Preparation and Characterization of Bio-Based PLA/PBAT and Cinnamon Essential Oil Polymer Fibers and Life-Cycle Assessment from Hydrolytic Degradation

 ,
,  ,
,  ,
, 
Abstract
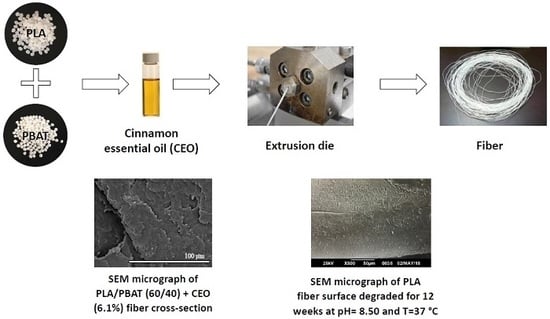
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Blend and Fiber Preparation
2.3. Scanning Electron Microscopy (SEM)
2.4. Fourier Transform Infrared Spectroscopy–Attenuated Total Reflection (FTIR-ATR)
2.5. Differential Scanning Calorimetry (DSC)
2.6. Mechanical Properties
2.7. Fiber Hydrolytic Degradation
2.8. Statistical Analysis
3. Results and Discussion
3.1. Scanning Electron Microscopy (SEM)
3.2. Fourier Transform Infrared Spectroscopy–Attenuated Total Reflection (FTIR- ATR)
3.3. Differential Scanning Calorimetry (DSC)
3.4. Mechanical Properties
3.5. Fiber Hydrolytic Degradation
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Garavand, F.; Rouhi, M.; Razavi, S.H.; Cacciotti, I.; Mohammadi, R. Improving the integrity of natural biopolymer films used in food packaging by crosslinking approach: A review. Int. J. Biol. Macromol. 2017, 104, 687–707. [Google Scholar] [CrossRef] [PubMed]
- Cacciotti, I.; Mori, S.; Cherubini, V.; Nanni, F. Eco-sustainable systems based on poly (lactic acid), diatomite and coffee grounds extract for food packaging. Int. J. Biol. Macromol. 2018, 112, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Fang, X.; Chen, H.; Qin, Y.; Xu, F.; Jin, T. Physicochemical properties and food application of antimicrobial PLA film. Food Control 2017, 73, 1522–1531. [Google Scholar] [CrossRef]
- Elsawy, M.; Kim, K.; Park, J.; Deep, A. Hydrolytic degradation of (PLA) and its composites. Renew. Sustain. Energy Rev. 2017, 79, 1346–1352. [Google Scholar] [CrossRef]
- Ferreira, F.; Cividanes, F.; Gouveia, R.; Lona, L. An overview on properties and applications of poly (butylene adipate-co-terephthalate)-PBAT based composites. Polym. Eng. Sci. 2019, 59, E7–E15. [Google Scholar] [CrossRef]
- Farah, S.; Anderson, D.; Langer, R. Physical and mechanical properties of PLA, and their functions in widespread applications-A comprehensive review. Adv. Drug Deliv. Rev. 2016, 107, 367–392. [Google Scholar] [CrossRef]
- Fukushima, K.; Wu, M.; Bocchini, S.; Rasyida, A.; Yang, M. PBAT based nanocomposites for medical and industrial applications. Mater. Sci. Eng. C 2012, 32, 1331–1351. [Google Scholar] [CrossRef]
- Lu, X.; Zhao, J.; Yang, X.; Xiao, P. Morphology and properties of biodegradable poly (lactic acid)/poly (butylene adipate-co-terephthalate) blends with different viscosity ratio. Polym. Test. 2017, 60, 58–67. [Google Scholar] [CrossRef]
- Cardoso, L.; Magaton, M.; Suman, R.; Massayoshi, M. Influence of chain extender on mechanical, thermal and morphological properties of blown films of PLA/PBAT blends. Polym. Test. 2015, 43, 27–37. [Google Scholar]
- Hernández-López, M.; Correa-Pacheco, Z.; Bautista-Baños, S.; Zavaleta-Avejar, L.; Benítez-Jiménez, J.; Sabino-Gutiérrez, M.; Ortega-Gudiño, P. Bio-based composite fibers from pine essential oil and PLA/PBAT polymer blend. Morphological, physicochemical, thermal and mechanical characterization. Mater. Chem. Phys. 2019, 234, 345–353. [Google Scholar] [CrossRef]
- Gigante, V.; Canesi, I.; Cinelli, P.; Coltelli, M.; Lazzeri, A. Rubber toughening of polylactic acid (PLA) with poly (butylene adipate-coterephthalate) (PBAT): Mechanical properties, fracture mechanics and analysis of ductile-to-brittle behavior while varying temperature and test speed. Eur. Polym. J. 2019, 115, 125–137. [Google Scholar] [CrossRef]
- Turalija, M.; Bischof, S.; Budimir, A.; Gaan, S. Antimicrobial PLA films from environment friendly additives. Compos. Part. B 2016, 102, 94–99. [Google Scholar] [CrossRef]
- Wei, D.; Wang, H.; Ziaee, Z.; Chibante, F.; Zheg, A.; Xiao, H. Non-leaching antimicrobial biodegradable PBAT films through a facile and novel approach. Mater. Sci. Eng. C 2016, 58, 986–991. [Google Scholar] [CrossRef] [PubMed]
- Arrieta, M.; López, J.; Hernández, A.; Rayón, E. Ternary PLA–PHB–Limonene blends intended for biodegradable food packaging applications. Eur. Polym. J. 2014, 50, 255–270. [Google Scholar] [CrossRef]
- Atarés, L.; Chiralt, A. Essential oils as additives in biodegradable films and coatings for active food packaging. Trends Food Sci. Technol. 2016, 48, 51–62. [Google Scholar] [CrossRef]
- Noshirvani, N.; Ghanbarzadeh, B.; Gardrat, C.; Rezaei, M.; Hashemi, M.; Le Coz, C.; Coma, V. Cinnamon and ginger essential oils to improve antifungal, physical and mechanical properties of chitosan-carboxymethyl cellulose films. Food Hydrocoll. 2017, 70, 36–45. [Google Scholar] [CrossRef]
- Khaleque, M.; Keya, C.; Hasan, K.; Hoque, M.; Inatsu, Y.; Bari, M. Use of cloves and cinnamon essential oil to inactivate Listeria monocytogenes in ground beef at freezing and refrigeration temperatures. LWT-Food Sci. Technol. 2016, 74, 219–223. [Google Scholar] [CrossRef]
- Marcet, I.; Weng, S.; Sáez-Orviz, S.; Rendueles, M.; Díaz, M. Production and characterisation of biodegradable PLA nanoparticles loaded with thymol to improve its antimicrobial effect. J. Food Eng. 2018, 239, 26–32. [Google Scholar] [CrossRef]
- Marcos, B.; Sárraga, C.; Castellari, M.; Kappen, F.; Schennink, G.; Arnau, J. Development of biodegradable films with antioxidant properties based on polyesters containing a-tocopherol and olive leaf extract for food packaging applications. J. Food Packag. Shelf Life 2014, 1, 140–150. [Google Scholar] [CrossRef]
- Cardoso, L.; Santos, J.; Camilloto, G.; Miranda, A.; Druzian, J.; Guimarães, A. Development of active films poly (butylene adipate co-terephthalate)-PBAT incorporated with oregano essential oil and application in fish fillet preservation. Ind. Crops Prod. 2017, 108, 388–397. [Google Scholar] [CrossRef]
- Yahyaoui, M.; Gordobil, O.; Herrera, R.; Abderrabba, M.; Labidi, J. Development of novel antimicrobial films based on poly (lactic acid) and essential oils. React. Funct. Polym. 2016, 109, 1–8. [Google Scholar] [CrossRef]
- Arezoo, E.; Mohammadreza, E.; Maryam, M.; Abdorreza, M. The synergistic effects of cinnamon essential oil and nano TiO2 on antimicrobial and functional properties of sago starch films. Int. J. Biol. Macromol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Shu, C.; Chen, Q.; Cao, J.; Jiang, W. The multi-layer film system improved the release and retention properties of cinnamon essential oil and its application as coating in inhibition to penicillium expansion on apple fruit. Food Chem. 2019, 299, 125109. [Google Scholar] [CrossRef] [PubMed]
- Black-Solis, J.; Ventura-Aguilar, R.; Correa-Pacheco, Z.; Corona-Rangel, M.; Bautista-Baños, S. Preharvest use of biodegradable polyester nets added with cinnamon essential oil and the effect on the storage life of tomatoes and the development of Alternaria alternata. Sci. Hortic. 2019, 245, 65–73. [Google Scholar] [CrossRef]
- Weng, Y.; Jin, Y.; Meng, Q.; Wang, L.; Zhang, M.; Wang, Y. Biodegradation behavior of poly(butylene adipate-coterephthalate) (PBAT), poly(lactic acid) (PLA), and their blend under soil conditions. Polym. Test. 2013, 32, 918–926. [Google Scholar] [CrossRef]
- Scaffaro, R.; Maio, A.; Sutera, F.; Gulino, E.F.; Morreale, M. Degradation and recycling of films based on biodegradable polymers: A short review. Polymers 2019, 11, 651. [Google Scholar] [CrossRef]
- Beltrán, F.; Climent-Pascual, E.; de la Orden, M.; Martínez, J. Effect of solid-state polymerization on the structure and properties of mechanically recycled poly (lactic acid). Polym. Degrad. Stabil. 2019. [Google Scholar] [CrossRef]
- Finniss, A.; Agar, W.; Gupta, R. Retarding the hydrolytic degradation of polylactic acid: Effect of induced crystallinity and graphene addition. J. Appl. Polym. Sci. 2016, 133, 44166. [Google Scholar] [CrossRef]
- Huang, Y.; Zhang, C.; Pan, Y.; Zhou, Y.; Jiang, L.; Dan, Y. Effect of NR on the hydrolytic degradation of PLA. Polym. Degrad. Stabil. 2013, 98, 943–950. [Google Scholar] [CrossRef]
- Iñiguez-Franco, F.; Auras, R.; Rubino, M.; Dolan, K.; Soto-Valdez, H.; Selke, S. Effect of nanoparticles on the hydrolytic degradation of PLA-nanocomposites by water ethanol solutions. Polym. Degrad. Stabil. 2017, 146, 287–297. [Google Scholar] [CrossRef]
- Al-Itry, R.; Lamnawar, K.; Maazouz, A. Improvement of thermal stability, rheological and mechanical 710 properties of PLA, PBAT and their blends by reactive extrusion with functionalized epoxy. Polym. Degrad. Stabil. 2012, 97, 1898–1914. [Google Scholar] [CrossRef]
- Rojas, H.; Rosales, C.; González, C.; Perera, R.; Trujillo, M. Influencia de la degradación durante el procesado en las propiedades reológicas, térmicas y de tracción de compuestos de poli(ácido láctico). Revista Latinoamericana de Metalurgia y Materiales 2014, 34, 118–135. [Google Scholar]
- Al-Itry, R.; Lamnawar, K.; Maazouz, A. Reactive extrusion of PLA, PBAT with a multifunctional epoxide: Physico-chemical and rheological properties. Eur. Polym. J. 2014, 58, 90–102. [Google Scholar] [CrossRef]
- Silva, R.; de Oliveira, T.; da Conceicao, A.; Araque, L.; Alves, T.; Barbosa, R. Evaluation of hydrolytic degradation of bionanocomposites through fourier transform infrared spectroscopy. Polimeros 2018, 28, 348–354. [Google Scholar] [CrossRef]
- Utracki, L. Compatibilization of polymer blends. Can. J. Chem. Eng. 2002, 80, 1008–1016. [Google Scholar] [CrossRef]
- Deng, Y.; Yu, C.; Wongwiwattana, P.; Thomas, N. Optimising ductility of poly (lactic acid)/poly (buthylene adipate-co-terephthalate) blends through co-continuous phase morphology. J. Polym. Environ. 2018, 26, 3802–3816. [Google Scholar] [CrossRef]
- Hongdilokkul, P.; Keeratipinit, K.; Chawthai, S.; Hararak, B.; Seadan, M.; Suttiruengwong, S. A study on properties of PLA/PBAT from blown film process. IOP Conf. Ser.-Mater. Sci. 2015, 87, 012112. [Google Scholar] [CrossRef]
- Quero, E.; Müller, A.; Signori, F.; Coltelli, M.; Bronco, S. Isothermal cold-crystallization of PLA/PBAT 789 blends with and without the addition of acetyl tributyl citrate. Macromol. Chem. Phys. 2012, 213, 36–48. [Google Scholar] [CrossRef]
- Kumar, M.; Mohanty, S.; Nayak, S.; Rahail, M. Effect of glycidyl methacrylate (GMA) on the thermal, mechanical and morphological property of biodegradable PLA/PBAT blend and its nanocomposites. Bioresour. Technol. 2010, 101, 8406–8415. [Google Scholar] [CrossRef]
- Brito, Y.; Sabino, M.; Ronca, G.; Muller, A. Changes in crystalline morphology, thermal, and mechanical properties with hydrolytic degradation of immiscible biodegradable PPDX/PCL blends. J. Appl. Polym. Sci. 2008, 110, 3848–3858. [Google Scholar] [CrossRef]
- Moustafa, H.; El Kissi, N.; Abou-Kandil, A.; Abdel-Aziz, M.; Dufresne, A. PLA/PBAT bionanocomposites with antimicrobial natural rosin for green packaging. ACS Appl. Mater. Interfaces 2017, 9, 20132–20141. [Google Scholar] [CrossRef] [PubMed]
- Teamsinsungvon, A.; Ruksakulpiwat, Y.; Jarukumjorn, K. Preparation and characterization of poly (lactic acid)/poly (butylene adipate-co-terephthalate) blends and their composite. Polym.-Plast. Technol. 2013, 52, 1362–1367. [Google Scholar] [CrossRef]
- Dil, E.; Carreau, P.; Favis, B. Morphology, miscibility and continuity development in poly (lactic acid)/poly (butylene adipate-co-terephthalate) blends. Polymer 2015, 68, 202–212. [Google Scholar]
- Jeyaratnam, N.; Nour, A.; Kanthasamy, R.; Nour, A.; Yuvaraj, A.; Akindovo, J. Essential oil from Cinnamomum cassia bark through hydrodistillation and advanced microwave assisted hydrodistillation. Ind. Crop. Prod. 2016, 92, 57–66. [Google Scholar] [CrossRef]
- Mofokeng, J.P.; Luyt, A.S.; Tábi, T.; Kovacs, J. Comparison of injection moulded, natural fibre-reinforced composites with PP and PLA as matrices. J. Thermoplast. Compos. 2012, 25, 927–948. [Google Scholar] [CrossRef]
- Nikolic, L.; Ristic, I.; Adnadjevic, B.; Nikolic, V.; Jovanovic, J.; Stankovic, M. Novel microwave-assisted synthesis of poly (d,l-lactide): The influence of monomer/initiator molar ratio on the product properties. Sensors 2010, 10, 5063–5073. [Google Scholar] [CrossRef]
- Zhao, P.; Liu, W.; Wu, Q.; Ren, J. Preparation, mechanical, and thermal properties of biodegradable polyesters/poly (lactic acid) blends. J. Nanomater. 2010, 2010, 4. [Google Scholar] [CrossRef]
- Sim, J.Y.; Raj, C.J.; Yu, K.H. Poly (butylene adipate-co-terephthalate) (PBAT)/Antimony-doped Tin oxide polymer composite for near infrared absorption coating applications. Bull. Korean Chem. Soc. 2019, 40, 674–679. [Google Scholar] [CrossRef]
- Kim, Y.; Lee, J.; Kim, S.; Baek, K.; Lee, J. Cinnamon bark oil and its components inhibit biofilm formation 800 and toxin production. Int. J. Food Microbiol. 2015, 195, 30–39. [Google Scholar] [CrossRef]
- Black-Solis, J.; Ventura-Aguilar, R.; Barrera-Necha, L.; Bautista-Baños, S. Chemical characterization, compositional variability and mathematical modelling of the effect of essential oils in Alternaria alternata. Rev. Mex. Fitopatol. 2017, 35, 204–226. [Google Scholar]
- Wei, D.; Wang, H.; Xiao, H.; Zheng, A.; Yang, Y. Morphology and mechanical properties of poly (butylene adipate-co-terephthalate)/potato starch blends in the presence of synthesized reactive compatibilizer or modified poly (butylene adipate-co-terephthalate). Carbohydr. Polym. 2015, 123, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Lv, S.; Zhang, Y.; Tan, H. Thermal and thermo-oxidative degradation kinetics and characteristics of poly (lactic acid) and its composites. Waste Manag. 2019, 87, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Cuadri, A.; Martín-Alfonso, J. Thermal, thermo-oxidative and thermomechanical degradation of PLA: A comparative study based on rheological, chemical and thermal properties. Polym. Degrad. Stabil. 2018, 150, 37–45. [Google Scholar] [CrossRef]
- Qin, Y.; Lin, W.; Liu, D.; Yuan, M.; Li, L. Development of active packaging film made from poly (lactic acid) incorporated essential oil. Prog. Org. Coat. 2017, 103, 76–82. [Google Scholar] [CrossRef]
- Turek, C.; Stintzing, F. Stability of Essential Oils: A Review. Compr. Rev. Food Sci. Food Saf. 2013, 12, 40–53. [Google Scholar] [CrossRef]
- Yu, W.; Wang, X.; Ferraris, E.; Zhang, J. Melt crystallization of PLA/talc in fused filament fabrication. Mater. Des. 2019, 182, 108013. [Google Scholar] [CrossRef]
- Shi, X.; Jing, Z.; Zhang, G. Influence of PLA stereocomplex crystals and thermal treatment temperature on the rheology and crystallization behavior of asymmetric poly (l-Lactide)/poly (d-lactide) blends. J. Polym. Res. 2018, 25, 71. [Google Scholar] [CrossRef]
- Sousa, J.; Arruda, S.; Lima, J.; Wellen, R.; Canedo, E.; Almeida, Y. Crystallization kinetics of poly (butylene adipate terephthalate) in biocomposite with coconut fiber. Matéria 2018, 24, 1–17. [Google Scholar] [CrossRef]
- Wei, B.; Zhao, Y.; Wei, Y.; Yao, J.; Chen, X.; Shao, Z. Morphology and properties of a new biodegradable material prepared from zein and poly (butylene adipate-terephthalate) by reactive blending. ACS Omega 2019, 4, 5609–5616. [Google Scholar] [CrossRef]
- Shojaeiarani, J.; Bajwa, D.; Rehovsky, C.; Bajwa, S.; Vahidi, G. Deterioration and physico-chemical and thermal properties of biopolymers due to reprocessing. Polymers 2019, 11, 58. [Google Scholar] [CrossRef]
- Lins, L.; Livi, S.; Duchet-Rumeau, J.; Gerard, J. Phosphonium ionic liquids as new compatibilizing agents of biopolymer blends composed of poly (butylene-adipate-co-terephtalate)/poly (lactic acid) (PBAT/PLA). RSC Adv. 2015, 5, 59082–59092. [Google Scholar] [CrossRef]
- Hesami, M.; Jalali-Arani, A. Cold crystallization behavior of poly (lactic acid) in its blend with acrylic rubber; the effect of acrylic rubber content. Polym. Int. 2017, 66, 1564–1571. [Google Scholar] [CrossRef]
- Carbonell-Verdu, A.; Ferri, J.; Dominici, F.; Boronat, T.; Sanchez-Nacher, L.; Balart, R.; Torre, L. Manufacturing and compatibilization of PLA/PBAT binary blends by cottonseed oil-based derivatives. Express Polym. Lett. 2018, 12, 808–823. [Google Scholar] [CrossRef]
- Phetwarotai, W.; Phusunti, N.; Aht-Ong, D. Preparation and Characteristics of poly (butylene adipate-co-terephthalate)/polylactide blend films via synergistic efficiency of plasticization and compatibilization. Chin. J. Polym. Sci. 2019, 37, 68–78. [Google Scholar] [CrossRef]
- Righetti, M. Crystallization of polymers investigated by temperature-modulated DSC. Materials 2017, 10, 442. [Google Scholar] [CrossRef] [PubMed]
- Signori, F.; Coltelli, M.; Bronco, S. Thermal degradation of poly (lactic acid) (PLA) and poly (butylene adipate-co-terephthalate) (PBAT) and their blends upon melt processing. Polym. Degrad. Stab. 2009, 94, 74–82. [Google Scholar] [CrossRef]
- Botta, L.; Scaffaro, R.; Sutera, F.; Mistretta, M. Reprocessing of PLA/graphene nanoplatelets nanocomposites. Polymers 2018, 10, 18. [Google Scholar] [CrossRef]
- Smith, E.; Dea, P. Application of Calorimetry in a Wide Context-Differential Scanning Calorimetry, Isothermal Titration Calorimetry and Microcalorimetry; InTech: London, UK, 2013. [Google Scholar]
- Perďochová, D.; Tomanová, K.; Alexy, P.; Bockaj, J.; Feranc, J.; Plavec, R.; Omaníková, L.; Jurkovic, P.; Prikryl, R. The influence of additives on the crystallization of blends based on polylactic acid. IOP Conf. Ser. Mater. Sci. Eng. 2017, 266, 012014. [Google Scholar] [CrossRef]
- Diep, P.T.N.; Takagi, H.; Shimizu, N.; Igarashi, N.; Sasaki, S.; Sakurai, S. Effects of loading amounts of plasticizers on improved crystallization of poly (l-lactic acid). J. Fiber Sci. Technol. 2019, 75, 99–111. [Google Scholar] [CrossRef]
- Chiu, H.; Huang, S.; Chen, Y.; Kuo, M.; Chiang, T.; Chang, C.; Wang, Y. Heat treatment effects on the mechanical properties and morphologies of poly (lactic acid)/poly (butylene adipate co-terephthalate) blends. Int. J. Polym. Sci. 2013, 2013, 951696. [Google Scholar] [CrossRef]
- Kashi, S.; Gupta, R.; Baum, T.; Kao, N.; Bhattacharya, S. Dielectric properties and electromagnetic interference shielding effectiveness of graphene-based biodegradable nanocomposites. Mater. Des. 2016, 109, 68–78. [Google Scholar] [CrossRef]
- Shi, N.; Cai, J.; Dou, Q. Crystallization, morphology and mechanical properties of PLA/PBAT/CaCO3 composites. Adv. Mater. Res. 2013, 602, 768–771. [Google Scholar]
- Jiang, L.; Wolcott, M.; Zhang, J. Study of biodegradable polylactide/poly (butylene adipate-co-terephthalate) blends. Biomacromolecules 2006, 7, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Roudaut, G.; Simatos, D.; Champion, D.; Contreras-Lopez, E.; Le Meste, M. Molecular mobility around the glass transition temperature: A mini review. Innov. Food Sci. Emerg. 2004, 5, 127–134. [Google Scholar] [CrossRef]
- Antunes, M.; Gago, C.; Cavaco, A.; Miguel, M. Edible coatings enriched with essential oils and their compounds for fresh and fresh-cut fruit. Recent Pat. Food Nutr. Agric. 2012, 4, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Siegenthaler, K.; Künkel, A.; Skupin, G.; Yamamoto, M. Synthetic Biodegradable Polymers; SpringerLink: New York, NY, USA, 2012. [Google Scholar]
- Bianco, A.; Calderone, M.; Cacciotti, I. Electrospun PHBV/PEO co-solution blends: Microstructure, thermal and mechanical properties. Mater. Sci. Eng. C-Mater. 2013, 33, 1067–1077. [Google Scholar] [CrossRef]
- Pan, H.; Li, Z.; Yang, J.; Li, X.; Ai, X.; Hao, Y.; Zhang, H.; Dong, L. The effect of MDI on the structure and mechanical properties of poly (lactic acid) and poly (butylene adipate-co- butylene terephthalate) blends. RSC Adv. 2018, 8, 4610–4623. [Google Scholar] [CrossRef]
- Tcharkhtchi, A.; Farzaneh, S.; Abdallah-Elhirtsi, S.; Esmaeillou, B.; Nony, F.; Baron, A. Thermal aging effect on mechanical properties of polyurethane. Int. J. Polym. Anal. Charact. 2014, 19, 571–584. [Google Scholar] [CrossRef]
- Chieng, B.; Ibrahim, N.; Then, Y.; Loo, Y. Epoxidized vegetable oils plasticized poly (lactic acid) biocomposites: Mechanical, thermal and morphology properties. Molecules 2014, 19, 16024–16038. [Google Scholar] [CrossRef]
- Giita, V.; Ibrahim, N.; Yunus, W.; Hassan, H.; Woei, C. A comparative study on the mechanical, thermal and morphological characterization of poly (lactic acid)/epoxidized palm oil blend. Int. J. Mol. Sci. 2012, 13, 5878–5898. [Google Scholar]
- Armentano, I.; Fortunati, E.; Burgos, N.; Dominici, F.; Luzi, F.; Fiori, S.; Jiménez, A.; Yoon, K.; Ahn, J.; Kang, S.; et al. Processing and characterization of plasticized PLA/PHB blends for biodegradable multiphase systems. Express Polym. Lett. 2015, 9, 583–596. [Google Scholar] [CrossRef]
- Amin, A.; Sauid, S.; Musa, M.; Hamid, K. The effect of glycerol content on the mechanical properties, surface morphology and water absorption of thermoplastic films from Tacca leontopetaloides starch. Jurnal Teknologi 2017, 79, 53–59. [Google Scholar]
- Farsetti, S.; Cioni, B.; Lazzeri, A. Physico-chemical properties of biodegradable rubber toughened polymers. Macromol. Symp. 2011, 301, 82–89. [Google Scholar] [CrossRef]
- Dou, Q.; Cai, J. Investigation of poly (lactide) (PLA)/poly (butylene adipate co-terephthalate) (PBAT)/bark flour of plane tree (PF) eco-composites. Materials 2016, 9, 393. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Pan, H.; Li, X.; Sun, S.; Zhang, H.; Dong, L. A study on the mechanical, thermal properties and crystallization behavior of poly (lactic acid)/thermoplastic poly (propylene carbonate) polyurethane blends. RSC Adv. 2017, 7, 46183–46194. [Google Scholar] [CrossRef]
- Lim, H.; Hoag, S. Plasticizer effects on physical-mechanical properties of solvent cast Soluplus® films. AAPS PharmSciTech 2013, 14, 903–910. [Google Scholar] [CrossRef]
- Pivsa-Art, W.; Chaiyasat, A.; Pivsa-Art, S.; Yamane, H.; Ohara, H. Preparation of polymer blends between poly (lactic acid) and poly (butylene adipate-co-terephthalate) and biodegradable polymers as compatibilizers. Energy Procedia 2013, 34, 549–554. [Google Scholar] [CrossRef]
- Sabino, M.; Albuerne, J.; Müller, A.; Brisson, J.; Prud’homme, R. Influence of in vitro hydrolytic degradation on the morphology and crystallization behavior of poly (p-dioxanone). Biomacromolecules 2004, 5, 358–370. [Google Scholar] [CrossRef]
- Fukushima, K.; Camino, G. Poly (Lactic Acid) Science and Technology; RSC Polymer Chemistry Series; Royal Society of Chemistry: London, UK, 2015. [Google Scholar]
- Sabino, M.; González, S.; Márquez, L.; Feijoó, J. Study of the hydrolytic degradation of polydioxanone (PPDX). Polym. Degrad. Stabil. 2000, 69, 209–216. [Google Scholar] [CrossRef]
- Rowe, M.; Eyiler, E.; Walters, K. Hydrolytic degradation of bio-based polyesters: Effect of pH and time. Polym. Test. 2016, 52, 192–199. [Google Scholar] [CrossRef]
- Gabbot, P. Principles and Applications of Thermal Analysis; Blackwell Publishing: Oxford, UK, 2008. [Google Scholar]
- Su, S.; Kopitzky, R.; Tolga, S.; Kabasci, S. Polylactide (PLA) and its blends with poly (butylene succinate) (PBS): A brief review. Polymers 2019, 11, 1193. [Google Scholar] [CrossRef] [PubMed]
- Limsukon, W.; Auras, R.; Selke, S. Hydrolytic degradation and lifetime prediction of poly (lactic acid) modified with a functional epoxy-based chain extender. Polym. Test. 2019, 80, 106108. [Google Scholar] [CrossRef]
- Rodríguez, E.; Marcos, B.; Huneault, M. Hydrolysis of polylactide in aqueous media. J. Appl. Polym. Sci. 2016, 44152. [Google Scholar] [CrossRef]
- Sabino, M.; Morales, D.; Ronca, G.; Feijoo, J. Estudio de la degradación hidrolítica de un copolímero biodegradable. Acta Científica Venezolana 2003, 54, 18–27. [Google Scholar] [PubMed]
- Kaavessina, M.; Chafidz, A.; Ali, I.; Al-Zahrani, S. Chractrization of poly (lactic acid)/hydroxyapatite prepared by a solvent-blending technique: Viscoelasticity and in vitro hydrolytic degradation. J. Elastom. Plast. 2015, 47, 753–768. [Google Scholar] [CrossRef]
- Tsuji, H.; Eto, T.; Sakamoto, Y. Synthesis and hydrolytic degradation of substituted of poly (DL-lactic acid)s. Materials 2011, 4, 1384–1398. [Google Scholar] [CrossRef]
- Höglund, A.; Hakkarainen, M.; Albertsson, A. Migration and Hydrolysis of Hydrophobic Polylactide Plasticizer. Biomacromolecules 2010, 11, 277–283. [Google Scholar] [CrossRef]
- Menčík, P.; Přikryl, R.; Stehnová, I.; Melčová, V.; Kontárová, S.; Figalla, S.; Alexy, P.; Bočkaj, J. Effect of Selected Commercial Plasticizers on Mechanical, Thermal, and Morphological Properties of Poly (3-hydroxybutyrate)/Poly (lactic acid)/Plasticizer Biodegradable Blends for Three-Dimensional (3D) Print. Materials 2018, 11, 1893. [Google Scholar] [CrossRef]
- Przybytek, A.; Sienkiewicz, M.; Kucińska-Lipka, J.; Janik, H. Preparation and characterization of biodegradable and compostable PLA/TPS/ESO compositions. Ind. Crop. Prod. 2018, 122, 375–383. [Google Scholar] [CrossRef]
- Rocca-Smith, J.; Whyte, O.; Brachais, C.; Champion, D.; Piasente, F.; Marcuzzo, E.; Sensidoni, A.; Debeaufort, F.; Karbowiak, T. Beyond biodegradability of poly(lactic acid): Physical and chemical stability in humid environments. ACS Sustain. Chem. Eng. 2017, 5, 2751–2762. [Google Scholar] [CrossRef]
- Zhang, X.; Espiritu, M.; Bilyk, A.; Kurniawan, L. Morphological behaviour of poly (lactic acid) during hydrolytic degradation. Polym. Degrad. Stabil. 2008, 93, 1964–1970. [Google Scholar] [CrossRef]
- Scaffaro, R.; Sutera, F.; Mistretta, M.; Botta, L.; La Mantia, F. Structure-properties relationship in melt reprocessed in PLA/hydrotalcites nanocomposites. Express Polym. Lett. 2017, 11, 555–564. [Google Scholar] [CrossRef]
- Mysiukiewicz, O.; Barczewski, M.; Skórczewska, K.; Szulc, J.; Kloziński, A. Accelerated weathering of polylactide-based composites filled with linseed cake: The influence of time and oil content within the filler. Polymers 2019, 11, 1495. [Google Scholar] [CrossRef] [PubMed]
- Ndazi, B.; Larlsson, S. Characterization of hydrolytic degradation of polylactic acid/rice hulls composites in water at different temperatures. Express Polym. Lett. 2011, 5, 119–131. [Google Scholar] [CrossRef]
- Andersson, S.; Hakkarainen, M.; Innkinen, S.; Södergård, A.; Albertsson, A. Customizing the hydrolytic degradation rate of stereocomplex PLA through different PDLA architectures. Biomacromolecules 2012, 13, 1212–1222. [Google Scholar] [CrossRef] [PubMed]
- Benali, S.; Aouadi, S.; Dechief, A.; Murariu, M.; Dubois, P. Key factor for tunning hydrolytic degradation of polylactide/zinc oxide nanocomposites. Nanocomposites 2015, 1, 51–61. [Google Scholar] [CrossRef]
- Xu, L.; Cawford, K.; Gorman, C. Effects of temperature and pH on the degradation of poly (lactic acid) brushes. Macromolecules 2011, 44, 4777–4782. [Google Scholar] [CrossRef]
- Woodard, L.; Grunlan, M. Hydrolytic degradation and erosion of polyester biomaterials. ACS Macro Lett. 2018, 7, 976–982. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Blend | PLA (wt %) | PBAT (wt %) |
|---|---|---|
| PLA100 | 100 | 0 |
| PLA70 | 70 | 30 |
| PLA60 | 60 | 40 |
| PLA50 | 50 | 50 |
| PBAT100 | 0 | 100 |
| Formulation | PLA60 (wt %) | CEO (wt %) | Feeding Speed CEO (g min−1) | Extruder (rpm) | Take-off Unit (rpm) |
|---|---|---|---|---|---|
| FC | 100.0 | 0.0 | 0.00 | 20 | 32 |
| F3 | 93.9 | 6.1 | 0.30 | 50 | 47 |
| FG | 95.7 | 0.0 (4.3 a) | 0.05 | 20 | 32 |
| Buffer | pH |
|---|---|
| Acetic acid | 4.98 |
| Phosphate | 7.40 |
| Ammonium | 8.50 |
| Compounds | Frequencies (Wavenumber in cm−1) | Assignment |
|---|---|---|
| CEO [44] | 3267–2697 | O-H stretching |
| 1672 and 1625 | C=O stretching in aldehyde | |
| 1574 | C=C stretching skeletal vibration of the aromatic ring | |
| 1452 | C-OH bending | |
| 1292 | -CH2 swing in alkanes and =C-H in-plane bending of the aromatic ring | |
| 1242 | symmetric expansion of C-O-C of aromatic acid ester and vibrational stretching of C-OH from phenolics in eugenol from essential oils | |
| 1122 | C-O stretching and C-OH deformation | |
| 971 | C-H bending | |
| 750 | –CH bending out-of-plane in aromatic ring | |
| 683 | CH=CH bending out-of-plane in alkenes | |
| PLA100 [45,46] | 3034–2874 | symmetrical and asymmetrical stretching of -CH in -CH3 |
| 1745 | C=O stretching | |
| 1448 | –CH in -CH3 | |
| 1178 | O-C-O stretching | |
| 1122, 1075, and 1050 | C-O-C | |
| 758 | C-C- stretching | |
| PBAT100 [47,48] | 3003–2819 | stretching of -CH in -CH3 |
| 1709 | C=O stretching | |
| 1522–1336 | C=O stretching for the phenolic group | |
| 1187–982 | C-O and C-O-C stretching vibrations | |
| 730 | –C-C- stretching |
| Compound | Tg (°C) | Tc (°C) | Tm (°C) | ΔHm (J/g) | Xc (%) | |
|---|---|---|---|---|---|---|
| PBAT | PLA | |||||
| PLA100 | 62.4 | 43.0 | - | 150.3 | 12.3 | 13.3 |
| PLA70 | 61.8 | 65.0 | 123.7 | 149.2 | 11.1 | 8.3 |
| PLA60 | 61.9 | 68.7 | 116.8 | 150.7 | 12.8 | 8.2 |
| PLA50 | 61.4 | 75.1 | 126.0 | 149.5 | 13.2 | 7.1 |
| PBAT100 | −34.0 [42] | 43.3 | 124.1 | - | 17.3 | 15.1 |
| FC | 61.2 | 74.8 | 130.5 | 149.6 | 12.2 | 7.9 |
| F3 | 59.2 | 69.0 | 125.5 | 148.1 | 12.7 | 8.2 |
| Compound | Young’s Modulus E (MPa) a | Tensile Strength at Break σ (MPa) a | Elongation at Break Ɛ (%) | Tenacity (kJ m−3) a |
|---|---|---|---|---|
| PLA100 | 792.6 ± 29.8 e | 52.4 ± 2.2 d | 8.8 ± 0.2 | 72.3 ± 4.1 a |
| PLA70 | 534.8 ± 13.5 d | 29.0 ± 2.0 c | 14.8 ± 2.8 | 108.1 ± 1.8 a |
| PLA60 | 319.7 ± 32.6 c | 13.5 ± 1.3 a | 17.9 ± 1.9 | 45.2 ± 1.8 a |
| PLA50 | 232.2 ± 20.0 b | 15.1 ± 1.2 ab | 27.4 ± 1.2 | 101.4 ± 2.7 a |
| PBAT100 | 1.6 ± 0.2 a | 17.7 ± 2.3 b | 1207.6 ± 84.1 | 115,368.0 ± 6466.8 b |
| F3 | 2057.4 ± 62.5 b | 32.9 ± 1.2 b | 9.8 ± 0.1 | 261.0 ± 14.7 b |
| FC | 2025.9 ± 23.4 b | 34.6 ± 1.7 b | 9.2 ± 0.8 | 193.4 ± 8.3 a |
| FG | 1651.9 ± 73.3 a | 26.6 ± 2.8 a | 9.2 ± 1.1 | 173.3 ± 16.9 a |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Correa-Pacheco, Z.N.; Black-Solís, J.D.; Ortega-Gudiño, P.; Sabino-Gutiérrez, M.A.; Benítez-Jiménez, J.J.; Barajas-Cervantes, A.; Bautista-Baños, S.; Hurtado-Colmenares, L.B. Preparation and Characterization of Bio-Based PLA/PBAT and Cinnamon Essential Oil Polymer Fibers and Life-Cycle Assessment from Hydrolytic Degradation. Polymers 2020, 12, 38. https://doi.org/10.3390/polym12010038
Correa-Pacheco ZN, Black-Solís JD, Ortega-Gudiño P, Sabino-Gutiérrez MA, Benítez-Jiménez JJ, Barajas-Cervantes A, Bautista-Baños S, Hurtado-Colmenares LB. Preparation and Characterization of Bio-Based PLA/PBAT and Cinnamon Essential Oil Polymer Fibers and Life-Cycle Assessment from Hydrolytic Degradation. Polymers. 2020; 12(1):38. https://doi.org/10.3390/polym12010038
Chicago/Turabian StyleCorrea-Pacheco, Zormy Nacary, Jaime Daniel Black-Solís, Pedro Ortega-Gudiño, Marcos Antonio Sabino-Gutiérrez, José Jesús Benítez-Jiménez, Alfonso Barajas-Cervantes, Silvia Bautista-Baños, and Liliana Beyalith Hurtado-Colmenares. 2020. "Preparation and Characterization of Bio-Based PLA/PBAT and Cinnamon Essential Oil Polymer Fibers and Life-Cycle Assessment from Hydrolytic Degradation" Polymers 12, no. 1: 38. https://doi.org/10.3390/polym12010038
APA StyleCorrea-Pacheco, Z. N., Black-Solís, J. D., Ortega-Gudiño, P., Sabino-Gutiérrez, M. A., Benítez-Jiménez, J. J., Barajas-Cervantes, A., Bautista-Baños, S., & Hurtado-Colmenares, L. B. (2020). Preparation and Characterization of Bio-Based PLA/PBAT and Cinnamon Essential Oil Polymer Fibers and Life-Cycle Assessment from Hydrolytic Degradation. Polymers, 12(1), 38. https://doi.org/10.3390/polym12010038