Theoretical Modeling of Chemical Equilibrium in Weak Polyelectrolyte Layers on Curved Nanosystems


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Chemical Equilibrium
3. Theoretical Methods
3.1. Molecular Theory
3.2. Self-Consistent Field Approach
Scheutjens–Fleer Lattice Theory
3.3. Analytical Approaches
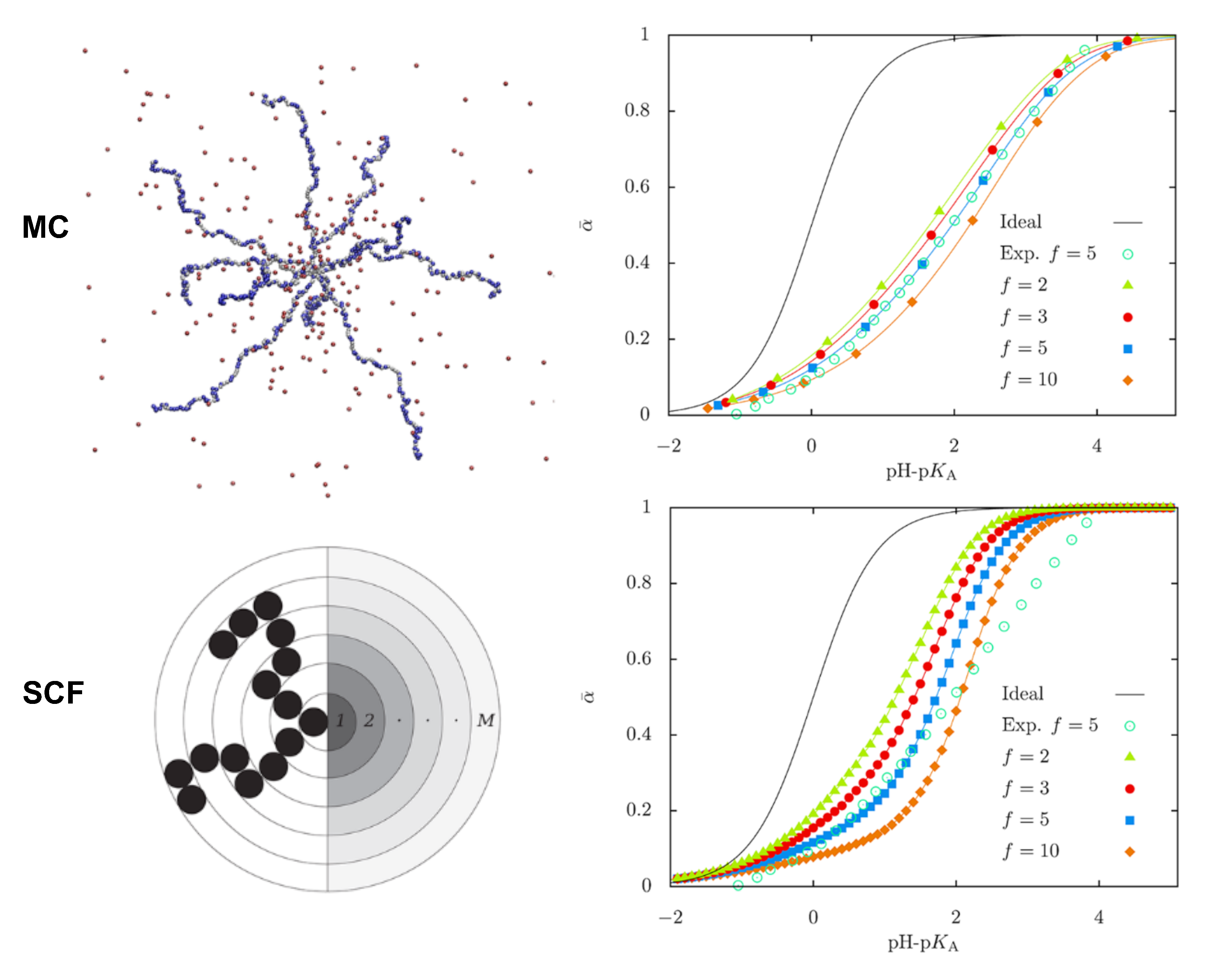
3.4. Simulations
4. Applications
4.1. Nanoparticles
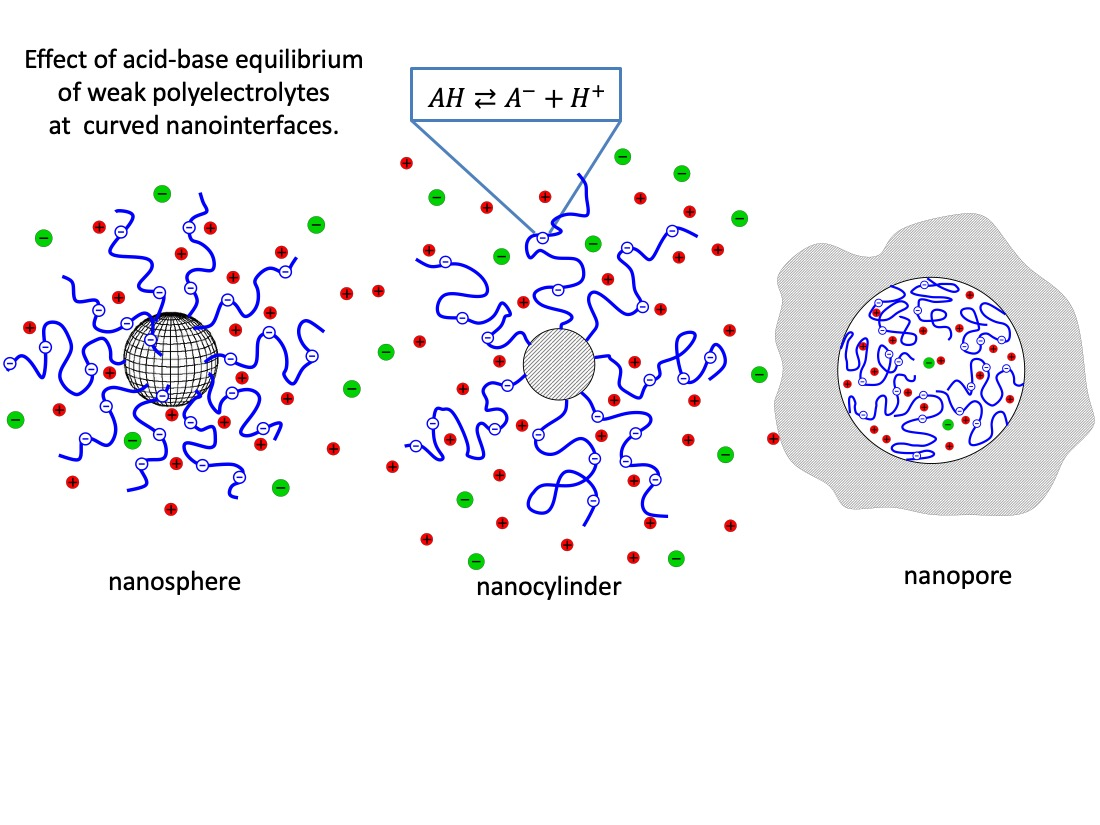
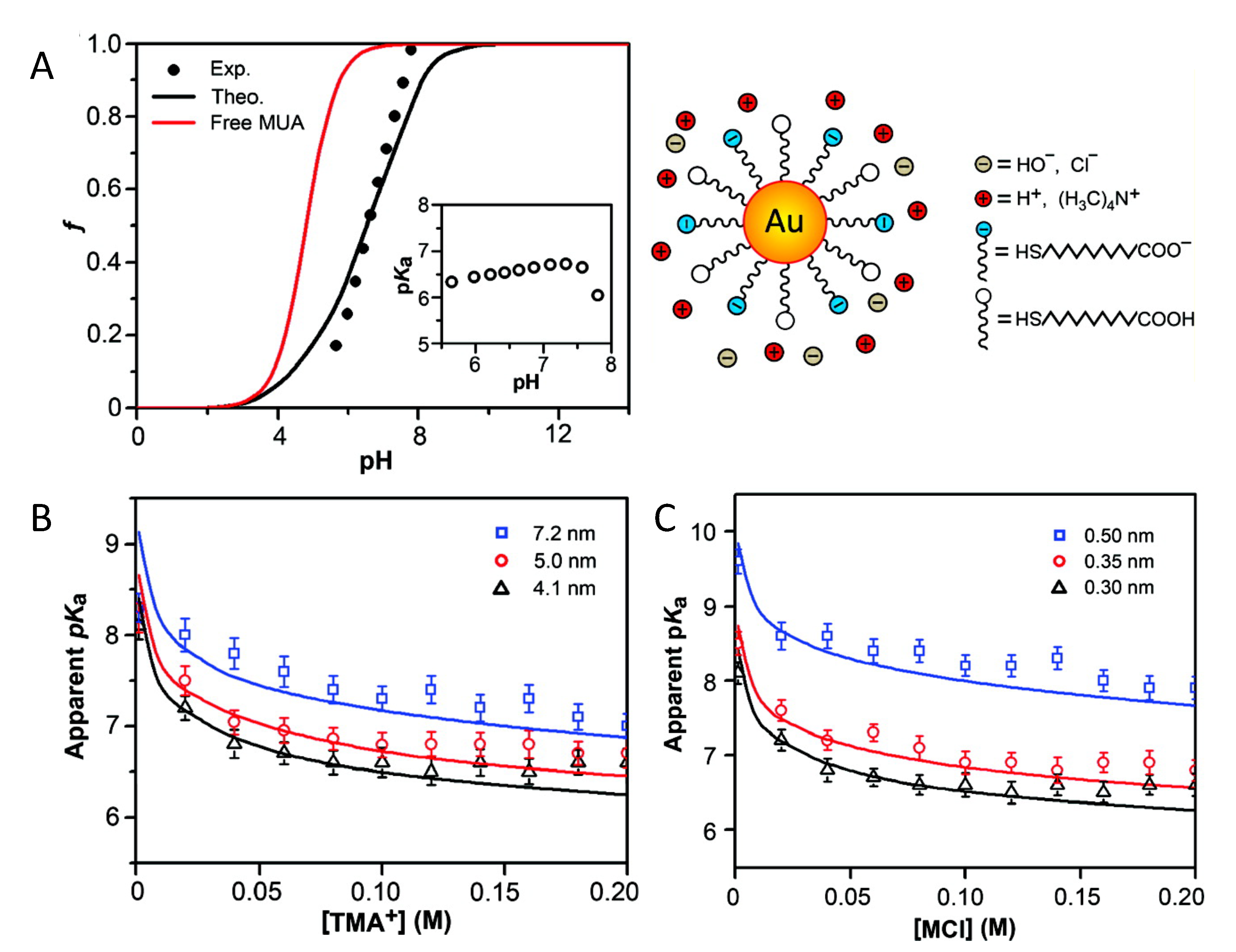
4.1.1. Charge Regulation & Curvature
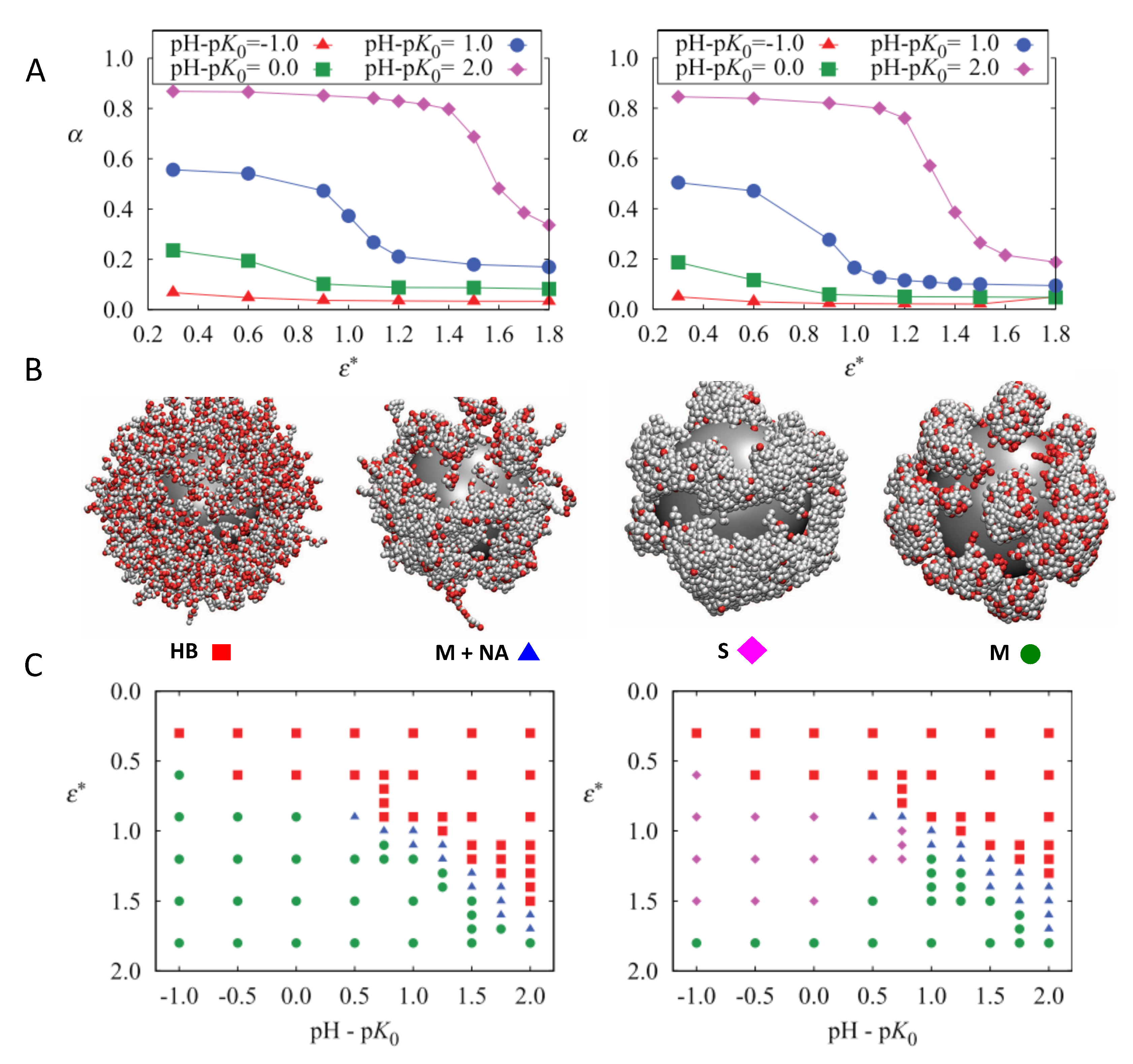
4.1.2. Conformational Properties of Grafted Weak PE
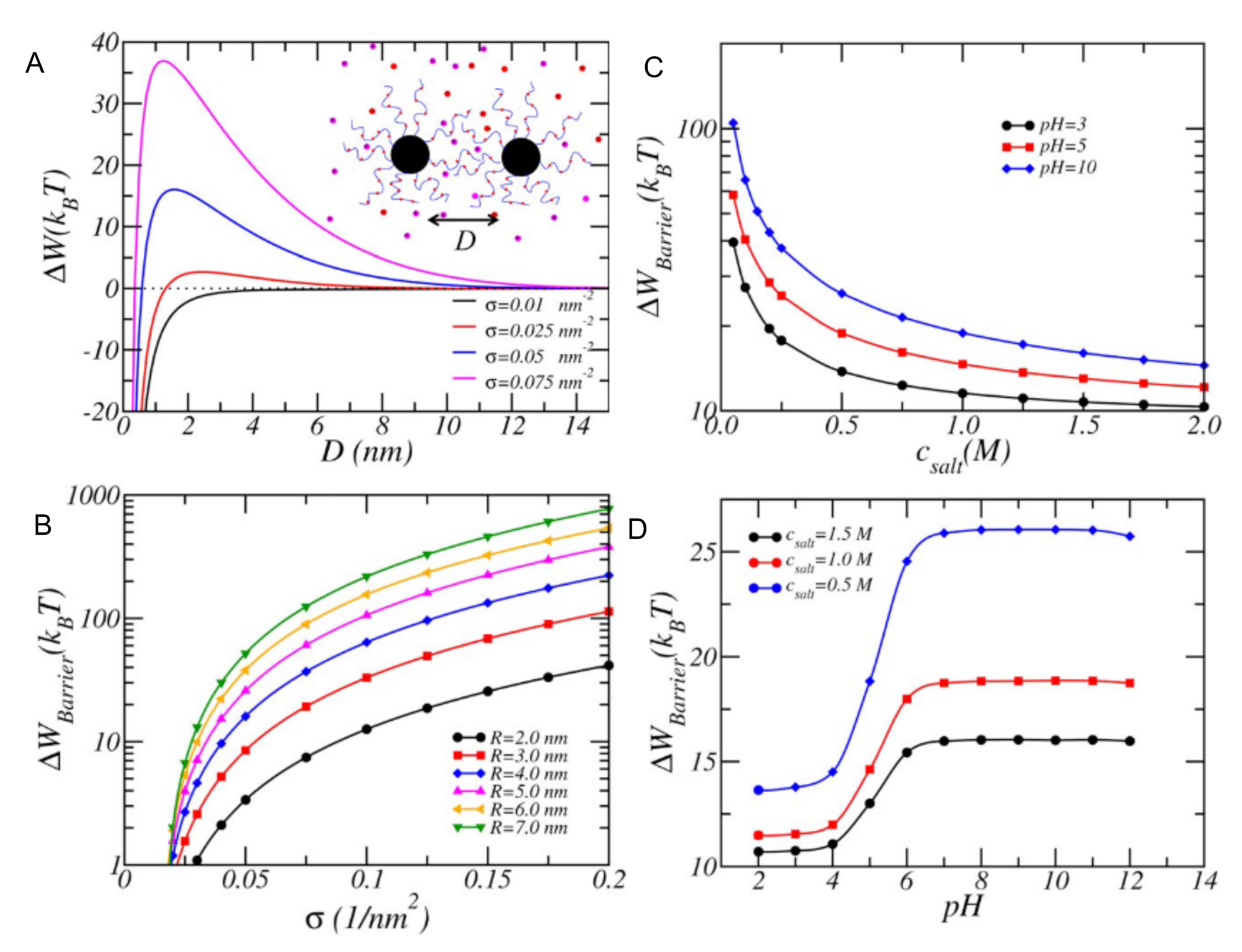
4.1.3. Interactions between Weak PE Coated NPs
4.1.4. Interactions between Weak PE-Coated NPs and Surfaces
4.1.5. Interactions between Weak PE + NPs and Surfaces
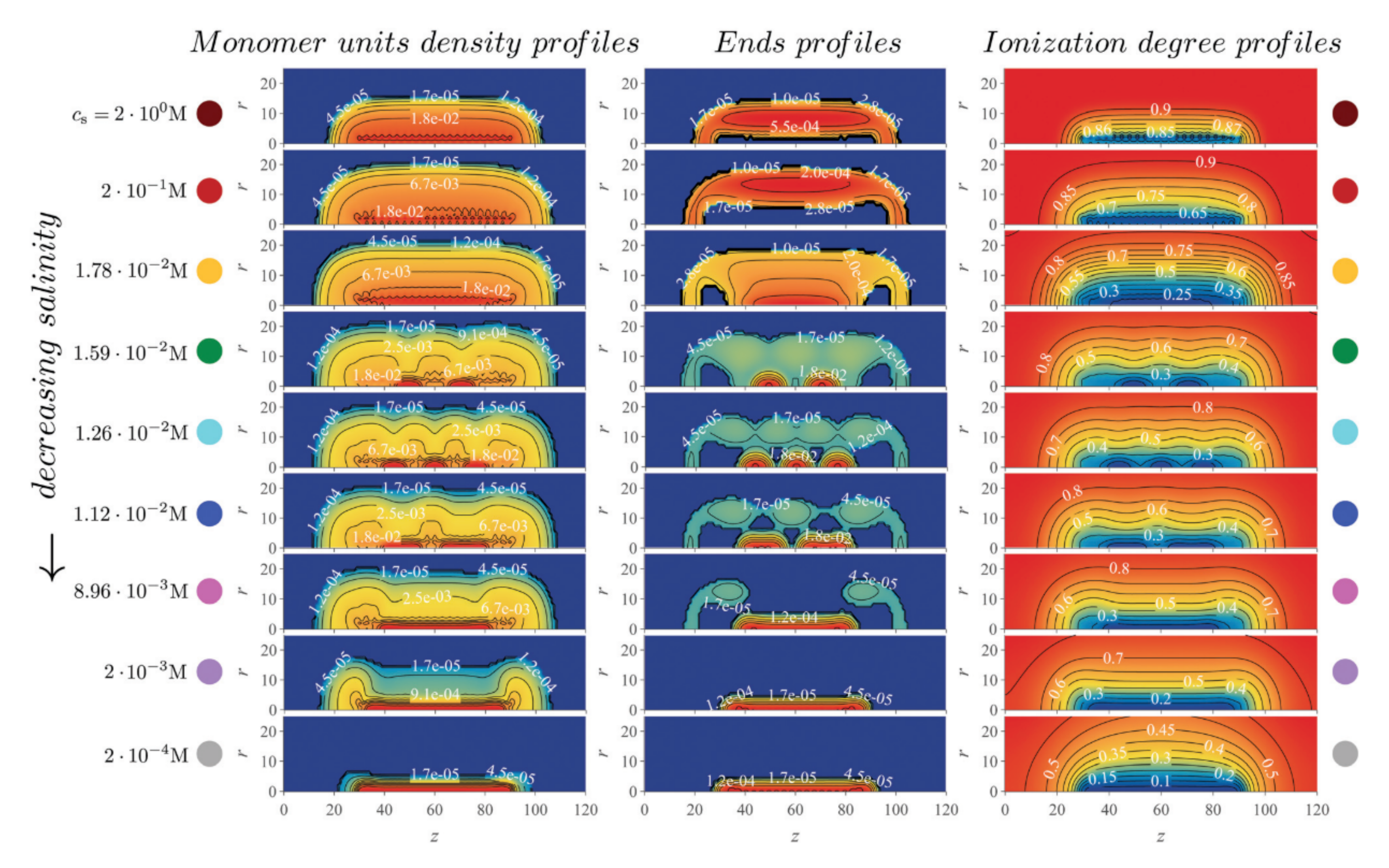
4.2. Star and Bottle-Brush Weak PE
4.3. Biomacromolecules
4.3.1. Neurofilaments
4.3.2. Aggrecans
4.3.3. Viral Capsids and Gene Vectors
4.3.4. Proteins
4.4. Beyond Acid–Base Equilibrium
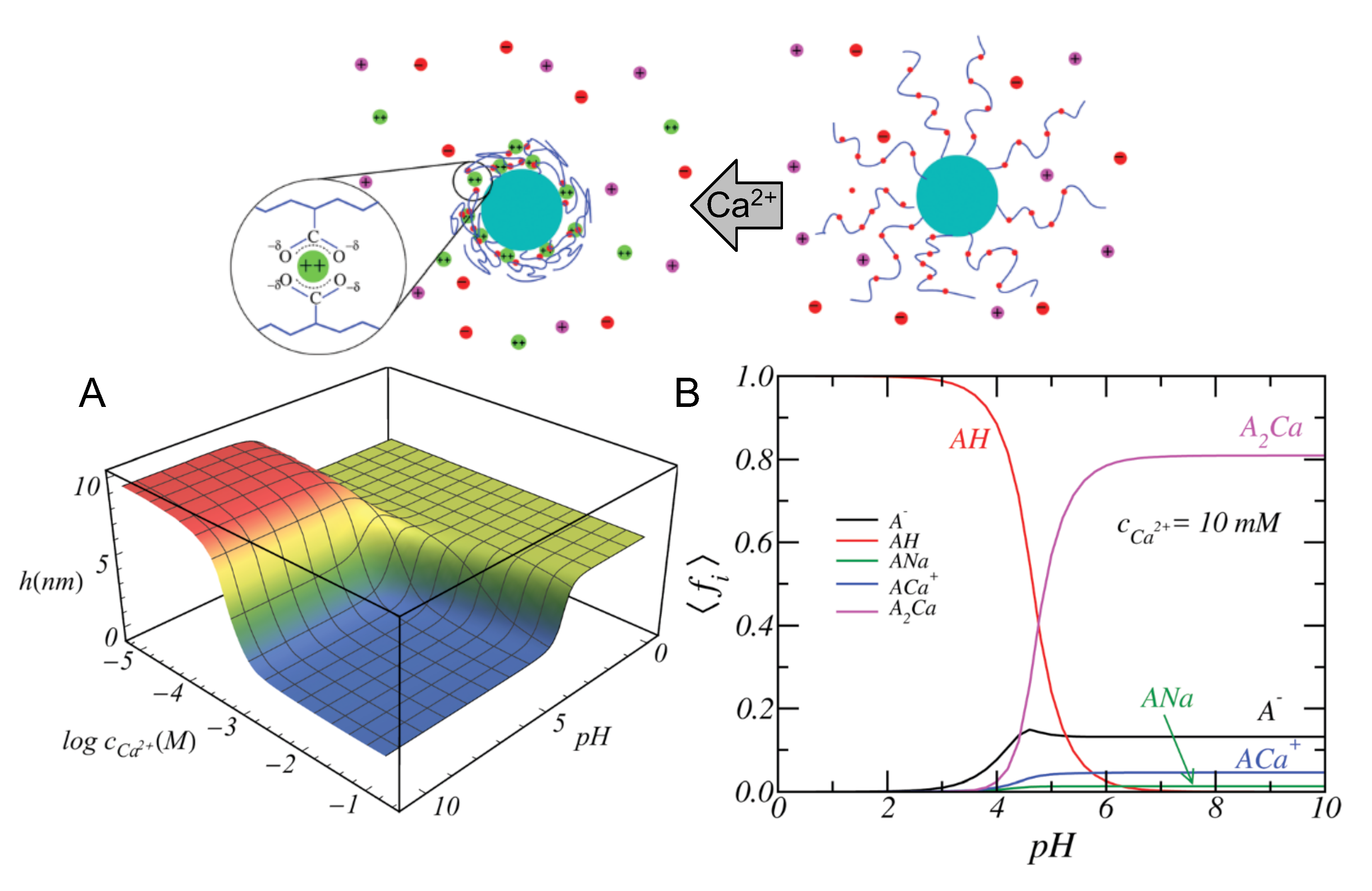
4.4.1. Ion Binding
4.4.2. Ligand–Receptor Binding
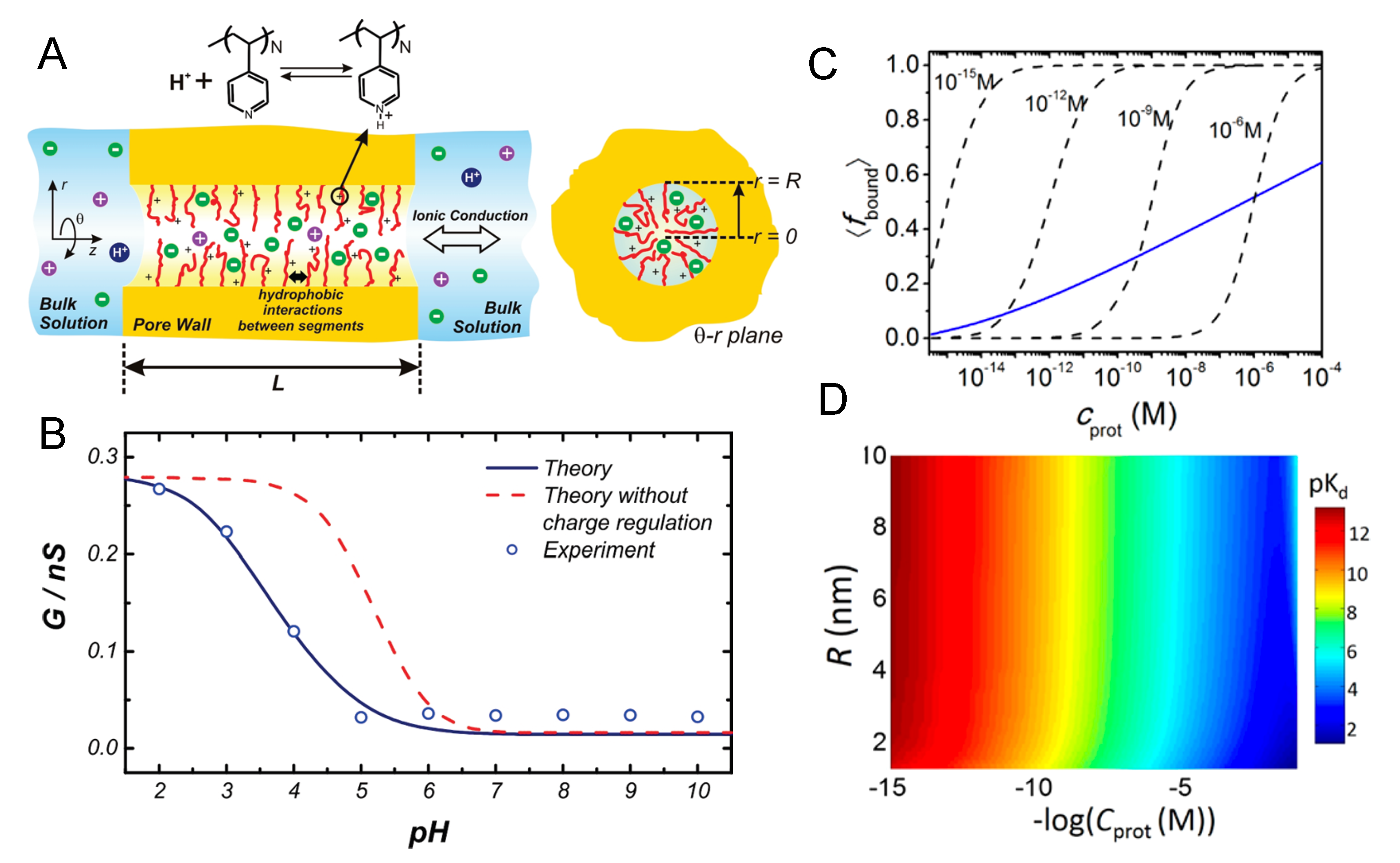
4.5. Artificial Nanopores
4.6. The Nuclear Pore Complex
5. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MD | Molecular Dynamics |
| MC | Monte Carlo |
| SCF | Self-consistent field |
| SCFT | Self-consistent field theory |
| SF-SCF | Scheujtens–Fleer Self-consistent field |
| MT | Molecular Theory |
| Probability Distribution Function | |
| DFT | Density Functional Theory |
| PE | Polyelectrolyte |
| NP | Nanoparticle |
| AFM | Atomic Force Microscopy |
References
- Tadmor, R.; Janik, J.; Klein, J.; Fetters, L.J. Sliding Friction with Polymer Brushes. Phys. Lett. 2003, 91, 115503. [Google Scholar] [CrossRef] [PubMed]
- Raviv, U.; Giasson, S.; Kampf, N.; Gohy, J.F.; Jérôme, R.; Klein, J. Normal and Frictional Forces between Surfaces Bearing Polyelectrolyte Brushes. Langmuir 2008, 24, 8678–8687. [Google Scholar] [CrossRef] [PubMed]
- Raviv, U.; Giasson, S.; Kampf, N.; Gohy, J.F.; Jérôme, R.; Klein, J. Lubrication by charged polymers. Nature 2003, 425, 163–165. [Google Scholar] [CrossRef] [PubMed]
- Cohen Stuart, M.A.; Huck, W.T.S.; Genzer, J.; Müller, M.; Ober, C.; Stamm, M.; Sukhorukov, G.B.; Szleifer, I.; Tsukruk, V.V.; Urban, M.; et al. Emerging applications of stimuli-responsive polymer materials. Nat. Mater. 2010, 9, 101–113. [Google Scholar] [CrossRef]
- Pendergast, M.M.; Hoek, E.M. A review of water treatment membrane nanotechnologies. Energy Environ. Sci. 2011, 4, 1946–1971. [Google Scholar] [CrossRef]
- Haywood, D.G.; Saha-Shah, A.; Baker, L.A.; Jacobson, S.C. Fundamental Studies of Nanofluidics: Nanopores, Nanochannels, and Nanopipets. Anal. Chem. 2015, 87, 172–187. [Google Scholar] [CrossRef]
- Shi, W.; Friedman, A.K.; Baker, L.A. Nanopore Sensing. Anal. Chem. 2017, 89, 157–188. [Google Scholar] [CrossRef]
- Ramos, A.P.; Cruz, M.A.; Tovani, C.B.; Ciancaglini, P. Biomedical applications of nanotechnology. Biophys. Rev. 2017, 9, 79–89. [Google Scholar] [CrossRef]
- Movassaghian, S.; Merkel, O.M.; Torchilin, V.P. Applications of polymer micelles for imaging and drug delivery. WIREs Nanomed. Nanobiotechnol. 2015, 7, 691–707. [Google Scholar] [CrossRef]
- Tanaka, M.; Sato, K.; Kitakami, E.; Kobayashi, S.; Hoshiba, T.; Fukushima, K. Design of biocompatible and biodegradable polymers based on intermediate water concept. Polym. J. 2015, 47, 114–121. [Google Scholar] [CrossRef]
- Kocak, G.; Tuncer, C.; Bütün, V. pH-Responsive polymers. Polym. Chem. 2017, 8, 144–176. [Google Scholar] [CrossRef]
- Netz, R.R.; Andelman, D. Neutral and charged polymers at interfaces. Phys. Rep. 2003, 380, 1–95. [Google Scholar] [CrossRef]
- Naji, A.; Seidel, C.; Netz, R.R. Theoretical Approaches to Neutral and Charged polymers. Adv. Polym. Sci. 2006, 198, 149–183. [Google Scholar] [CrossRef]
- Katchalsky, A. Polyelectrolytes. Pure Appl. Chem. 1971, 26, 327–373. [Google Scholar] [CrossRef]
- Katchalsky, A.; Shavit, N.; Eisenberg, H. Dissociation of weak polymeric acids and bases. J. Polym. Sci. 1954, 13, 69–84. [Google Scholar] [CrossRef]
- Katchalsky, A.; Spitnik, P. Potentiometric titrations of polymethacrylic acid. J. Polym. Sci. 1947, 2, 432–446. [Google Scholar] [CrossRef]
- Alexandrowicz, Z.; Katchalsky, A. Colligative properties of polyelectrolyte solutions in excess of salt. J. Polym. Sci. A Gen. Pap. 1963, 1, 3231–3260. [Google Scholar] [CrossRef]
- Nap, R.; Gong, P.; Szleifer, I. Weak polyelectrolytes tethered to surfaces: Effect of geometry, acid-base equilibrium and electrical permittivity. J. Polym. Sci. Part B Polym. Phys. 2006, 44, 2638–2662. [Google Scholar] [CrossRef]
- Nap, R.J.; Tagliazucchi, M.; Gonzalez Solveyra, E.; Ren, C.L.; Uline, M.J.; Szleifer, I. Modeling of Chemical Equilibria in Polymer and Polyelectrolyte Brushes. In Polymer and Biopolymer Brushes; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2017; Chapter 6; pp. 161–221. [Google Scholar] [CrossRef]
- Schwinger, J.; DeRaad, L.L., Jr.; Milton, K.A.; Tsai, W.Y. Classical Electrodynamics; Persus Books: Reading, MA, USA, 1998. [Google Scholar]
- Wang, Z.G. Variational electrostatics for charge solvation. J. Theor. Comput. Chem. 2008, 7, 397–419. [Google Scholar] [CrossRef]
- Raphael, E.; Joanny, J. Annealed and Quenched Polyelectrolytes. Europhys. Lett. 1990, 13, 623–628. [Google Scholar] [CrossRef]
- Szleifer, I.; Carignano, M.A. Tethered polymer layers. Adv. Chem. Phys. 1996, 94, 165–260. [Google Scholar]
- Gong, P.; Wu, T.; Genzer, J.; Szleifer, I. Behavior of Surface-Anchored Poly(acrylic acid) Brushes with Grafting Density Gradients on Solid Substrates: 2. Theory. Macromolecules 2007, 40, 8765–8773. [Google Scholar] [CrossRef]
- Wu, T.; Gong, P.; Szleifer, I.; Vlček, P.; Šubr, V.; Genzer, J. Behavior of Surface-Anchored Poly(acrylic acid) Brushes with Grafting Density Gradients on Solid Substrates: 1. Experiment. Macromolecules 2007, 40, 8756–8764. [Google Scholar] [CrossRef]
- Wang, D.; Nap, R.J.; Lagzi, I.; Kowalczyk, B.; Han, S.; Grzybowski, B.A.; Szleifer, I. How and why nanoparticle’s curvature regulates the apparent pKa of the coating ligands. J. Am. Chem. Soc. 2011, 133, 2192–2197. [Google Scholar] [CrossRef] [PubMed]
- Tagliazucchi, M.; Azzaroni, O.; Szleifer, I. Responsive Polymers End-Tethered in Solid-State Nanochannels: When Nanoconfinement Really Matters. J. Am. Chem. Soc. 2010, 132, 12404–12411. [Google Scholar] [CrossRef] [PubMed]
- Tagliazucchi, M.; Calvo, E.J.; Szleifer, I. Molecular theory of chemically modified electrodes by redox polyelectrolytes under equilibrium conditions: Comparison with experiment. J. Phys. Chem. C 2008, 112, 458–471. [Google Scholar] [CrossRef]
- Hehmeyer, O.J.; Arya, G.; Panagiotopoulos, A.Z.; Szleifer, I. Monte Carlo simulation and molecular theory of tethered polyelectrolytes. J. Chem. Phys. 2007, 126, 244902. [Google Scholar] [CrossRef]
- Nap, R.J.; Park, S.H.; Szleifer, I. Competitive calcium ion binding to end-tethered weak polyelectrolytes. Soft Matter 2018, 14, 2365–2378. [Google Scholar] [CrossRef]
- Doi, M.; Edwards, S.F. The Theory of Polymer Dynamcis; Clarendon Press: Oxford, UK, 1986. [Google Scholar]
- Fredrickson, G.H. The Equilibrium Theory of Inhomogeneous Polymers; Clarendon Press: Oxford, UK, 2006. [Google Scholar]
- Matsen, M.W. The standard Gaussian model for block copolymer melts. J. Phys. Condens. Matter 2002, 14, R21–R47. [Google Scholar] [CrossRef]
- Schmid, F. Self-consistent-field theories for complex fluids. Phys. Condens. Matter 1998, 10, 8105–8138. [Google Scholar] [CrossRef]
- Netz, R.R.; Schick, M. Polymer brushes:From Self-consistent field theory to classical theory. Macromolecules 1998, 31, 5105–5122. [Google Scholar] [CrossRef] [PubMed]
- Milner, S.T. Polymer brushes. Science 1991, 251, 905–914. [Google Scholar] [CrossRef] [PubMed]
- Milner, S.T.; Witten, T.A.; Cates, M.E. Theory of the grafted polymer brush. Macromolecules 1988, 21, 2610–2619. [Google Scholar] [CrossRef]
- Netz, R.R.; Schick, M. Classical theory of polymer brushes. Europhys. Lett. 1997, 38, 37–42. [Google Scholar] [CrossRef]
- Edwards, S.F. The statistical mechanics of polymers with excluded volume. Proc. Phys. Soc. 1965, 85, 613–624. [Google Scholar] [CrossRef]
- Kim, J.U.; Matsen, M.W. Finite-stretching corrections to the Milner-Witten-Cates theory for polymer brushes. Eur. Phys. J. E 2007, 23, 135–144. [Google Scholar] [CrossRef]
- Reddy, M.S. Biomineralization of calcium carbonates and their engineered applications: A review. Front. Microbiol. 2013, 4, 314. [Google Scholar] [CrossRef]
- Witte, K.N.; Won, Y.Y. Effect of Interfacial Curvature on the Miscibility of Laterally Mobile, Mixed Polyelectrolyte and Neutral Polymer Brushes: An SCF Numerical Analysis. Macromolecules 2008, 41, 2735–2738. [Google Scholar] [CrossRef]
- Witte, K.N.; Kim, S.; Won, Y.Y. Self-Consistent Field Theory Study of the Effect of Grafting Density on the Height of a Weak Polyelectrolyte Brush. J. Phys. Chem. B 2009, 113, 11076–11084. [Google Scholar] [CrossRef]
- Kumar, R.; Sumpter, B.G.; Kilbey, S.M., II. Charge regulation and local dielectric function in planar polyelectrolyte brushes. J. Chem. Phys. 2012, 136, 234901. [Google Scholar] [CrossRef]
- Léonforte, F.; Welling, U.; Müller, M. Single-chain-in-mean-field simulations of weak polyelectrolyte brushes. J. Chem. Phys. 2016, 145, 224902. [Google Scholar] [CrossRef] [PubMed]
- Shi, A.C.; Noolandi, J. Theory of inhomogeneous weakly charged polyelectrolytes. Macromol. Theory Simul. 1999, 8, 214–229. [Google Scholar] [CrossRef]
- Vilgis, T. Polymer theory: Path integrals and scaling. Phys. Rep. 2000, 336, 167–254. [Google Scholar] [CrossRef]
- Fleer, G.J. Polymers at interfaces and in colloidal dispersions. Adv. Colloid Interface Sci. 2010, 159, 99–116. [Google Scholar] [CrossRef] [PubMed]
- Fleer, G.; Cohen Stuart, M.A.; Scheutjens, J.M.H.M.; Cosgrove, T.; Vincent, B. Polymers at Interfaces; Chapman and Hall: London, UK, 1993. [Google Scholar]
- Israëls, R.; Leermakers, F.A.M.; Fleer, G.J.; Zhulina, E.B. Charged Polymeric Brushes: Structure and Scaling Relations. Macromolecules 1994, 27, 3249–3261. [Google Scholar] [CrossRef]
- Israëls, R.; Leermakers, F.A.M.; Fleer, G.J. On the theory of grafted weak polyacids. Macromolecules 1994, 27, 3087–3093. [Google Scholar] [CrossRef]
- Okrugin, B.M.; Richter, R.P.; Leermakers, F.A.M.; Neelov, I.M.; Zhulina, E.B.; Borisov, O.V. Electroresponsive Polyelectrolyte Brushes Studied by Self-Consistent Field Theory. Polymers 2020, 12, 898. [Google Scholar] [CrossRef] [PubMed]
- Leermakers, F.A. Self-Consistent Field Modeling of Pulling a Test-Chain away from or Pushing It into a Polymer Adsorption Layer. Polymers 2020, 12, 1684. [Google Scholar] [CrossRef]
- Borisov, O.V.; Zhulina, E.B.; Leermakers, F.A.M.; Ballauff, M.; Müller, A.H.E. Conformations and Solution Properties of Star-Branched Polyelectrolytes. In Self Organized Nanostructures of Amphiphilic Block Copolymers I; Springer: Berlin/Heidelberg, Germany, 2010; pp. 1–55. [Google Scholar] [CrossRef]
- Borisov, O.V.; Zhulina, E.B.; Leermakers, F.A.M.; Müller, A.H.E. Self-Assembled Structures of Amphiphilic Ionic Block Copolymers: Theory, Self-Consistent Field Modeling and Experiment. In Self Organized Nanostructures of Amphiphilic Block Copolymers I; Springer: Berlin/Heidelberg, Germany, 2011; pp. 57–129. [Google Scholar] [CrossRef]
- De Gennes, P.G. Scaling Concepts in Polymer Physics; Cornell University Press: Ithaca, NY, USA, 1979. [Google Scholar]
- Ballauff, M.; Borisov, O. Polyelectrolyte brushes. Curr. Opin. Colloid Interface Sci. 2006, 11, 316–323. [Google Scholar] [CrossRef]
- Alexander, S. Adsorption of chain molecules with a polar head: A scaling description. J. Phys. France 1977, 38, 983–987. [Google Scholar] [CrossRef]
- De Gennes, P.G. Conformations of Polymers Attached to an Interface. Macromolecules 1980, 13, 1069–1075. [Google Scholar] [CrossRef]
- Lyatskaya, Y.V.; Leermakers, F.A.M.; Fleer, G.J.; Zhulina, E.B.; Birshstein, T.M. Analytical self-consistent-field model of weak polyacid brushes. Macromolecules 1995, 28, 3562–3569. [Google Scholar] [CrossRef]
- Lebedeva, I.O.; Zhulina, E.B.; Borisov, O.V. Self-consistent field theory of polyelectrolyte brushes with finite chain extensibility. J. Chem. Phys. 2017, 146, 214901. [Google Scholar] [CrossRef] [PubMed]
- Pincus, P. Colloid stabilization with grafted polyelectrolytes. Macromolecules 1991, 24, 2912–2919. [Google Scholar] [CrossRef]
- Semenov, A.N. Contribution to the theory of microphase layering in block-copolymer melts. Sov. Phys. JETP 1985, 26, 733–742. [Google Scholar]
- Sachar, H.S.; Sivasankar, V.S.; Das, S. Revisiting the strong stretching theory for pH-responsive polyelectrolyte brushes: Effects of consideration of excluded volume interactions and an expanded form of the mass action law. Soft Matter 2019, 15, 559–574. [Google Scholar] [CrossRef]
- Skvortsov, A.; Pavlushkov, I.; Gorbunov, A.; Zhulina, Y.; Borisov, O.; Pryamitsyn, V. Structure of densely grafted polymeric monolayers. Polymer Sci. USSR 1988, 30, 1706–1715. [Google Scholar] [CrossRef]
- Zhulina, Y.; Pryamitsyn, V.; Borisov, O. Structure and conformational transitions in grafted polymer chain layers. A new theory. Polym. Sci. USSR 1989, 31, 205–216. [Google Scholar] [CrossRef]
- Borisov, O.V.; Birshtein, T.M.; Zhulina, E.B. Collapse of grafted polyelectrolyte layer. J. Phys. II 1991, 1, 521–526. [Google Scholar] [CrossRef]
- Landsgesell, J.; Nová, L.; Rud, O.; Uhlík, F.; Sean, D.; Hebbeker, P.; Holm, C.; Košovan, P. Simulations of ionization equilibria in weak polyelectrolyte solutions and gels. Soft Matter 2019, 15, 1155–1185. [Google Scholar] [CrossRef]
- Donnini, S.; Tegeler, F.; Groenhof, G.; Grubmüller, H. Constant pH Molecular Dynamics in Explicit Solvent with lambda-Dynamics. J. Chem. Theory Comput. 2011, 7, 1962–1978. [Google Scholar] [CrossRef] [PubMed]
- Barr, S.A.; Panagiotopoulos, A.Z. Conformational transitions of weak polyacids grafted to nanoparticles. J. Chem. Phys. 2012, 137, 144704. [Google Scholar] [CrossRef] [PubMed]
- Mercurieva, A.A.; Birshtein, T.M.; Zhulina, E.B.; Iakovlev, P.; van Male, J.; Leermakers, F.A.M. An annealed polyelectrolyte brush in a polar-nonpolar binary solvent: Effect of pH and ionic strength. Macromolecules 2002, 35, 4739–4752. [Google Scholar] [CrossRef]
- Mercurieva, A.A.; Birshtein, T.M.; Zhulina, E.B.; Iakovlev, P.; van Male, J.; Leermakers, F.A.M. Erratum: An annealed polyelectrolyte brush in a polar-nonpolar binary solvent (Effect of pH and Ionic Strength (2002) 35 (4739–4752)). Macromolecules 2002, 35, 7166. [Google Scholar] [CrossRef][Green Version]
- Zhulina, E.B.; Borisov, O.V. Poisson-Boltzmann Theory of pH-Sensitive (Annealing) Polyelectrolyte Brush. Langmuir 2011, 27, 10615–10633. [Google Scholar] [CrossRef]
- Israels, R.; Gersappe, D.; Fasolka, M.; Roberts, V.A.; Balazs, A.C. pH-Controlled Gating in Polymer Brushes. Macromolecules 1994, 27, 6679–6682. [Google Scholar] [CrossRef]
- Murdoch, T.J.; Willott, J.D.; de Vos, W.M.; Nelson, A.; Prescott, S.W.; Wanless, E.J.; Webber, G.B. Influence of Anion Hydrophilicity on the Conformation of a Hydrophobic Weak Polyelectrolyte Brush. Macromolecules 2016, 49, 9605–9617. [Google Scholar] [CrossRef]
- Tagliazucchi, M.; Olvera de la Cruz, M.; Szleifer, I. Self-organization of grafted polyelectrolyte layers via the coupling of chemical equilibrium and physical interactions. Proc. Natl. Acad. Sci. USA 2010, 107, 5300. [Google Scholar] [CrossRef]
- Nap, R.J.; Tagliazucchi, M.; Szleifer, I. Born energy, acid-base equilibrium, structure and interactions of end-grafted weak polyelectrolyte layers. J. Chem. Phys. 2014, 140, 024910. [Google Scholar] [CrossRef]
- Walker, D.A.; Leitsch, E.K.; Nap, R.J.; Szleifer, I.; Grzybowski, B.A. Geometric curvature of nanoparticles controls their chemical patchiness and self-assembly. Nat. Nanotechnol. 2013, 8, 676–681. [Google Scholar] [CrossRef]
- Rathee, V.S.; Sikora, B.J.; Sidky, H.; Whitmer, J.K. Simulating the thermodynamics of charging in weak polyelectrolytes: The Debye–Hückel limit. Mater. Res. Express 2018, 5, 014010. [Google Scholar] [CrossRef]
- Rathee, V.; Sidky, H.; Sikora, B.; Whitmer, J. Explicit Ion Effects on the Charge and Conformation of Weak Polyelectrolytes. Polymers 2019, 11, 183. [Google Scholar] [CrossRef] [PubMed]
- Blanco, P.M.; Madurga, S.; Narambuena, C.F.; Mas, F.; Garcés, J.L. Role of Charge Regulation and Fluctuations in the Conformational and Mechanical Properties of Weak Flexible Polyelectrolytes. Polymers 2019, 11, 1962. [Google Scholar] [CrossRef] [PubMed]
- Mella, M.; Izzo, L. Modulation of ionization and structural properties of weak polyelectrolytes due to 1D, 2D, and 3D confinement. J. Polym. Sci. Part B Polym. Phys. 2017, 55, 1088–1102. [Google Scholar] [CrossRef]
- Landsgesell, J.; Holm, C.; Smiatek, J. Simulation of weak polyelectrolytes: A comparison between the constant pH and the reaction ensemble method. Eur. Phys. J. Spec. Top. 2017, 226, 725–736. [Google Scholar] [CrossRef]
- Rathee, V.S.; Zervoudakis, A.J.; Sidky, H.; Sikora, B.J.; Whitmer, J.K. Weak polyelectrolyte complexation driven by associative charging. J. Chem. Phys. 2018, 148, 114901. [Google Scholar] [CrossRef] [PubMed]
- Zhulina, E.B.; Borisov, O.V. Polyelectrolytes Grafted to Curved Surfaces. Macromolecules 1996, 29, 2618–2626. [Google Scholar] [CrossRef]
- Bartels, C.; Ronis, D. Competition between Conformational and Chemical Equilibrium in Suspensions of Polyelectrolyte-Coated Particles. Macromolecules 2011, 44, 3174–3178. [Google Scholar] [CrossRef]
- Tong, C. Numerical Study of Weak Polybase Brushes Grafted on Neutral or Charged Spherical Surface by the Self-Consistent Field Theory. Langmuir 2014, 30, 15301–15308. [Google Scholar] [CrossRef]
- Jelínek, K.; Limpouchová, Z.; Uhlí k, F.; Procházka, K. SCF Study of Amphiphilic Micellar Shells Containing Polyelectrolyte and Hydrophobic Sequences. Macromolecules 2007, 40, 7656–7664. [Google Scholar] [CrossRef]
- Uhlík, F.; Jelí nek, K.; Limpouchová, Z.; Procházka, K. Stimuli-Responsive Amphiphilic Shells of Kinetically Frozen Polymeric Micelles in Aqueous Media: Monte Carlo Simulations and Comparison to Self-Consistent Field Calculations. Macromolecules 2008, 41, 3711–3719. [Google Scholar] [CrossRef]
- Lozsán, R.A.E.; Romero-Cano, M.S. Carboxylated core-shell particles: II. Experimental and theoretical comparison of salt-induced swelling. J. Colloid Interface Sci. 2011, 354, 70–75. [Google Scholar] [CrossRef]
- Popov, K.I.; Nap, R.J.; Szleifer, I.; Olvera de la Cruz, M. Interacting Nanoparticles with Functional Surface Groups. J. Polym. Sci. Part B Polym. Phys. 2012, 50, 852–862. [Google Scholar] [CrossRef]
- Park, Y.; Whitaker, R.D.; Nap, R.J.; Paulsen, J.L.; Mathiyazhagan, V.; Doerrer, L.H.; Song, Y.Q.; Hürlimann, M.D.; Szleifer, I.; Wong, J.Y. Stability of Superparamagnetic Iron Oxide Nanoparticles at Different pH Values: Experimental and Theoretical Analysis. Langmuir 2012, 28, 6246–6255. [Google Scholar] [CrossRef] [PubMed]
- Nap, R.J.; Park, S.H.; Szleifer, I. On the stability of nanoparticles coated with polyelectrolytes in high salinity solutions. J. Polym. Sci. Part B Polym. Phys. 2014, 52, 1689–1699. [Google Scholar] [CrossRef]
- Yu, J.; Nap, R.J.; Szleifer, I.; Wong, J.Y. Effect of Polymer Surface Modification of Superparamagnetic Iron Oxide Nanoparticle Dispersions in High Salinity Environments. Langmuir 2019. [Google Scholar] [CrossRef]
- Saito, T.; Koopal, L.K.; Nagasaki, S.; Tanaka, S. Adsorption of Heterogeneously Charged Nanoparticles on a Variably Charged Surface by the Extended Surface Complexation Approach: Charge Regulation, Chemical Heterogeneity, and Surface Complexation. J. Phys. Chem. B 2008, 112, 1339–1349. [Google Scholar] [CrossRef]
- Nap, R.J.; Park, Y.; Wong, J.Y.; Szleifer, I. Adsorption of Acid and Polymer Coated Nanoparticles: A Statistical Thermodymamics Approach. Langmuir 2013, 29, 14482–14493. [Google Scholar] [CrossRef]
- De Oliveira, V.M.; de Carvalho, S.J. Adsorption of pH-responsive polyelectrolyte chains onto spherical macroions. Eur. Phys. J. E 2014, 37, 75. [Google Scholar] [CrossRef]
- Stornes, M.; Linse, P.; Dias, R.S. Monte Carlo Simulations of Complexation between Weak Polyelectrolytes and a Charged Nanoparticle. Influence of Polyelectrolyte Chain Length and Concentration. Macromolecules 2017, 50, 5978–5988. [Google Scholar] [CrossRef]
- Ulrich, S.; Seijo, M.; Stoll, S. The many facets of polyelectrolytes and oppositely charged macroions complex formation. Curr. Opin. Colloid Interface Sci. 2006, 11, 268–272. [Google Scholar] [CrossRef]
- Ulrich, S.; Seijo, M.; Carnal, F.; Stoll, S. Formation of Complexes between Nanoparticles and Weak Polyampholyte Chains. Monte Carlo Simulations. Macromolecules 2011, 44, 1661–1670. [Google Scholar] [CrossRef]
- Carnal, F.; Stoll, S. Adsorption of Weak Polyelectrolytes on Charged Nanoparticles. Impact of Salt Valency, pH, and Nanoparticle Charge Density. Monte Carlo Simulations. J. Phys. Chem. B 2011, 115, 12007–12018. [Google Scholar] [CrossRef] [PubMed]
- Wolterink, J.K.; Koopal, L.; Stuart, M.C.; Van-Riemsdijk, W. Surface charge regulation upon polyelectrolyte adsorption, hematite, polystyrene sulfonate, surface charge regulation: Theoretical calculations and hematite-poly(styrene sulfonate) system. Colloids Surfaces A Physicochem. Eng. Asp. 2006, 291, 13–23. [Google Scholar] [CrossRef]
- Gilles, F.M.; Boubeta, F.M.; Azzaroni, O.; Szleifer, I.; Tagliazucchi, M. Modulation of Polyelectrolyte Adsorption on Nanoparticles and Nanochannels by Surface Curvature. J. Phys. Chem. C 2018, 122, 6669–6677. [Google Scholar] [CrossRef]
- Winkler, R.G.; Cherstvy, A.G. Strong and Weak Polyelectrolyte Adsorption onto Oppositely Charged Curved Surfaces. In Polyelectrolyte Complexes in the Dispersed and Solid State I; Springer: Berlin/Heidelberg, Germany, 2013; pp. 1–56. [Google Scholar] [CrossRef]
- Vermeer, A.W.P.; Leermakers, F.A.M.; Koopal, L.K. Adsorption of Weak Polyelectrolytes on Surfaces with a Variable Charge. Self-Consistent-Field Calculations. Langmuir 1997, 13, 4413–4421. [Google Scholar] [CrossRef]
- Tong, C. The numerical study of the adsorption of flexible polyelectrolytes with the annealed charge distribution onto an oppositely charged sphere by the self-consistent field theory. J. Chem. Phys. 2013, 139, 084903. [Google Scholar] [CrossRef]
- Klein Wolterink, J.; van Male, J.; Cohen Stuart, M.A.; Koopal, L.K.; Zhulina, E.B.; Borisov, O.V. Annealed star-branched polyelectrolytes in solution. Macromolecules 2002, 35, 9176–9190. [Google Scholar] [CrossRef]
- Lewis, T.; Pryamitsyn, V.; Ganesan, V. Mean field theory of charged dendrimer molecules. J. Chem. Phys. 2011, 135, 204902. [Google Scholar] [CrossRef]
- Uhlík, F.; Košovan, P.; Limpouchová, Z.; Prochá¡zka, K.; Borisov, O.V.; Leermakers, F.A.M. Modeling of Ionization and Conformations of Starlike Weak Polyelectrolytes. Macromolecules 2014, 47, 4004–4016. [Google Scholar] [CrossRef]
- Rud, O.V.; Birshtein, T.M. Conformational properties and interaction of polyelectrolyte pH-sensitive stars. Polym. Sci. Ser. A 2013, 55, 757–771. [Google Scholar] [CrossRef]
- Rud, O.V.; Leermakers, F.A.M.; Birshtein, T.M. Interaction of a Hydrophobic Weak Polyelectrolyte Star with an Apolar Surface. Langmuir 2014, 30, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Borisov, O.V.; Zhulina, E.B. Conformations of polyelectrolyte molecular brushes: A mean-field theory. J. Chem. Phys. 2018, 149, 184904. [Google Scholar] [CrossRef] [PubMed]
- Prokacheva, V.M.; Rud, O.V.; Uhlík, F.; Borisov, O.V. Intramolecular micellization and nanopatterning in pH- and thermo-responsive molecular brushes. Soft Matter 2020, 16, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Beck, R.; Deek, J.; Safinya, C.R. Structures and interactions in ‘bottlebrush’ neurofilaments: The role of charged disordered proteins in forming hydrogel networks. Biochem. Soc. Trans. 2012, 40, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Laser-Azogui, A.; Kornreich, M.; Malka-Gibor, E.; Beck, R. Neurofilament assembly and function during neuronal development. Curr. Opin. Cell Biol. 2015, 32, 92–101. [Google Scholar] [CrossRef]
- Pregent, S.; Lichtenstein, A.; Avinery, R.; Laser-Azogui, A.; Patolsky, F.; Beck, R. Probing the Interactions of Intrinsically Disordered Proteins Using Nanoparticle Tags. Nano Lett. 2015, 15, 3080–3087. [Google Scholar] [CrossRef]
- Srinivasan, N.; Bhagawati, M.; Ananthanarayanan, B.; Kumar, S. Stimuli-sensitive intrinsically disordered protein brushes. Nat. Commun. 2014, 5, 5145. [Google Scholar] [CrossRef]
- Zhulina, E.B.; Leermakers, F.A.M. A Self-Consistent Field Analysis of the Neurofilament Brush with Amino-Acid Resolution. Biophys. J. 2007, 93, 1421–1430. [Google Scholar] [CrossRef]
- Zhulina, E.B.; Leermakers, F.A.M. Effect of the Ionic Strength and pH on the Equilibrium Structure of a Neurofilament Brush. Biophys. J. 2007, 93, 1452–1463. [Google Scholar] [CrossRef]
- Zhulina, E.B.; Leermakers, F.A.M. The Polymer Brush Model of Neurofilament Projections: Effect of Protein Composition. Biophys. J. 2010, 98, 462–469. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Leermakers, F.; Zhulina, E. How the projection domains of NF-L and α-internexin determine the conformations of NF-M and NF-H in neurofilaments. Eur. Biophys. J. 2010, 39, 1323–1334. [Google Scholar] [CrossRef] [PubMed]
- Leermakers, F.A.M.; Zhulina, E.B. Self-consistent field modeling of the neurofilament network. Biophys. Rev. Lett. 2008, 3, 459–489. [Google Scholar] [CrossRef]
- Stevens, M.J.; Hoh, J.H. Conformational Dynamics of Neurofilament Side-Arms. J. Phys. Chem. B 2010, 114, 8879–8886. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chang, R.; Teunissen, C.; Gebremichael, Y.; Petzold, A. Neurofilament stoichiometry simulations during neurodegeneration suggest a remarkable self-sufficient and stable in vivo protein structure. J. Neurol. Sci. 2011, 307, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.; Zhou, X.; Zhulina, E.B.; Jho, Y. Monte Carlo Simulation of the Neurofilament Brush. Israel J. Chem. 2016, 56, 599–606. [Google Scholar] [CrossRef]
- Lee, J.; Kim, S.; Chang, R.; Jayanthi, L.; Gebremichael, Y. Effects of molecular model, ionic strength, divalent ions, and hydrophobic interaction on human neurofilament conformation. J. Chem. Phys. 2013, 138, 015103. [Google Scholar] [CrossRef]
- Jayanthi, L.; Stevenson, W.; Kwak, Y.; Chang, R.; Gebremichael, Y. Conformational properties of interacting neurofilaments: Monte Carlo simulations of cylindrically grafted apposing neurofilament brushes. J. Biol. Phys. 2013, 39, 343–362. [Google Scholar] [CrossRef][Green Version]
- Kiani, C.; Chen, L.; Wu, Y.J.; Yee, A.J.; Yang, B.B. Structure and function of aggrecan. Cell Res. 2002, 12, 19–32. [Google Scholar] [CrossRef]
- Nap, R.J.; Szleifer, I. Structure and Interactions of Aggrecans: Statistical Thermodynamics Approach. Biophys. J. 2008, 95, 4570–4583. [Google Scholar] [CrossRef]
- Šiber, A.; Lošdorfer Božič, A.; Podgornik, R. Energies and pressures in viruses: Contribution of nonspecific electrostatic interactions. Phys. Chem. Chem. Phys. 2012, 14, 3746–3765. [Google Scholar] [CrossRef] [PubMed]
- Ni, P.; Wang, Z.; Ma, X.; Das, N.C.; Sokol, P.; Chiu, W.; Dragnea, B.; Hagan, M.; Kao, C.C. An Examination of the Electrostatic Interactions between the N-Terminal Tail of the Brome Mosaic Virus Coat Protein and Encapsidated RNAs. J. Mol. Biol. 2012, 419, 284–300. [Google Scholar] [CrossRef] [PubMed]
- Prinsen, P.; van der Schoot, P.; Gelbart, W.M.; Knobler, C.M. Multishell Structures of Virus Coat Proteins. J. Phys. Chem. B 2010, 114, 5522–5533. [Google Scholar] [CrossRef] [PubMed]
- Kegel, W.K.; van der Schoot, P. Physical Regulation of the Self-Assembly of Tobacco Mosaic Virus Coat Protein. Biophys. J. 2006, 91, 1501–1512. [Google Scholar] [CrossRef] [PubMed]
- Nap, R.J.; Lošdorfer Božič, A.; Szleifer, I.; Podgornik, R. The role of solution conditions in the bacteriophage PP7 capsid charge regulation. Biophys. J. 2014, 107, 1970–1979. [Google Scholar] [CrossRef] [PubMed]
- Barroso daSilva, F.L.; Dias, L.G. Development of constant-pH simulation methods in implicit solvent and applications in biomolecular systems. Biophys. Rev. 2017, 9, 699–728. [Google Scholar] [CrossRef]
- Gonzalez Solveyra, E.; Szleifer, I. What is the role of curvature on the properties of nanomaterials for biomedical applications? Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2016, 8, 334–354. [Google Scholar] [CrossRef]
- De Vos, W.M.; Leermakers, F.A.M.; de Keizer, A.; Cohen Stuart, M.A.; Kleijn, J.M. Field Theoretical Analysis of Driving Forces for the Uptake of Proteins by Like-Charged Polyelectrolyte Brushes: Effects of Charge Regulation and Patchiness. Langmuir 2010, 26, 249–259. [Google Scholar] [CrossRef]
- Boubeta, F.M.; Soler-Illia, G.J.A.A.; Tagliazucchi, M. Electrostatically Driven Protein Adsorption: Charge Patches versus Charge Regulation. Langmuir 2018, 34, 15727–15738. [Google Scholar] [CrossRef]
- Narambuena, C.F.; Longo, G.S.; Szleifer, I. Lysozyme adsorption in pH-responsive hydrogel thin-films: The non-trivial role of acid-base equilibrium. Soft Matter 2015, 11, 6669–6679. [Google Scholar] [CrossRef]
- Hagemann, A.; Giussi, J.M.; Longo, G.S. Use of pH Gradients in Responsive Polymer Hydrogels for the Separation and Localization of Proteins from Binary Mixtures. Macromolecules 2018, 51, 8205–8216. [Google Scholar] [CrossRef]
- Jiang, T.; Wu, J. Self-organization of multivalent counterions in polyelectrolyte brushes. J. Chem. Phys. 2008, 129, 084903. [Google Scholar] [CrossRef] [PubMed]
- Guptha, V.S.; Hsiao, P.Y. Polyelectrolyte brushes in monovalent and multivalent salt solutions. Polymer 2014, 55, 2900–2912. [Google Scholar] [CrossRef]
- Yu, J.; Jackson, N.E.; Xu, X.; Brettmann, B.K.; Ruths, M.; de Pablo, J.J.; Tirrell, M. Multivalent ions induce lateral structural inhomogeneities in polyelectrolyte brushes. Sci. Adv. 2017, 3, eaao1497. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Jackson, N.E.; Xu, X.; Morgenstern, Y.; Kaufman, Y.; Ruths, M.; de Pablo, J.J.; Tirrell, M. Multivalent counterions diminish the lubricity of polyelectrolyte brushes. Science 2018, 360, 1434–1438. [Google Scholar] [CrossRef] [PubMed]
- Birshtein, T.M.; Zhulina, E.B. The effect of polyvalent salt ions on the properties of annealed polyelectrolyte brushes. Ber. Bunsenges. Phys. Chem. 1996, 100, 929–935. [Google Scholar] [CrossRef]
- Zhulina, E.B.; Borisov, O.V.; Birshtein, T.M. Polyelectrolyte Brush Interaction with Multivalent Ions. Macromolecules 1999, 32, 8189–8196. [Google Scholar] [CrossRef]
- Uline, M.J.; Rabin, Y.; Szleifer, I. Effects of the Salt Concentration on Charge Regulation in Tethered Polyacid Monolayers. Langmuir 2011, 27, 4679–4689. [Google Scholar] [CrossRef]
- Jahan, M.; Uline, M. Quantifying Mg2+ Binding to ssDNA Oligomers: A Self-Consistent Field Theory Study at Varying Ionic Strengths and Grafting Densities. Polymers 2018, 10, 1403. [Google Scholar] [CrossRef]
- Lauw, Y.; Leermakers, F.A.M.; Cohen Stuart, M.A.; Pinheiro, J.P.; Custers, J.P.A.; van den Broeke, L.J.P.; Keurentjes, J.T.F. On the Binding of Calcium by Micelles Composed of Carboxy-Modified Pluronics Measured by Means of Differential Potentiometric Titration and Modeled with a Self-Consistent-Field Theory. Langmuir 2006, 22, 10932–10941. [Google Scholar] [CrossRef]
- Nap, R.J.; Gonzalez Solveyra, E.; Szleifer, I. Interplay of nanointerface curvature and calcium bindingin weak polyelectrolytes coated nanoparticles. Biomater. Sci. 2018, 6, 1048–1058. [Google Scholar] [CrossRef]
- Lopez, L.G.; Nap, R.J. Highly sensitive gating in pH-responsive nanochannels as a result of ionic bridging and nanoconfinement. Phys. Chem. Chem. Phys. 2018, 20, 16657–16665. [Google Scholar] [CrossRef] [PubMed]
- Longo, G.; Szleifer, I. Ligand-Receptor Interactions in Tethered Polymer Layers. Langmuir 2005, 21, 11342–11351. [Google Scholar] [CrossRef] [PubMed]
- Ren, C.l.; Carvajal, D.; Shull, K.R.; Szleifer, I. Streptavidin-biotin binding in the presence of a polymer spacer. A theoretical description. Langmuir 2009, 25, 12283–12292. [Google Scholar] [CrossRef] [PubMed]
- Malaspina, D.C.; Longo, G.; Szleifer, I. Behavior of ligand binding assays with crowded surfaces: Molecular model of antigen capture by antibody-conjugated nanoparticles. PLoS ONE 2017, 12, e0185518. [Google Scholar] [CrossRef] [PubMed]
- Tagliazucchi, M.; Szleifer, I. How Does Confinement Change Ligand-Receptor Binding Equilibrium? Protein Binding in Nanopores and Nanochannels. J. Am. Chem. Soc. 2015, 137, 12539–12551. [Google Scholar] [CrossRef] [PubMed]
- Nap, R.J.; Szleifer, I. How to optimize Binding of Coated Nanoparticles: Coupling of Physical Interactions, Molecular Organization and Chemical state. Biomater. Sci. 2013, 1, 814–823. [Google Scholar] [CrossRef]
- Martinez-Veracoechea, F.J.; Frenkel, D. Designing super selectivity in multivalent nano-particle binding. Proc. Natl. Acad. Sci. USA 2011, 108, 10963–10968. [Google Scholar] [CrossRef]
- Huang, K.; Szlufarska, I. Friction and Slip at the Solid/Liquid Interface in Vibrational Systems. Langmuir 2012, 28, 17302–17312. [Google Scholar] [CrossRef]
- Huang, K.; Szlufarska, I. Green-Kubo relation for friction at liquid-solid interfaces. Phys. Rev. E 2014, 89, 032119. [Google Scholar] [CrossRef]
- Falk, K.; Sedlmeier, F.; Joly, L.; Netz, R.R.; Bocquet, L. Molecular Origin of Fast Water Transport in Carbon Nanotube Membranes: Superlubricity versus Curvature Dependent Friction. Nano Lett. 2010, 10, 4067–4073. [Google Scholar] [CrossRef] [PubMed]
- Stein, D.; Kruithof, M.; Dekker, C. Surface-Charge-Governed Ion Transport in Nanofluidic Channels. Phys. Rev. Lett. 2004, 93, 035901. [Google Scholar] [CrossRef] [PubMed]
- Vlassiouk, I.; Smirnov, S.; Siwy, Z. Ionic selectivity of single nanochannels. Nano. Lett. 2008, 8, 1978–1985. [Google Scholar] [CrossRef] [PubMed]
- Daiguji, H. Ion transport in nanofluidic channels. Chem. Soc. Rev. 2010, 39, 901–911. [Google Scholar] [CrossRef]
- Antila, H.S.; Luijten, E. Dielectric Modulation of Ion Transport near Interfaces. Phys. Rev. Lett. 2018, 120, 135501. [Google Scholar] [CrossRef]
- Hou, X.; Jiang, L. Learning from Nature: Building Bio-Inspired Smart Nanochannels. ACS Nano 2009, 3, 3339–3342. [Google Scholar] [CrossRef]
- Hou, X.; Liu, Y.; Dong, H.; Yang, F.; Li, L.; Jiang, L. A pH-Gating Ionic Transport Nanodevice: Asymmetric Chemical Modification of Single Nanochannels. Adv. Mater. 2010, 22, 2440–2443. [Google Scholar] [CrossRef]
- Peleg, O.; Tagliazucchi, M.; Kroeger, M.; Rabin, Y.; Szleifer, I. Morphology Control of Hairy Nanopores. ACS Nano 2011, 5, 4737–4747. [Google Scholar] [CrossRef]
- Tagliazucchi, M.; Huang, K.; Szleifer, I. Routes for nanoparticle translocation through polymer-brush-modified nanopores. J. Phys Condens. Matter 2018, 30, 274006. [Google Scholar] [CrossRef]
- Dimitrov, D.I.; Milchev, A.; Binder, K. Polymer brushes in cylindrical pores: Simulation versus scaling theory. J. Chem. Phys. 2006, 125, 034905. [Google Scholar] [CrossRef]
- Li, C.W.; Merlitz, H.; Wu, C.X.; Sommer, J.U. Nanopores as Switchable Gates for Nanoparticles: A Molecular Dynamics Study. Macromolecules 2018, 51, 6238–6247. [Google Scholar] [CrossRef]
- Chen, G.; Das, S. Thermodynamics, electrostatics, and ionic current in nanochannels grafted with pH-responsive end-charged polyelectrolyte brushes: General. Electrophoresis 2017, 38, 720–729. [Google Scholar] [CrossRef] [PubMed]
- Sachar, H.S.; Sivasankar, V.S.; Etha, S.A.; Chen, G.; Das, S. Ionic current in nanochannels grafted with pH-responsive polyelectrolyte brushes modeled using augmented strong stretching theory. Electrophoresis 2020, 41, 554–561. [Google Scholar] [CrossRef] [PubMed]
- Sachar, H.S.; Sivasankar, V.S.; Das, S. Electrokinetic energy conversion in nanochannels grafted with pH-responsive polyelectrolyte brushes modelled using augmented strong stretching theory. Soft Matter 2019, 15, 5973–5986. [Google Scholar] [CrossRef] [PubMed]
- Zhulina, E.B.; Birshtein, T.M.; Borisov, O.V. Theory of Ionizable Polymer Brushes. Macromolecules 1995, 28, 1491. [Google Scholar] [CrossRef]
- Zhulina, E.B.; Rubinstein, M. Ionic strength dependence of polyelectrolyte brush thickness. Soft Matter 2012, 8, 9376–9383. [Google Scholar] [CrossRef]
- Yameen, B.; Ali, M.; Neumann, R.; Ensinger, W.; Knoll, W.; Azzaroni, O. Synthetic Proton-Gated Ion Channels via Single Solid-State Nanochannels Modified with Responsive Polymer Brushes. Nano Lett. 2009, 9, 2788–2793. [Google Scholar] [CrossRef]
- Yin, J.; Yang, R.; Yan, S.; Luan, S. Surfaces functionalized with hierarchical polymer brushes and their biomedical applications. Sci. Sin. Chim. 2020, 50, 447–462. [Google Scholar] [CrossRef]
- Badi, N.; Lutz, J.F. Sequence control in polymer synthesis. Chem. Soc. Rev. 2009, 38, 3383–3390. [Google Scholar] [CrossRef]
- Tong, X.M.; Guo, B.H.; Huang, Y.B. Toward the synthesis of sequence-controlled vinyl copolymers. Chem. Commun. 2011, 47, 1455–1457. [Google Scholar] [CrossRef]
- Lutz, J.F. Sequence-controlled polymerizations: The next Holy Grail in polymer science? Polym. Chem. 2010, 1, 55–62. [Google Scholar] [CrossRef]
- Ananth, A.N.; Mishra, A.; Frey, S.; Dwarkasing, A.; Versloot, R.; van der Giessen, E.; Görlich, D.; Onck, P.; Dekker, C. Spatial structure of disordered proteins dictates conductance and selectivity in nuclear pore complex mimics. eLife 2018, 7, e31510. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Ainsa, S.; Keyser, U.F. DNA origami nanopores: Developments, challenges and perspectives. Nanoscale 2014, 6, 14121–14132. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.A.; Gao, H.; Ouyang, X. Advances in DNA Origami Nanopores: Fabrication, Characterization and Applications: Advances in DNA Origami Nanopores: Fabrication, Characterization and Applications. Chin. J. Chem. 2018, 36, 875–885. [Google Scholar] [CrossRef]
- Cheng, L.; Cao, D. Designing a Thermo-switchable Channel for Nanofluidic Controllable Transportation. ACS Nano 2011, 5, 1102–1108. [Google Scholar] [CrossRef]
- Huang, K.; Szleifer, I. Design of Multifunctional Nanogate in Response to Multiple External Stimuli Using Amphiphilic Diblock Copolymer. J. Am. Chem. Soc. 2017, 139, 6422–6430. [Google Scholar] [CrossRef]
- Rout, M.P.; Aitchison, J.D.; Suprapto, A.; Hjertaas, K.; Zhao, Y.M.; Chait, B.T. The yeast nuclear pore complex: Composition, architecture, and transport mechanism. J. Cell Biol. 2000, 148, 635–651. [Google Scholar] [CrossRef]
- Patel, S.S.; Belmont, B.J.; Sante, J.M.; Rexach, M.F. Natively unfolded nucleoporins gate protein diffusion across the nuclear pore complex. Cell 2007, 129, 83–96. [Google Scholar] [CrossRef]
- Hoelz, A.; Debler, E.W.; Blobel, G. The Structure of the Nuclear Pore Complex. Annu. Rev. Biochem. 2011, 80, 613–643. [Google Scholar] [CrossRef]
- Kim, S.J.; Fernandez-Martinez, J.; Nudelman, I.; Shi, Y.; Zhang, W.; Raveh, B.; Herricks, T.; Slaughter, B.D.; Hogan, J.A.; Upla, P.; et al. Integrative structure and functional anatomy of a nuclear pore complex. Nature 2018, 555, 475–482. [Google Scholar] [CrossRef]
- Rout, M.P.; Aitchison, J.D.; Magnasco, M.O.; Chait, B.T. Virtual gating and nuclear transport: The hole picture. Trends Cell Biol. 2003, 13, 622–628. [Google Scholar] [CrossRef] [PubMed]
- Peters, R. Translocation through the nuclear pore complex: Selectivity and speed by reduction-of-dimensionality. Traffic 2005, 6, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Hülsmann, B.B.; Labokha, A.; Görlich, D. The Permeability of Reconstituted Nuclear Pores Provides Direct Evidence for the Selective Phase Model. Cell 2012, 150, 738–751. [Google Scholar] [CrossRef] [PubMed]
- Yamada, J.; Phillips, J.L.; Patel, S.; Goldfien, G.; Calestagne-Morelli, A.; Huang, H.; Reza, R.; Acheson, J.; Krishnan, V.V.; Newsam, S.; et al. A Bimodal Distribution of Two Distinct Categories of Intrinsically Disordered Structures with Separate Functions in FG Nucleoporins. Mol. Cell. Proteom. 2010, 9, 2205–2224. [Google Scholar] [CrossRef]
- Huang, K.; Szleifer, I. Modeling the nucleoporins that form the hairy pores. Biochem. Soc. Trans. 2020. [Google Scholar] [CrossRef]
- Mincer, J.S.; Simon, S.M. Simulations of nuclear pore transport yield mechanistic insights and quantitative predictions. Proc. Natl. Acad. Sci. USA 2011, 108, E351–E358. [Google Scholar] [CrossRef]
- Moussavi-Baygi, R.; Jamali, Y.; Karimi, R.; Mofrad, M.R.K. Brownian Dynamics Simulation of Nucleocytoplasmic Transport: A Coarse-Grained Model for the Functional State of the Nuclear Pore Complex. PLoS Comput. Biol. 2011, 7, e1002049. [Google Scholar] [CrossRef]
- Osmanović, D.; Fassati, A.; Ford, I.J.; Hoogenboom, B.W. Physical modeling of the nuclear pore complex. Soft Matter 2013, 9, 10442. [Google Scholar] [CrossRef]
- Peyro, M.; Soheilypour, M.; Ghavami, A.; Mofrad, M.R.K. Nucleoporin’s Like Charge Regions Are Major Regulators of FG Coverage and Dynamics Inside the Nuclear Pore Complex. PLoS ONE 2015, 10, e0143745. [Google Scholar] [CrossRef]
- Tagliazucchi, M.; Peleg, O.; Kroger, M.; Rabin, Y.; Szleifer, I. Effect of charge, hydrophobicity, and sequence of nucleoporins on the translocation of model particles through the nuclear pore complex. Proc. Natl. Acad. Sci. USA 2013, 110, 10336–10337. [Google Scholar] [CrossRef]
- Ghavami, A.; Veenhoff, L.M.; van der Giessen, E.; Onck, P.R. Probing the Disordered Domain of the Nuclear Pore Complex through Coarse-Grained Molecular Dynamics Simulations. Biophys. J. 2014, 107, 1393–1402. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Tagliazucchi, M.; Park, S.H.; Rabin, Y.; Szleifer, I. Nanocompartmentalization of the Nuclear Pore Lumen. Biophys. J. 2020, 118, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Vovk, A.; Gu, C.; Opferman, M.G.; Kapinos, L.E.; Lim, R.Y.; Coalson, R.D.; Jasnow, D.; Zilman, A. Simple biophysics underpins collective conformations of the intrinsically disordered proteins of the Nuclear Pore Complex. eLife 2016, 5, e10785. [Google Scholar] [CrossRef] [PubMed]
- Osmanović, D.; Rabin, Y. Effect of non-specific interactions on formation and stability of specific complexes. J. Chem. Phys. 2016, 144, 205104. [Google Scholar] [CrossRef]
- Zahn, R.; Osmanović, D.; Ehret, S.; Araya Callis, C.; Frey, S.; Stewart, M.; You, C.; Görlich, D.; Hoogenboom, B.W.; Richter, R.P. A physical model describing the interaction of nuclear transport receptors with FG nucleoporin domain assemblies. eLife 2016, 5, e14119. [Google Scholar] [CrossRef]
- Gamini, R.; Han, W.; Stone, J.E.; Schulten, K. Assembly of Nsp1 Nucleoporins Provides Insight into Nuclear Pore Complex Gating. PLoS Comput. Biol. 2014, 10, e1003488. [Google Scholar] [CrossRef]
- Milles, S.; Mercadante, D.; Aramburu, I.V.; Jensen, M.R.; Banterle, N.; Koehler, C.; Tyagi, S.; Clarke, J.; Shammas, S.L.; Blackledge, M.; et al. Plasticity of an Ultrafast Interaction between Nucleoporins and Nuclear Transport Receptors. Cell 2015, 163, 734–745. [Google Scholar] [CrossRef]
- Raveh, B.; Karp, J.M.; Sparks, S.; Dutta, K.; Rout, M.P.; Sali, A.; Cowburn, D. Slide-and-exchange mechanism for rapid and selective transport through the nuclear pore complex. Proc. Natl. Acad. Sci. USA 2016, 113, E2489–E2497. [Google Scholar] [CrossRef]
- Ando, D.; Zandi, R.; Kim, Y.; Colvin, M.; Rexach, M.; Gopinathan, A. Nuclear Pore Complex Protein Sequences Determine Overall Copolymer Brush Structure and Function. Biophys. J. 2014, 106, 1997–2007. [Google Scholar] [CrossRef]
- Ma, J.; Goryaynov, A.; Sarma, A.; Yang, W. Self-regulated viscous channel in the nuclear pore complex. Proc. Natl. Acad. Sci. USA 2012, 109, 7326–7331. [Google Scholar] [CrossRef]
- Ader, C.; Frey, S.; Maas, W.; Schmidt, H.B.; Gorlich, D.; Baldus, M. Amyloid-like interactions within nucleoporin FG hydrogels. Proc. Natl. Acad. Sci. USA 2010, 107, 6281–6285. [Google Scholar] [CrossRef] [PubMed]
- Rajoo, S.; Vallotton, P.; Onischenko, E.; Weis, K. Stoichiometry and compositional plasticity of the yeast nuclear pore complex revealed by quantitative fluorescence microscopy. Proc. Natl. Acad. Sci. USA 2018, 115, E3969–E3977. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Ginzburg, V.V.; Wang, Z.G. Density functional theory for charged fluids. Soft Matter 2018, 14, 5878–5887. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, K.S.; Curro, J.G. PRISM theory of the structure, thermodynamics, and phase transitions of polymer liquids and alloys. In Atomistic Modeling of Physical Properties; Monnerie, L., Suter, U.W., Eds.; Springer: Berlin/Heidelberg, Germany, 1994; pp. 319–377. [Google Scholar] [CrossRef]
- Jiang, T.; Wu, J. Ionic Effects in Collapse of Polyelectrolyte Brushes. J. Phys. Chem. B 2008, 112, 7713–7720. [Google Scholar] [CrossRef]
- González-Mozuelos, P.; Olvera de la Cruz, M. Ion condensation in salt-free dilute polyelectrolyte solutions. J. Chem. Phys. 1995, 103, 3145–3157. [Google Scholar] [CrossRef]
- Solis, F.J.; Olvera de la Cruz, M. Attractive interactions between rodlike polyelectrolytes: Polarization, crystallization, and packing. Phys. Rev. E 1999, 60, 4496. [Google Scholar] [CrossRef]
- Borue, V.Y.; Erukhimovich, I.Y. A statistical theory of weakly charged polyelectrolytes: Fluctuations, equation of state and microphase separation. Macromolecules 1988, 21, 3240–3249. [Google Scholar] [CrossRef]
- Borue, V.Y.; Erukhimovich, I.Y. A statistical theory of globular polyelectrolyte complexes. Macromolecules 1990, 23, 3625–3632. [Google Scholar] [CrossRef]
- Marko, J.F.; Rabin, Y. Microphase separation of charged diblock copolymers: Melts and solutions. Macromolecules 1992, 25, 1503–1509. [Google Scholar] [CrossRef]
- Santangelo, C.D.; Lau, A.W.C. Effects of counterion fluctuations in a polyelectrolyte brush. Eur. Phys. J. E 2004, 13, 335–344. [Google Scholar] [CrossRef]
- Sing, C.E.; Zwanikken, J.W.; Olvera de la Cruz, M. Electrostatic control of blockcopolymer morphology. Nat. Mater. 2014, 13, 694–698. [Google Scholar] [CrossRef] [PubMed]












© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonzalez Solveyra, E.; Nap, R.J.; Huang, K.; Szleifer, I. Theoretical Modeling of Chemical Equilibrium in Weak Polyelectrolyte Layers on Curved Nanosystems. Polymers 2020, 12, 2282. https://doi.org/10.3390/polym12102282
Gonzalez Solveyra E, Nap RJ, Huang K, Szleifer I. Theoretical Modeling of Chemical Equilibrium in Weak Polyelectrolyte Layers on Curved Nanosystems. Polymers. 2020; 12(10):2282. https://doi.org/10.3390/polym12102282
Chicago/Turabian StyleGonzalez Solveyra, Estefania, Rikkert J. Nap, Kai Huang, and Igal Szleifer. 2020. "Theoretical Modeling of Chemical Equilibrium in Weak Polyelectrolyte Layers on Curved Nanosystems" Polymers 12, no. 10: 2282. https://doi.org/10.3390/polym12102282
APA StyleGonzalez Solveyra, E., Nap, R. J., Huang, K., & Szleifer, I. (2020). Theoretical Modeling of Chemical Equilibrium in Weak Polyelectrolyte Layers on Curved Nanosystems. Polymers, 12(10), 2282. https://doi.org/10.3390/polym12102282