Crystallization and Microstructure Evolution of Microinjection Molded Isotactic Polypropylene with the Assistance of Poly(Ethylene Terephthalate)



Abstract

1. Introduction
2. Experimental
2.1. Materials
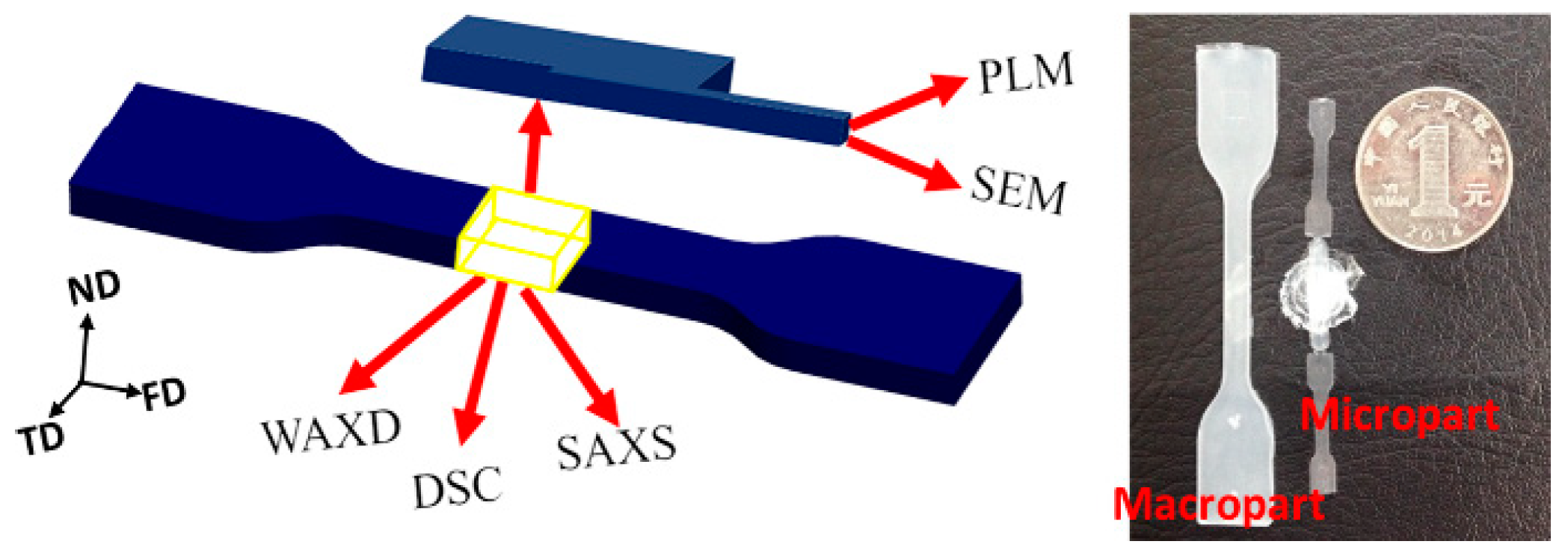
2.2. Sample Preparation
2.3. Characterization
2.3.1. Differential Scanning Calorimetry (DSC)
2.3.2. Scanning Electron Microscope (SEM)
2.3.3. Polarized Light Microscope (PLM)
2.3.4. Dynamic Rheology Measurements
2.3.5. Synchrotron Two-Dimensional Wide-Angle X-ray Diffraction (2D-WAXD) and Small-Angle X-ray Scattering (2D-SAXS)
2.3.6. Tensile Test
3. Results and Discussion
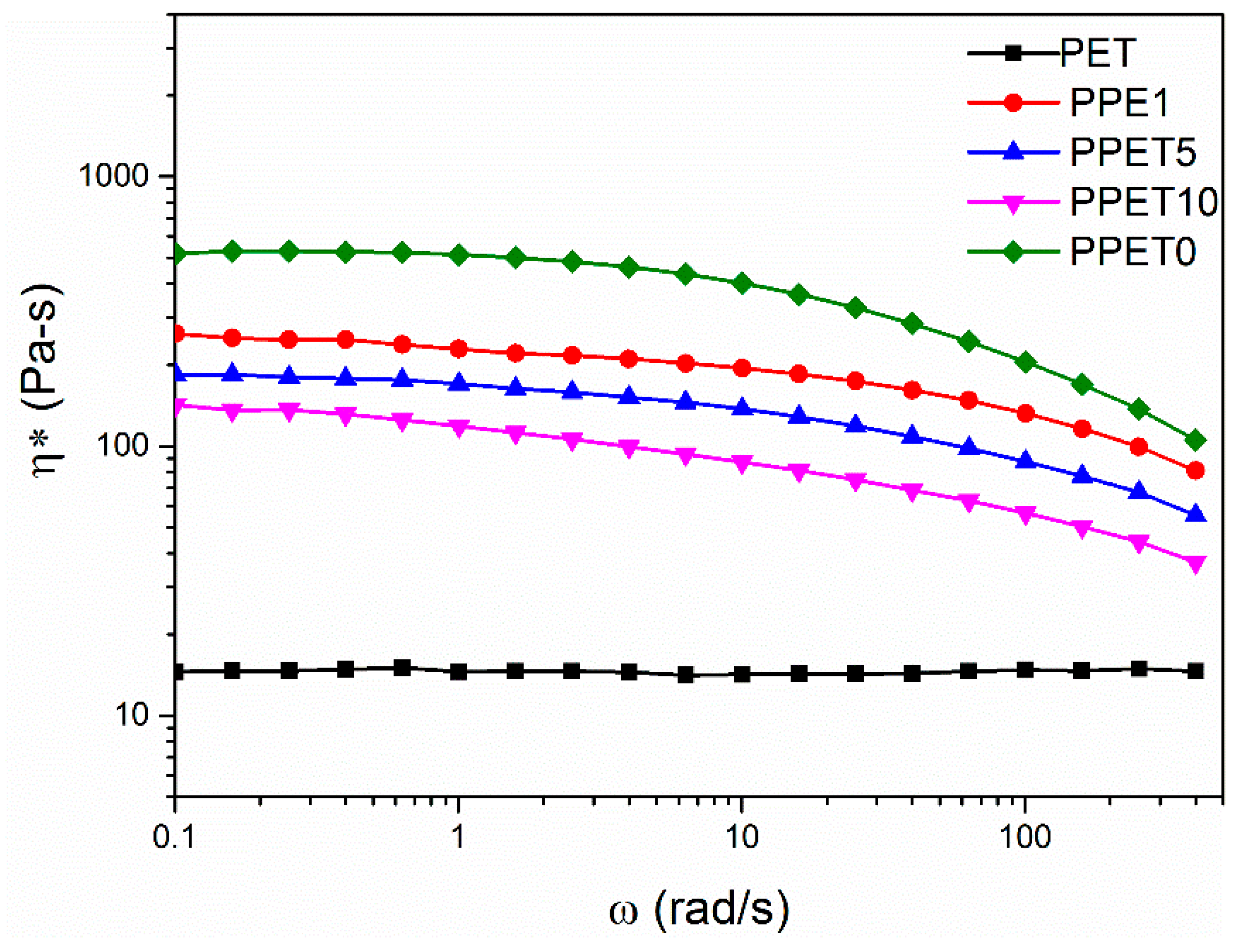
3.1. Dynamic Rheology Analyses
3.2. Morphological Observation
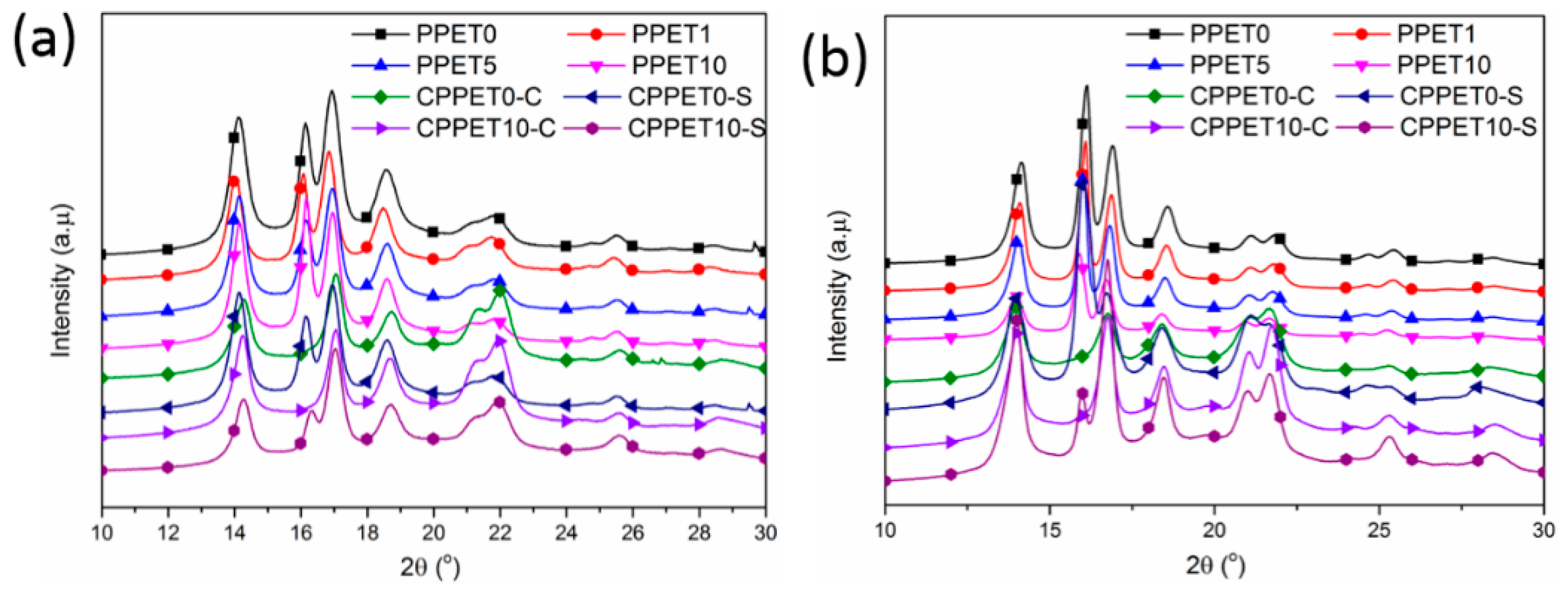
3.3. Crystalline Structure
3.4. Orientation Analysis
3.5. Crystalline Structure Analysis
3.6. Mechanical Properties
4. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Lu, Z.; Zhang, K. Morphology and mechanical properties of polypropylene micro-arrays by micro-injection molding. Int. J. Adv. Manuf. Tech. 2009, 40, 490–496. [Google Scholar] [CrossRef]
- Abbasi, S.; Carreau, P.J.; Derdouri, A. Flow induced orientation of multiwalled carbon nanotubes in polycarbonate nanocomposites: Rheology, conductivity and mechanical properties. Polymer 2010, 51, 922–935. [Google Scholar] [CrossRef]
- Giboz, J.; Copponnex, T.; Mélé, P. Microinjection molding of thermoplastic polymers: A review. J. Micromech. Microeng. 2007, 17, R96. [Google Scholar] [CrossRef]
- Tseng, S.-C.; Chen, Y.-C.; Kuo, C.-L.; Shew, B.-Y. A study of integration of liga and m-edm technology on the microinjection molding of ink-jet printers’ nozzle plates. Microsyst. Technol. 2005, 12, 116–119. [Google Scholar] [CrossRef]
- Kim, H.; Colton, J.S. Fabrication and analysis of plastic hypodermic needles. J. Med. Eng. Technol. 2005, 29, 181–186. [Google Scholar] [CrossRef]
- Liu, Z.; Chen, Y.; Ding, W.; Zhang, C. Filling behavior, morphology evolution and crystallization behavior of microinjection molded poly (lactic acid)/hydroxyapatite nanocomposites. Compos. Part A Appl. Sci. Manuf. 2015, 72, 85–95. [Google Scholar] [CrossRef]
- Wallrabe, U.; Dittrich, H.; Friedsam, G.; Hanemann, T.; Mohr, J.; Müller, K.; Piotter, V.; Ruther, P.; Schaller, T.; Zissler, W. Micromolded easy-assembly multi fiber connector: Ribcon®. Microsyst. Technol. 2002, 8, 83–87. [Google Scholar] [CrossRef]
- Li, M.; Qi, Y.; Zhao, Z.; Xiang, Z.; Liao, X.; Niu, Y. Morphology evolution and crystalline structure of controlled-rheology polypropylene in micro-injection molding. Polym. Adv. Technol. 2016, 27, 494–503. [Google Scholar] [CrossRef]
- Barhoumi, N.; Jaziri, M.; Massardier, V.; Cassagnau, P. Valorization of poly (butylene terephthalate) wastes by blending with virgin polypropylene: Effect of the composition and the compatibilization. Polym. Eng. Sci. 2008, 48, 1592–1599. [Google Scholar] [CrossRef]
- Saujanya, C.; Radhakrishnan, S. Structure development and properties of pet fibre filled pp composites. Polymer 2001, 42, 4537–4548. [Google Scholar] [CrossRef]
- Wu, S. Chain structure, phase morphology, and toughness relationships in polymers and blends. Polym. Eng. Sci. 1990, 30, 753–761. [Google Scholar] [CrossRef]
- Xu, L.; Huang, Y.F.; Xu, J.Z.; Ji, X.; Li, Z.M. Improved performance balance of polyethylene by simultaneously forming oriented crystals and blending ultrahigh-molecular-weight polyethylene. RSC Adv. 2014, 4, 1512–1520. [Google Scholar] [CrossRef]
- Jayanarayanan, K.; Ravichandran, A.; Rajendran, D.; Sivathanupillai, M.; Venkatesan, A.; Thomas, S.; Joseph, K. Morphology and mechanical properties of normal blends and in-situ microfibrillar composites from low-density polyethylene and poly (ethylene terephthalate). Polym. Plast. Technol. Eng. 2010, 49, 442–448. [Google Scholar] [CrossRef]
- Jayanarayanan, K.; Thomas, S.; Joseph, K. Morphology, static and dynamic mechanical properties of in situ microfibrillar composites based on polypropylene/poly (ethylene terephthalate) blends. Compos. Part A Appl. Sci. Manuf. 2008, 39, 164–175. [Google Scholar] [CrossRef]
- Jayanarayanan, K.; Thomas, S.; Joseph, K. In situ microfibrillar blends and composites of polypropylene and poly (ethylene terephthalate): Morphology and thermal properties. J. Polym. Res. 2011, 18, 1–11. [Google Scholar] [CrossRef]
- Li, Z.M.; Li, L.B.; Shen, K.Z.; Yang, M.B.; Huang, R. In-situ microfibrillar pet/ipp blend via slit die extrusion, hot stretching, and quenching: Influence of hot stretch ratio on morphology, crystallization, and crystal structure of ipp at a fixed pet concentration. J. Polym. Sci. Part B Polym. Phys. 2004, 42, 4095–4106. [Google Scholar] [CrossRef]
- Zhao, Z.; Yang, Q.; Xiang, Z.; Kong, M.; Tang, D.; Huang, Y. Effect of in situ poly (ethylene terephthalate)(PET) microfibrils on the morphological structure and crystallization behavior of isotactic polypropylene (iPP) under an intensive shear rate. Polym. Adv. Technol. 2015, 26, 1275–1284. [Google Scholar] [CrossRef]
- Zhao, Z.; Yang, Q.; Gong, P.; Sun, H.; Wu, P.; Huang, Y. Effects of Process Temperatures on the Flow-Induced Crystallization of Isotactic Polypropylene/Poly (ethylene terephthalate) Blends in Microinjection Molding. Ind. Eng. Chem. Res. 2017, 56, 9467–9477. [Google Scholar] [CrossRef]
- Zhong, G.-J.; Li, L.; Mendes, E.; Byelov, D.; Fu, Q.; Li, Z.-M. Suppression of skin-core structure in injection-molded polymer parts by in situ incorporation of a microfibrillar network. Macromolecules 2006, 39, 6771–6775. [Google Scholar] [CrossRef]
- Yi, X.; Chen, C.; Zhong, G.-J.; Xu, L.; Tang, J.-H.; Ji, X.; Hsiao, B.S.; Li, Z.-M. Suppressing the skin–core structure of injection-molded isotactic polypropylene via combination of an in situ microfibrillar network and an interfacial compatibilizer. J. Phys. Chem. B 2011, 115, 7497–7504. [Google Scholar] [CrossRef]
- Wang, K.; Guo, M.; Zhao, D.; Zhang, Q.; Du, R.; Fu, Q. Facilitating transcrystallization of polypropylene/glass fiber composites by imposed shear during injection molding. Polymer 2006, 47, 8374–8379. [Google Scholar] [CrossRef]
- Jin, M.; La, R.; Zhang, Y.; Liu, K.; Li, X.; Zhang, J. Stratiform β crystals in ultrahigh molecular weight polyethylene and β-nucleating agent-nucleated isotactic polypropylene at micro-injection molding condition. Polym. Test. 2015, 42, 135–143. [Google Scholar] [CrossRef]
- Xu, X.; Li, X.P.; Jin, B.Q.; Sheng, Q.; Wang, T.; Zhang, J. Influence of morphology evolution on the mechanical properties of beta nucleated isotactic polypropylene in presence of polypropylene random copolymer. Polym. Test. 2016, 51, 13–19. [Google Scholar] [CrossRef]
- Shangguan, Y.; Song, Y.; Peng, M.; Li, B.; Zheng, Q. Formation of β-crystal from nonisothermal crystallization of compression-molded isotactic polypropylene melt. Eur. Polym. J. 2005, 41, 1766–1771. [Google Scholar] [CrossRef]
- Zhao, Z.; Yang, Q.; Kong, M.; Tang, D.; Chen, Q.; Liu, Y.; Lou, F.; Huang, Y.; Liao, X. Unusual hierarchical structures of micro-injection molded isotactic polypropylene in presence of an in situ microfibrillar network and a β-nucleating agent. RSC Adv. 2015, 5, 43571–43580. [Google Scholar] [CrossRef]
- Picken, S.J.; Aerts, J.; Visser, R.; Northolt, M.G. Structure and rheology of aramid solutions: X-ray scattering measurements. Macromolecules 1990, 23, 3849–3854. [Google Scholar] [CrossRef]
- Turner-Jones, A.; Cobbold, A. The β crystalline form of isotactic polypropylene. J. Polym. Sci. Part B Polym. Lett. 1968, 6, 539–546. [Google Scholar] [CrossRef]
- Isayev, A.I.; Modic, M. Self-Reinforced melt processible polymer composites: Extrusion, compression, and injection molding. Polym. Compos. 1987, 8, 158–175. [Google Scholar] [CrossRef]
- Mehta, A.; Isayev, A.I. The dynamic properties, temperature transitions, and thermal stability of poly (etherether ketone)-thermotropic liquid crystalline polymer blends. Polym. Eng. Sci. 2004, 31, 963–970. [Google Scholar] [CrossRef]
- Liu, F.; Guo, C.; Wu, X.; Qian, X.; Zhang, J. Morphological comparison of isotactic polypropylene parts prepared by micro-njection molding and conventional injection molding. Polym. Adv. Technol. 2012, 23, 686–694. [Google Scholar] [CrossRef]
- Pesneau, I.; Kadi, A.A.; Bousmina, M.; Cassagnau, P.; Michel, A. From polymer blends to in situ polymer/polymer composites: Morphology control and mechanical properties. Polym. Eng. Sci. 2002, 42, 1990–2004. [Google Scholar] [CrossRef]
- Tadmor, Z. Molecular orientation in injection molding. J. Appl. Polym. Sci. 1974, 18, 1753–1772. [Google Scholar] [CrossRef]
- Varga, J. Supermolecular structure of isotactic polypropylene. J. Mater. Sci. 1992, 27, 2557–2579. [Google Scholar] [CrossRef]
- Varga, J. β-modification of isotactic polypropylene: Preparation, structure, processing, properties, and application. J. Macromol. Sci. Part B 2002, 41, 1121–1171. [Google Scholar] [CrossRef]
- Somani, R.H.; Yang, L.; Hsiao, B.S.; Agarwal, P.K.; Fruitwala, H.A.; Tsou, A.H. Shear-induced precursor structures in isotactic polypropylene melt by in-situ rheo-saxs and rheo-waxd studies. Macromolecules 2002, 35, 9096–9104. [Google Scholar] [CrossRef]
- Fujiyama, M.; Wakino, T.; Kawasaki, Y. Structure of skin layer in injection-molded polypropylene. J. Appl. Polym. Sci. 1988, 35, 29–49. [Google Scholar] [CrossRef]
- Lotz, B.; Wittmann, J. Structural relationships in blends of isotactic polypropylene and polymers with aliphatic sequences. J. Polym. Sci. Part B Polym. Phys. 1986, 24, 1559–1575. [Google Scholar] [CrossRef]
- He, B.; Yuan, X.; Yang, H.; Tan, H.; Qian, L.; Zhang, Q.; Fu, Q. Ultrasonic measurement of orientation in hdpe/ipp blends obtained by dynamic packing injection molding. Polymer 2006, 47, 2448–2454. [Google Scholar] [CrossRef]
- Zhao, Z.; Yang, Q.; Xi, S.; Kong, M.; Huang, Y.; Liao, X. New understanding of the hierarchical distribution of isotactic polypropylene blends formed by microinjection-molded poly (ethylene terephthalate) and β-nucleating agent. RSC Adv. 2015, 5, 61127–61136. [Google Scholar] [CrossRef]
- Na, B.; Zhang, Q.; Wang, K.; Li, L.; Fu, Q. Origin of various lamellar orientations in high-density polyethylene/isotactic polypropylene blends achieved via dynamic packing injection molding: Bulk crystallization vs. Epitaxy. Polymer 2005, 46, 819–825. [Google Scholar] [CrossRef]
- Fujiyama, M.; Azuma, K. Skin/core morphology and tensile impact strength of injection-molded polypropylene. J. Appl. Polym. Sci. 1979, 23, 2807–2811. [Google Scholar] [CrossRef]
- Varga, J. β-modification of polypropylene and its two-component systems. J. Therm. Anal. Calorim. 1989, 35, 1891–1912. [Google Scholar] [CrossRef]
- Housmans, J.-W.; Gahleitner, M.; Peters, G.W.; Meijer, H.E. Structure–property relations in molded, nucleated isotactic polypropylene. Polymer 2009, 50, 2304–2319. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Designation | Composition |
|---|---|
| PPET0 | iPP |
| PPET1 | 99 wt % iPP + 1 wt % PET |
| PPET5 | 95 wt % iPP + 5 wt % PET |
| PPET10 | 90 wt % iPP + 10 wt % PET |
| Samples | Tm = 80 °C | Tm = 120 °C | ||
|---|---|---|---|---|
| fwaxd | fsaxs | fwaxd | fsaxs | |
| PPET0 | 0.912 | 0.856 | 0.874 | 0.826 |
| PPET1 | 0.925 | 0.872 | 0.893 | 0.842 |
| PPET5 | 0.956 | 0.937 | 0.926 | 0.897 |
| PPET10 | 0.941 | 0.915 | 0.911 | 0.872 |
| Samples | Tm = 120 °C | Tm = 80 °C | ||
|---|---|---|---|---|
| DSC | 1D-WAXD | DSC | 1D-WAXD | |
| Kβ | φβ | Kβ | φβ | |
| PPET0 | 8.1 | 41.7 | --- | 24.9 |
| PPET1 | 8.4 | 42.8 | --- | 28.1 |
| PPET5 | 9.3 | 43.9 | --- | 34.3 |
| PPET10 | 7.9 | 39.6 | --- | 32.7 |
| CPPET0-C | 4.2 | -- | --- | --- |
| CPPET0-S | 21.3 | 46.9 | --- | 17.5 |
| CPPET10-C | --- | --- | --- | --- |
| CPPET10-S | -- | 13.6 | --- | 7.8 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Z.; Zhang, X.; Yang, Q.; Ai, T.; Jia, S.; Zhou, S. Crystallization and Microstructure Evolution of Microinjection Molded Isotactic Polypropylene with the Assistance of Poly(Ethylene Terephthalate). Polymers 2020, 12, 219. https://doi.org/10.3390/polym12010219
Zhao Z, Zhang X, Yang Q, Ai T, Jia S, Zhou S. Crystallization and Microstructure Evolution of Microinjection Molded Isotactic Polypropylene with the Assistance of Poly(Ethylene Terephthalate). Polymers. 2020; 12(1):219. https://doi.org/10.3390/polym12010219
Chicago/Turabian StyleZhao, Zhongguo, Xin Zhang, Qi Yang, Taotao Ai, Shikui Jia, and Shengtai Zhou. 2020. "Crystallization and Microstructure Evolution of Microinjection Molded Isotactic Polypropylene with the Assistance of Poly(Ethylene Terephthalate)" Polymers 12, no. 1: 219. https://doi.org/10.3390/polym12010219
APA StyleZhao, Z., Zhang, X., Yang, Q., Ai, T., Jia, S., & Zhou, S. (2020). Crystallization and Microstructure Evolution of Microinjection Molded Isotactic Polypropylene with the Assistance of Poly(Ethylene Terephthalate). Polymers, 12(1), 219. https://doi.org/10.3390/polym12010219