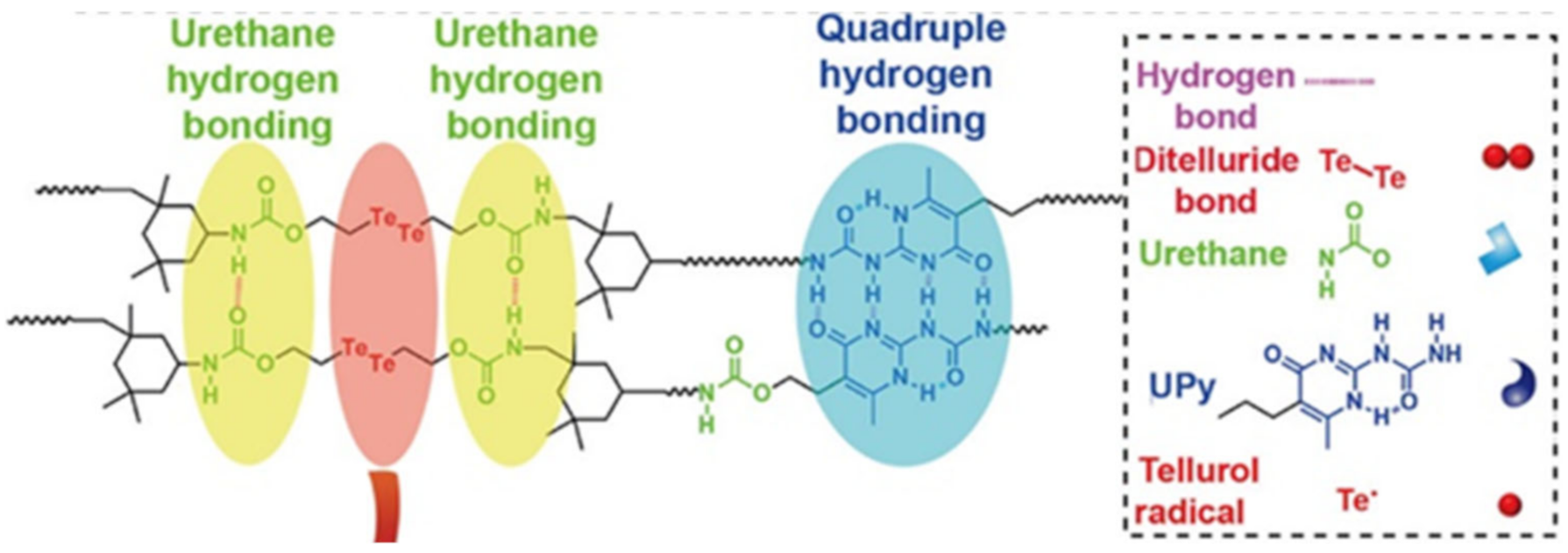
- Fan, W.; Jin, Y.; Shi, L.; Du, W.; Zhou, R.; Lai, S.; Shen, Y.; Li, Y. Simultaneously Achieving Fast Self-Healing and Reprocessing of Supertough Water-Dispersed “Living” Supramolecular Polymers Containing Dynamic Ditelluride Bonds under Visible Light. ACS Appl. Mater. Interfaces 2020, 12, 6383–6395. [Google Scholar] [CrossRef]
- Wei, X.; Ma, K.; Cheng, Y.; Sun, L.; Chen, D.; Zhao, X.; Lu, H.; Song, B.; Yang, K.; Jia, P. Adhesive, Conductive, Self-Healing, and Antibacterial Hydrogel Based on Chitosan–Polyoxometalate Complexes for Wearable Strain Sensor. ACS Appl. Polym. Mater. 2020, 2, 2541–2549. [Google Scholar] [CrossRef]
- Wang, T.; Zhang, Y.; Liu, Q.; Cheng, W.; Wang, X.; Pan, L.; Xu, B.; Xu, H. A Self-Healable, Highly Stretchable, and Solution Processable Conductive Polymer Composite for Ultrasensitive Strain and Pressure Sensing. Adv. Funct. Mater. 2018, 28, 1705551. [Google Scholar] [CrossRef]
- Akbar, Z.A.; Jeon, J.-W.; Jang, S.-Y. Intrinsically self-healable, stretchable thermoelectric materials with a large ionic Seebeck effect. Energy Environ. Sci. 2020, 13, 2915–2923. [Google Scholar] [CrossRef]
- Feng, E.; Gao, W.; Li, J.; Wei, J.; Yang, Q.; Li, Z.; Ma, X.; Zhang, T.; Yang, Z. Stretchable, Healable, Adhesive, and Redox-Active Multifunctional Supramolecular Hydrogel Based Flexible Supercapacitor. ACS Sustainable Chem. Eng. 2020, 8, 3311–3320. [Google Scholar] [CrossRef]
- Yang, R.; Yao, Y.; Duan, Z.; Yuan, Z.; Tai, H.; Jiang, Y.; Zheng, Y.; Wang, D. Constructing Electrically and Mechanically Self-Healing Elastomers by Hydrogen Bonded Intermolecular Network. Langmuir 2020, 36, 3029–3037. [Google Scholar] [CrossRef]
- Duan, N.; Sun, Z.; Ren, Y.; Liu, Z.; Liu, L.; Yan, F. Imidazolium-based ionic polyurethanes with high toughness, tunable healing efficiency and antibacterial activities. Polym. Chem. 2020, 11, 867–875. [Google Scholar] [CrossRef]
- Liu, S.; Qiu, Y.; Yu, W.; Zhang, X. Highly Stretchable and Self-Healing Strain Sensor Based on Gellan Gum Hybrid Hydrogel for Human Motion Monitoring. ACS Appl. Polym. Mater. 2020, 2, 1325–1334. [Google Scholar] [CrossRef]
- Lima, G.M.R.; Orozco, F.; Picchioni, F.; Moreno-Villoslada, I.; Pucci, A.; Bose, R.K.; Araya-Hermosilla, R. Electrically Self-Healing Thermoset MWCNTs Composites Based on Diels-Alder and Hydrogen Bonds. Polymers 2019, 11, 1885. [Google Scholar]
- Su, X.; Wang, H.; Tian, Z.; Duan, X.; Chai, Z.; Feng, Y.; Wang, Y.; Fan, Y.; Huang, J. A Solvent Co-cross-linked Organogel with Fast Self-Healing Capability and Reversible Adhesiveness at Extreme Temperatures. ACS Appl. Mater. Interfaces 2020, 12, 29757–29766. [Google Scholar] [CrossRef]
- Song, M.; Yu, H.-Y.; Zhu, J.; Ouyang, Z.; Abdalkarim, S.Y.H.; Tam, K.C.; Li, Y. Constructing stimuli-free self-healing, robust and ultrasensitive biocompatible hydrogel sensors with conductive cellulose nanocrystals. Chem. Eng. J. 2020, 398, 125547. [Google Scholar] [CrossRef]
- Liao, H.; Guo, X.; Wan, P.; Yu, G. Conductive MXene Nanocomposite Organohydrogel for Flexible, Healable, Low-Temperature Tolerant Strain Sensors. Adv. Funct. Mater. 2019, 29, 1904507. [Google Scholar] [CrossRef]
- Chen, Y.; Pu, X.; Liu, M.; Kuang, S.; Zhang, P.; Hua, Q.; Cong, Z.; Guo, W.; Hu, W.; Wang, Z.L. Shape-Adaptive, Self-Healable Triboelectric Nanogenerator with Enhanced Performances by Soft Solid-Solid Contact Electrification. ACS Nano 2019, 13, 8936–8945. [Google Scholar] [CrossRef]
- Wang, P.; Pei, D.; Wang, Z.; Li, M.; Ma, X.; You, J.; Li, C. Biocompatible and self-healing ionic gel skin as shape-adaptable and skin-adhering sensor of human motions. Chem. Eng. J. 2020, 398, 125540. [Google Scholar] [CrossRef]
- Shahidzadeh, M.; Varkaneh, Z.K.; Ramezanzadeh, B.; Pedram, M.Z.; Yarmohammadi, M. Self-healing dual cured polyurethane elastomeric coatings prepared by orthogonal reactions. Prog. Org. Coat. 2020, 140, 105503. [Google Scholar] [CrossRef]
- Dai, X.; Du, Y.; Wang, Y.; Liu, Y.; Xu, N.; Li, Y.; Shan, D.; Xu, B.B.; Kong, J. Stretchable Self-Healing Polymeric Networks with Recyclability and Dual Responsiveness. ACS Appl. Polym. Mater. 2020, 2, 1065–1072. [Google Scholar] [CrossRef]
- Li, Y.; Guo, W.; Li, W.; Liu, X.; Zhu, H.; Zhang, J.; Liu, X.; Wei, L.; Sun, A. Tuning hard phase towards synergistic improvement of toughness and self-healing ability of poly(urethane urea) by dual chain extenders and coordinative bonds. Chem. Eng. J. 2020, 393, 124583. [Google Scholar] [CrossRef]
- Chen, W.; Bu, Y.; Li, D.; Liu, Y.; Chen, G.; Wan, X.; Li, N. Development of high-strength, tough, and self-healing carboxymethyl guar gum-based hydrogels for human motion detection. J. Mater. Chem. C 2020, 8, 900–908. [Google Scholar] [CrossRef]
- Chen, Y.; Tang, Z.; Liu, Y.; Wu, S.; Guo, B. Mechanically robust, self-healable, and reprocessable elastomers enabled by dynamic dual cross-links. Macromolecules 2019, 52, 3805–3812. [Google Scholar] [CrossRef]
- Rao, V.K.; Shauloff, N.; Sui, X.; Wagner, H.D.; Jelinek, R. Polydiacetylene hydrogel self-healing capacitive strain sensor. J. Mater. Chem. C 2020, 8, 6034–6041. [Google Scholar] [CrossRef]
- Li, S.; Pan, H.; Wang, Y.; Sun, J. Polyelectrolyte complex-based self-healing, fatigue-resistant and anti-freezing hydrogels as highly sensitive ionic skins. J. Mater. Chem. A 2020, 8, 3667–3675. [Google Scholar] [CrossRef]
- Tie, J.; Rong, L.; Liu, H.; Wang, B.; Mao, Z.; Zhang, L.; Zhong, Y.; Feng, X.; Sui, X.; Xu, H. An autonomously healable, highly stretchable and cyclically compressible, wearable hydrogel as a multimodal sensor. Polym. Chem. 2020, 11, 1327–1336. [Google Scholar] [CrossRef]
- Zhao, P.; Yin, C.; Zhang, Y.; Chen, X.; Yang, B.; Xia, J.; Bian, L. Mussel cuticle-mimetic ultra-tough, self-healing elastomers with double-locked nanodomains exhibit fast stimuli-responsive shape transformation. J. Mater. Chem. A 2020, 8, 12463–12471. [Google Scholar] [CrossRef]
- Wu, X.; Luo, R.; Li, Z.; Wang, J.; Yang, S. Readily self-healing polymers at subzero temperature enabled by dual cooperative crosslink strategy for smart paint. Chem. Eng. J. 2020, 398, 125593. [Google Scholar] [CrossRef]
- Xu, S.; Sheng, D.; Zhou, Y.; Wu, H.; Xie, H.; Tian, X.; Sun, Y.; Liu, X.; Yang, Y. A dual supramolecular crosslinked polyurethane with superior mechanical properties and autonomous self-healing ability. New J. Chem. 2020, 44, 7395–7400. [Google Scholar] [CrossRef]
- Zhang, S.; Xu, B.; Lu, X.; Wang, L.; Li, Y.; Ma, N.; Wei, H.; Zhang, X.; Wang, G. Readily producing a Silly Putty-like hydrogel with good self-healing, conductive and photothermal conversion properties based on dynamic coordinate bonds and hydrogen bonds. J. Mater. Chem. C 2020, 8, 6763–6770. [Google Scholar] [CrossRef]
- Cao, C.; Yi, B.; Zhang, J.; Hou, C.; Wang, Z.; Lu, G.; Huang, X.; Yao, X. Sprayable Superhydrophobic Coating with High Processibility and Rapid Damage-Healing Nature. Chem. Eng. J. 2020, 392, 124834. [Google Scholar] [CrossRef]
- Xia, L.; Huang, L.; Qing, Y.; Zhang, X.; Wu, Y.; Jiang, W.; Lu, X. In situ filling of a robust carbon sponge with hydrogel electrolyte: A type of omni-healable electrode for flexible supercapacitors. J. Mater. Chem. A 2020, 8, 7746–7755. [Google Scholar] [CrossRef]
- Zhao, W.; Qu, X.; Xu, Q.; Lu, Y.; Yuan, W.; Wang, W.; Wang, Q.; Huang, W.; Dong, X. Ultrastretchable, Self-Healable, and Wearable Epidermal Sensors Based on Ultralong Ag Nanowires Composited Binary-Networked Hydrogels. Adv. Electron. Mater. 2020, 6, 2000267. [Google Scholar] [CrossRef]
- Gao, Z.; Kong, L.; Jin, R.; Liu, X.; Hu, W.; Gao, G. Mechanical, adhesive and self-healing ionic liquid hydrogels for electrolytes and flexible strain sensors. J. Mater. Chem. C 2020, 8, 11119–11127. [Google Scholar] [CrossRef]
- Liu, J.; Xiao, C.; Tang, J.; Liu, Y.; Hua, J. Construction of a Dual Ionic Network in Natural Rubber with High Self-healing Efficiency through Anionic Mechanism. Ind. Eng. Chem. Res. 2020, 59, 12755–12765. [Google Scholar] [CrossRef]
- Ge, G.; Yuan, W.; Zhao, W.; Lu, Y.; Zhang, Y.; Wang, W.; Chen, P.; Huang, W.; Si, W.; Dong, X. Highly stretchable and autonomously healable epidermal sensor based on multi-functional hydrogel frameworks. J. Mater. Chem. A 2019, 7, 5949–5956. [Google Scholar] [CrossRef]
- Ding, C.; Yang, Q.; Tian, M.; Guo, C.; Deng, F.; Dang, Y.; Zhang, M. Novel collagen-based hydrogels with injectable, self-healing, wound-healing properties via a dynamic crosslinking interaction. Polym. Int. 2020, 69, 858–866. [Google Scholar] [CrossRef]
- Jing, X.; Li, H.; Mi, H.-Y.; Liu, Y.-J.; Feng, P.-Y.; Tan, Y.-M.; Turng, L.-S. Highly Transparent, Stretchable, and Rapid Self-Healing Polyvinyl Alcohol/Cellulose Nanofibril Hydrogel Sensors for Sensitive Pressure Sensing and Human Motion Detection. Sens. Actuators B Chem. 2019, 195, 159–167. [Google Scholar] [CrossRef]
- Zhang, L.; Qiu, T.; Sun, X.; Guo, L.; He, L.; Ye, J.; Li, X. Achievement of Both Mechanical Properties and Intrinsic Self-Healing under Body Temperature in Polyurethane Elastomers: A Synthesis Strategy from Waterborne Polymers. Polymers 2020, 12, 989. [Google Scholar] [CrossRef]
- Ouyang, C.; Zhao, C.; Li, W.; Wu, X.; Le, X.; Chen, T.; Huang, W.; Gao, Q.; Shan, X.; Zhg, R.; et al. Super-Tough, Self-Healing Polyurethane Based on Diels-Alder Bonds and Dynamic Zinc–Ligand Interactions. Macromol. Mater. Eng. 2020, 305, 2000089. [Google Scholar] [CrossRef]
- Sun, X.; Yao, F.; Wang, C.; Qin, Z.; Zhang, H.; Yu, Q.; Zhang, H.; Dong, X.; Wei, Y.; Li, J. Ionically Conductive Hydrogel with Fast Self-Recovery and Low Residual Strain as Strain and Pressure Sensors. Macromol. Rapid Commun. 2020, 41, 2000185. [Google Scholar] [CrossRef]
- Ge, G.; Lu, Y.; Qu, X.; Zhao, W.; Ren, Y.; Wang, W.; Wang, Q.; Huang, W.; Dong, X. Muscle-Inspired Self-Healing Hydrogels for Strain and Temperature Sensor. ACS Nano 2020, 14, 218–228. [Google Scholar] [CrossRef]
- Deng, Y.; Zhang, Q.; Feringa, B.L.; Tian, H.; Qu, D.-H. Toughening a Self-Healable Supramolecular Polymer by Ionic Cluster-Enhanced Iron-Carboxylate Complexes. Angew. Chem. Int. Ed. 2020, 59, 5278–5283. [Google Scholar] [CrossRef]
- Xu, M.; Cheng, B.; Sheng, Y.; Zhou, J.; Wang, M.; Jiang, X.; Lu, X. High-Performance Cross-Linked Self-Healing Material Based on Multiple Dynamic Bonds. ACS Appl. Polym. Mater. 2020, 2, 2228–2237. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, Z.; Wu, X.; Guan, Q.; Chen, S.; Sun, L.; Guo, Y.; Wang, S.; Song, J.; Jeffries, E.M.; et al. A Highly Efficient Self-Healing Elastomer with Unprecedented Mechanical Properties. Adv. Mater. 2019, 31, 1901402. [Google Scholar] [CrossRef]
- Menon, A.V.; Madras, G.; Bose, S. The journey of self-healing and shape memory polyurethanes from bench to translational research. Polym. Chem. 2019, 10, 4370–4388. [Google Scholar] [CrossRef]
- Van Herck, N.; Du Prez, F.E. Fast Healing of Polyurethane Thermosets Using Reversible Triazolinedione Chemistry and Shape-Memory. Macromolecules 2018, 51, 3405–3414. [Google Scholar] [CrossRef]
- Yang, Y.; Davydovich, D.; Hornat, C.C.; Liu, X.; Urban, M.W. Leaf-Inspired Self-Healing Polymers. Chem 2018, 4, 1928–1936. [Google Scholar] [CrossRef]
- Fan, L.F.; Rong, M.Z.; Zhang, M.Q.; Chen, X.D. Repeated Intrinsic Self-Healing of Wider Cracks in Polymer via Dynamic Reversible Covalent Bonding Molecularly Combined with a Two-Way Shape Memory Effect. ACS Appl. Mater. Interfaces 2018, 10, 38538–38546. [Google Scholar] [CrossRef] [PubMed]
- Lai, H.-Y.; Wang, H.-Q.; Lai, J.-C.; Li, C.-H. A Self-Healing and Shape Memory Polymer that Functions at Body Temperature. Molecules 2019, 24, 3224. [Google Scholar] [CrossRef]
- Lu, W.; Le, X.; Zhang, J.; Huang, Y.; Chen, T. Supramolecular shape memory hydrogels: A new bridge between stimuli-responsive polymers and supramolecular chemistry. Chem. Soc. Rev. 2017, 46, 1284–1294. [Google Scholar] [CrossRef]
- Wang, K.; Jia, Y.-G.; Zhao, C.; Zhu, X. Multiple and two-way reversible shape memory polymers: Design strategies and applications. Prog. Mater. Sci. 2019, 105, 100572. [Google Scholar] [CrossRef]
- Ban, J.; Mu, L.; Yang, J.; Chen, S.; Zhuo, H. New stimulus-responsive shape-memory polyurethanes capable of UV light-triggered deformation, hydrogen bond-mediated fixation, and thermal-induced recovery. J. Mater. Chem. A 2017, 5, 14514–14518. [Google Scholar] [CrossRef]
- Chen, H.-M.; Wang, L.; Zhou, S.-B. Recent Progress in Shape Memory Polymers for Biomedical Applications. Chin. J. Polym. Sci. 2018, 36, 905–917. [Google Scholar] [CrossRef]
- Jiang, Z.-C.; Xiao, Y.-Y.; Kang, Y.; Pan, M.; Li, B.-J.; Zhang, S. Shape Memory Polymers Based on Supramolecular Interactions. ACS Appl. Mater. Interfaces 2017, 9, 20276–20293. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Luo, H.; Gao, D.; Zhou, X.; Cui, P.; Thangavel, G.; Parida, K.; Lee, P.S. Self-restoring, waterproof, tunable microstructural shape memory triboelectric nanogenerator for self-powered water temperature sensor. Nano Energy 2019, 61, 584–593. [Google Scholar] [CrossRef]
- Xu, W.; Wong, M.-C.; Guo, Q.; Jia, T.; Hao, J. Healable and shape-memory dual functional polymers for reliable and multipurpose mechanical energy harvesting devices. J. Mater. Chem. A 2019, 7, 16267–16276. [Google Scholar] [CrossRef]
- Lee, J.H.; Hinchet, R.; Kim, S.K.; Kim, S.; Kim, S.-W. Shape memory polymer-based self-healing triboelectric nanogenerator. Energy Environ. Sci. 2015, 8, 3605–3613. [Google Scholar] [CrossRef]
- Wu, W.; Kurup, S.N.; Ellingford, C.; Li, J.; Wan, C. Coupling Dynamic Covalent Bonds and Ionic Crosslinking Network to Promote Shape Memory Properties of Ethylene-vinyl Acetate Copolymers. Polymers 2020, 12, 983. [Google Scholar] [CrossRef]
- Zhao, L.; Jiang, B.; Huang, Y. Self-healable polysiloxane/graphene nanocomposite and its application in pressure sensor. J. Mater. Sci. 2019, 54, 5472–5483. [Google Scholar] [CrossRef]
- Liu, R.; Kuang, X.; Deng, J.; Wang, Y.-C.; Wang, A.C.; Ding, W.; Lai, Y.-C.; Chen, J.; Wang, P.; Lin, Z.; et al. Shape Memory Polymers for Body Motion Energy Harvesting and Self-Powered Mechanosensing. Adv. Mater. 2018, 30, 1705195. [Google Scholar] [CrossRef]
- Cui, H.; Tian, W.; Kang, Y.; Wang, Y. Characteristics of a novel thermal-induced epoxy shape memory polymer for smart device applications. Mater. Res. Express 2020, 7, 015706. [Google Scholar] [CrossRef]
- Garg, H.; Mohanty, J.; Gupta, P.; Das, A.; Tripathi, B.P.; Kumar, B. Polyethylenimine-Based Shape Memory Polyurethane with Low Transition Temperature and Excellent Memory Performance. Macromol. Mater. Eng. 2020, 305, 2000215. [Google Scholar] [CrossRef]
- Li, A.; Challapalli, A.; Li, G. 4d Print. Recycl. Lightweight Archit. Using High Recovery Stress Shape Mem. Polym. Sci. Rep. 2019, 9, 7621. [Google Scholar]
- Li, A.; Fan, J.; Li, G. Recyclable thermoset shape memory polymers with high stress and energy output via facile Uv-Curing. J. Mater. Chem. A 2018, 6, 11479–11487. [Google Scholar] [CrossRef]
- Konlan, J.; Mensah, P.; Ibekwe, S.; Crosby, K.; Li, G. Vitrimer based composite laminates with shape memory alloy Z-pins for repeated healing of impact induced delamination. Compos. Part. B Eng. 2020, 200, 108324. [Google Scholar] [CrossRef]
- Zhou, Q.; Dong, X.; Xiong, Y.; Zhang, B.; Lu, S.; Wang, Q.; Liao, Y.; Yang, Y.; Wang, H. Multi-Responsive Lanthanide-Based Hydrogel with Encryption, Naked Eye Sensing, Shape Memory, Self-Healing, and Antibacterial Activity. ACS Appl. Mater. Interfaces 2020, 12, 28539–28549. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Pham, H.Q.; Phung, D.T.T.; Truong, T.T.; Nguyen, H.T.; Doan, T.C.D.; Dang, C.M.; Tran, H.L.; Mai, P.T.; Tran, D.T.; et al. Macromolecular design of a reversibly crosslinked shape-memory material with thermo-healability. Polymer 2020, 188, 122144. [Google Scholar] [CrossRef]
- Chen, T.; Fang, L.; Li, X.; Gao, D.; Lu, C.; Xu, Z. Self-healing polymer coatings of polyurea-urethane/epoxy blends with reversible and dynamic bonds. Prog. Org. Coat. 2020, 147, 105876. [Google Scholar] [CrossRef]
- Chen, T.; Fang, L.; Lu, C.; Xu, Z. Effects of Blended Reversible Epoxy Domains on Structures and Properties of Self-Healing/Shape-Memory Thermoplastic Polyurethane. Macromol. Mater. Eng. 2020, 305, 1900578. [Google Scholar] [CrossRef]
- Jiang, L.; Liu, Z.; Lei, Y.; Yuan, Y.; Wu, B.; Lei, J. Sustainable Thermosetting Polyurea Vitrimers Based on a Catalyst-Free Process with Reprocessability, Permanent Shape Reconfiguration and Self-Healing Performance. ACS Appl. Polym. Mater. 2019, 1, 3261–3268. [Google Scholar] [CrossRef]
- Suslu, H.; Fan, J.; Ibekwe, S.; Jerro, D.; Mensah, P.; Li, G. Shape memory alloy reinforced vitrimer composite for healing wide-opened cracks. Smart Mater. Struct. 2020, 29, 065008. [Google Scholar] [CrossRef]
- Leeuwenburgh, S.C.G.; De Belie, N.; van der Zwaag, S. Self-Healing Materials are Coming of Age. Adv. Mater. Interfaces 2018, 5, 1800736. [Google Scholar] [CrossRef]
- Tsou, Y.-H.; Zhang, X.-Q.; Bai, X.; Zhu, H.; Li, Z.; Liu, Y.; Shi, J.; Xu, X. Dopant-Free Hydrogels with Intrinsic Photoluminescence and Biodegradable Properties. Adv. Funct. Mater. 2018, 28, 1802607. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, H.; Liu, R.; Wen, X.; Hou, T.-C.; Wang, Z.L. Fully enclosed triboelectric nanogenerators for applications in water and harsh environments. Adv. Energy Mater. 2013, 3, 1563–1568. [Google Scholar] [CrossRef]
- Qi, J.; Wang, A.C.; Yang, W.; Zhang, M.; Hou, C.; Zhang, Q.; Li, Y.; Wang, H. Hydrogel-based hierarchically wrinkled stretchable nanofibrous membrane for high performance wearable triboelectric nanogenerator. Nano Energy 2020, 67, 104206. [Google Scholar] [CrossRef]
- Wu, C.; Wang, A.C.; Ding, W.; Guo, H.; Wang, Z.L. Triboelectric Nanogenerator: A Foundation of the Energy for the New Era. Adv. Energy Mater. 2019, 9, 1802906. [Google Scholar] [CrossRef]
- Zhu, G.; Peng, B.; Chen, J.; Jing, Q.; Wang, Z.L. Triboelectric nanogenerators as a new energy technology: From fundamentals, devices, to applications. Nano Energy 2015, 14, 126–138. [Google Scholar] [CrossRef]
- Luo, N.; Feng, Y.; Wang, D.; Zheng, Y.; Ye, Q.; Zhou, F.; Liu, W. New Self-Healing Triboelectric Nanogenerator Based on Simultaneous Repair Friction Layer and Conductive Layer. ACS Appl. Mater. Interfaces 2020, 12, 30390–30398. [Google Scholar] [CrossRef]
- Park, S.; Ryu, H.; Park, S.; Hong, H.; Jung, H.Y.; Park, J.-J. Rotating Triboelectric Generator Using Sliding Contact and Noncontact from 1D Fiber Friction. Nano Energy 2017, 33, 184–194. [Google Scholar] [CrossRef]































































Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dzhardimalieva, G.I.; Yadav, B.C.; Kudaibergenov, S.E.; Uflyand, I.E. Basic Approaches to the Design of Intrinsic Self-Healing Polymers for Triboelectric Nanogenerators. Polymers 2020, 12, 2594. https://doi.org/10.3390/polym12112594
Dzhardimalieva GI, Yadav BC, Kudaibergenov SE, Uflyand IE. Basic Approaches to the Design of Intrinsic Self-Healing Polymers for Triboelectric Nanogenerators. Polymers. 2020; 12(11):2594. https://doi.org/10.3390/polym12112594
Chicago/Turabian StyleDzhardimalieva, Gulzhian I., Bal C. Yadav, Sarkyt E. Kudaibergenov, and Igor E. Uflyand. 2020. "Basic Approaches to the Design of Intrinsic Self-Healing Polymers for Triboelectric Nanogenerators" Polymers 12, no. 11: 2594. https://doi.org/10.3390/polym12112594
APA StyleDzhardimalieva, G. I., Yadav, B. C., Kudaibergenov, S. E., & Uflyand, I. E. (2020). Basic Approaches to the Design of Intrinsic Self-Healing Polymers for Triboelectric Nanogenerators. Polymers, 12(11), 2594. https://doi.org/10.3390/polym12112594