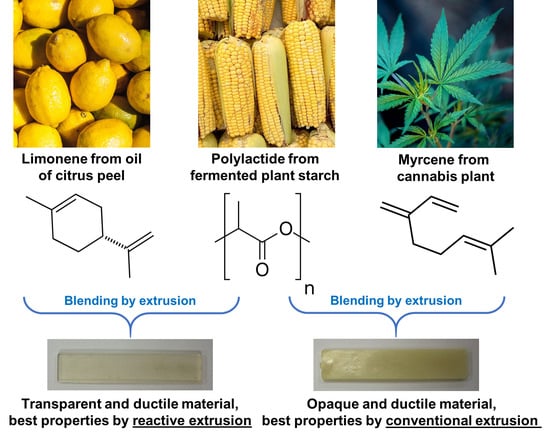
3.1. Processability
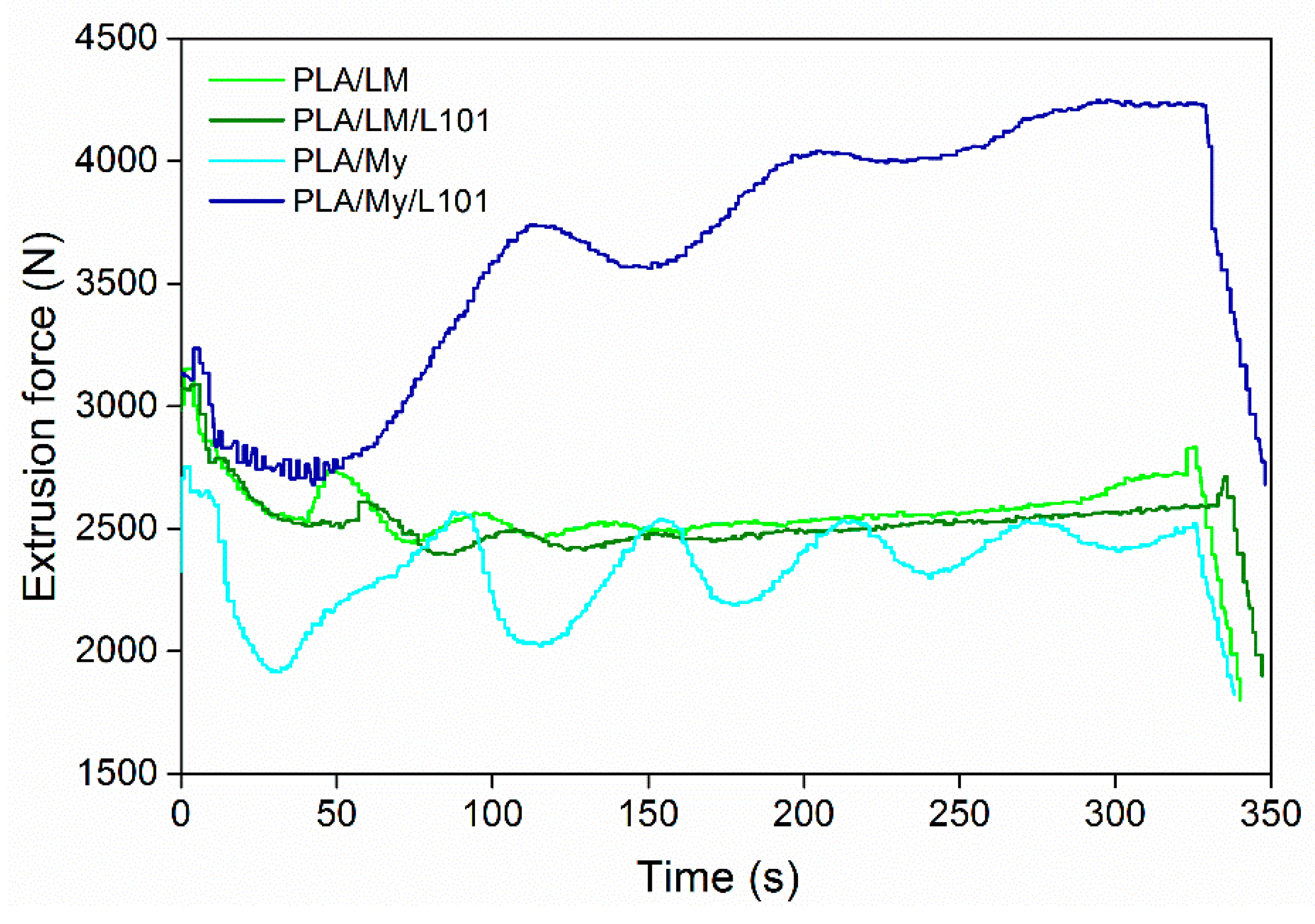
As starting point, it is important to verify the processability of the PLA-based materials by monitoring the extrusion force or torque as a function of the extrusion time, by quantifying degradation from molecular weight measurements after each processing step, and by observing the physical aspect of the final shaped materials. The evolution of the extrusion force with time can provide rough information about the material viscosity. In particular, if the force drastically increases during the extrusion, chemical reactions of the PLA matrix, and/or of the plasticizer, and/or between the PLA matrix and the plasticizer can be expected. In the opposite case, an important decrease of the force may be a sign of thermal degradation of PLA [
27] and/or the plasticizer. Extrusion force–time curves were provided in
Figure 1 for PLA/LM 80/20, PLA/LM/L101 79/20/1, PLA/My 80/20, and PLA/My/L101 79/20/1. Just after the injection of the liquid plasticizer or plasticizer/initiator mixture into the extruder, the recorded extrusion force decreased significantly, and then, exhibited some oscillations for all the materials. These oscillations tended to disappear with time, so that the force reached a final value slightly lower than the initial one prior liquid injection in the case of PLA/LM 80/20, PLA/LM/L101 79/20/1, PLA/My 80/20. In the case of PLA/My/L101, the extrusion force significantly increased after the initial step of force decrease.
Our results demonstrate that extrusion mainly involves a homogenization of PLA/LM, PLA/LM/L101, and PLA/My, without significant reaction or degradation modifying the viscosity. Nevertheless, in the case of PLA/My/L101, the extrusion force increase reflects some marked reactions inducing an increase of viscosity. As mentioned in the introduction section, My is highly reactive and can spontaneously polymerize by thermally activated radical polymerization [
17,
18]. It is also shown in the literature that My polymerization in the presence of a radical initiator yields poly(myrcene) (pMy) with different possible structures [
28]. Such a reaction may be dominant in the case of PLA/My/L101. The nature of the reactions occurring during reactive extrusion will be discussed in the next sections.
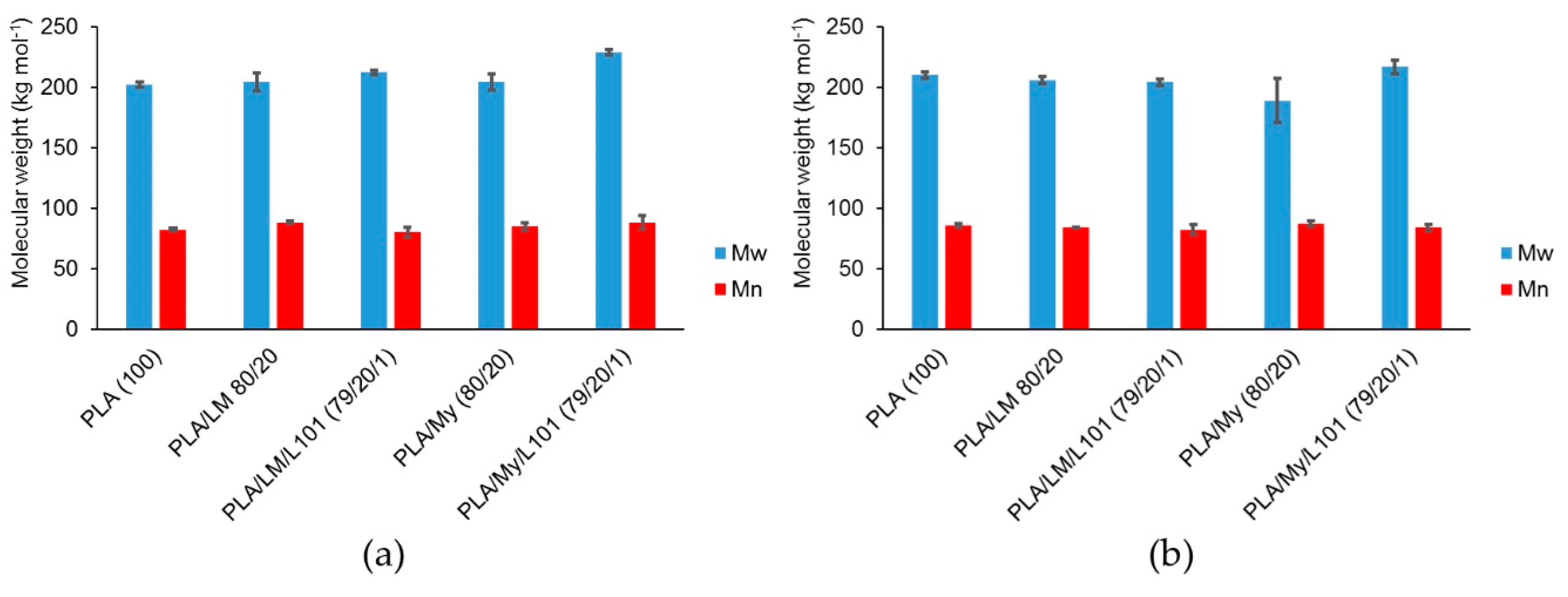
To further characterize the possible lack of degradation or the possible reactions occurring during the extrusion step, SEC testing was conducted to measure the molecular weight of PLA-based materials (
Figure 2a). Such measurements were also done after the injection step (
Figure 2b). In the case of PLA,
was found to be 202 kg·mol
−1 after extrusion (
Figure 2a) and 210 kg·mol
−1 after the injection step (
Figure 2b). Knowing that the initial molecular weight of PLA 4043D pellets was 199 kg·mol
−1 [
29], the inherent precision of SEC measurements being a few percent, and the standard deviation for each material being around 3%, no significant degradation occurred with the current processing conditions. Moreover, no influence of the addition of LM or My on PLA molecular weight was noted after the extrusion step, confirming that extrusion mainly involved homogenization of the blends without degradation (
Figure 1). In the case of the reactive extrusion in the presence of L101, a weak but significant increase of
was detected for PLA/LM/L101 (
= 212 kg·mol
−1) and PLA/My/L101 (
= 229 kg·mol
−1) compared to PLA/LM (
= 204 kg·mol
−1) and PLA/My (
= 204 kg·mol
−1), respectively.
According to previous works [
30], an increase of PLA molecular weight during reactive extrusion can be attributed to the free radical initiated branching of PLA. It can also be hypothesized that this potential increase of PLA molecular weight may be attributed to the grafting of the plasticizers onto PLA backbone. Nevertheless, based on the weak increase of
, such reactions may be quite limited.
A picture of the injected specimens was provided in
Figure 3. All the materials exhibited a homogeneous shape and aspect indicating good processing conditions and mixing. It can be seen that PLA/LM, and PLA/LM/L101 were quite transparent and slightly green, while PLA/My and PLA/My/L101 appeared opaque and slightly yellow.
The transparency of PLA/LM and PLA/LM/L101 can be explained by a low crystallinity level and no or very small inclusions of plasticizer that do not scatter light. The opposite case can also be supposed for PLA/My and PLA/My/L101. The microstructure of the plasticized PLAs will be studied in the next section to elucidate these aspects. It can however be concluded that PLA/My or PLA/My/L101 may be suitable for opaque packaging applications, while PLA/LM or PLA/LM/L101 may be suitable for transparent packaging applications.
3.2. Chemical Structure and Microstructure
To assess the chemical structure of PLA-based materials after reactive extrusion, it was suitable to combine direct methods such as Fourier transform infrared spectroscopy (FTIR) and 1H NMR, and indirect methods such as swelling testing and microstructural investigations to gather a maximum of information.
PLA and terpenes being soluble in chloroform, some swelling testing of the plasticized PLA was done with this solvent at room temperature to qualitatively characterize the presence of a gel, and hence, of a crosslinked network. In the case of PLA, PLA/LM, and PLA/LM/L101, the dissolution of the material was total in chloroform, while in the case of PLA/My and PLA/My/L101, dissolution in chloroform induced the formation of a turbid solution.
Therefore, a crosslinked molecular network in PLA/My and PLA/My/L101 is expected, resulting in a very low gel fraction, which can be due to crosslink formation in the PLA matrix or crosslink formation in the pMy inclusions [
28]. Concerning PLA/LM/L101, crosslinks in the PLA matrix or in the LM phase may not be sufficiently present to induce a gel formation in chloroform.
FTIR measurements in attenuated total reflectance (ATR) mode from 400 cm
−1 to 4000 cm
−1 were conducted to identify any potential change of chemical bonds of PLA matrix after reactive extrusion with LM or My (results not shown here). However, no significant change was noticed compared to neat PLA. Concerning
1H NMR investigation (curves provided in the
Supplementary Materials), in the case of the PLA-myrcene system, the spectra of the extruded samples with and without L101 showed no evidence of other signals than those arising from the starting materials or impurities contained therein (
Figure S1). One of these impurities was pMy [
31], and we can thus not conclude whether any polymerization of myrcene is taking place during the extrusion process. Comparison of the
1H NMR spectra of the PLA-limonene system with or without the accelerator L101 with those of the starting materials did not reveal any crosslinking between the two ingredients (
Figure S2).
Based on FTIR and 1H NMR results, direct methods enabling to identify the chemical structure of the PLA materials do not allow to extract information about the change in PLA and plasticizer structure during the extrusion. One explanation may be the total absence or very diluted structural changes, so that they cannot be detected.
The microstructure of PLA and plasticized PLA materials was observed by µCT testing (
Figure 4). In particular, the density variation of the materials was visualized in 3D. In the case of PLA/LM and PLA/LM/L101, no contrast was noted on the extracted 2D images proving that these plasticized PLA materials have a homogeneous microstructure at the µCT resolution (
Figure 4a,b). On the contrary, in the case of PLA/My (
Figure 4c), some black spots with a maximum size of 60 µm were observed. The number and size of these black spots significantly increased when adding L101 during the extrusion (
Figure 4d). In particular, the largest black spots reached a size of 200 µm. Obviously, these black spots had a lower density compared to the matrix and were considered as plasticizer inclusions.
As predicted by the Hansen Solubility Parameters (
Table 1), LM was more soluble in PLA compared to My, explaining the presence of micrometric inclusions in PLA plasticized with My that were not present in PLA plasticized with LM at the µCT resolution. Furthermore, the presence of L101 seems to promote the polymerization of myrcene.
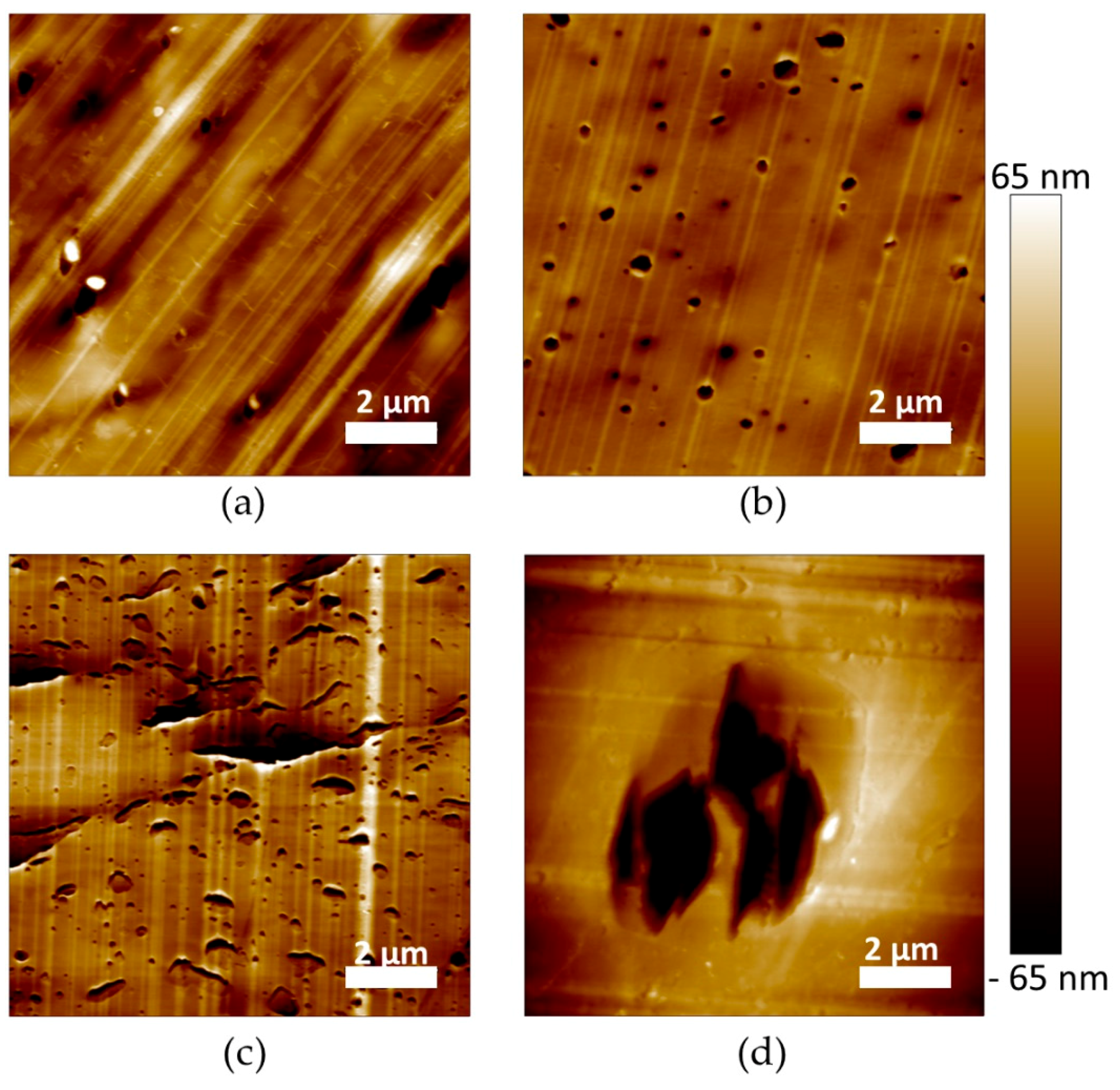
The microstructure of the materials was also characterized at a lower scale by AFM imaging (
Figure 5). The images were recorded with the topographical contrast mode. All the plasticized PLA materials exhibited inclusions with a heterogeneous size, considered as being the plasticizer phase (
Figure 5a–d). Very large inclusions were obtained for PLA/My/L101 confirming µCT observations (
Figure 4d and
Figure 5d). In some cases, the inclusions were properly cut by the cryo-ultramicrotome polishing procedure showing a homogeneous internal structure (
Figure 5c), while in the opposite case the plasticizer inclusions were totally removed by the preparation method conducting to holes (
Figure 5b). Intermediate cases were also noted for which the inclusion internal structure was damaged and exhibited some fibrils (
Figure 5d).
3.3. Thermal and Mechanical Behaviours
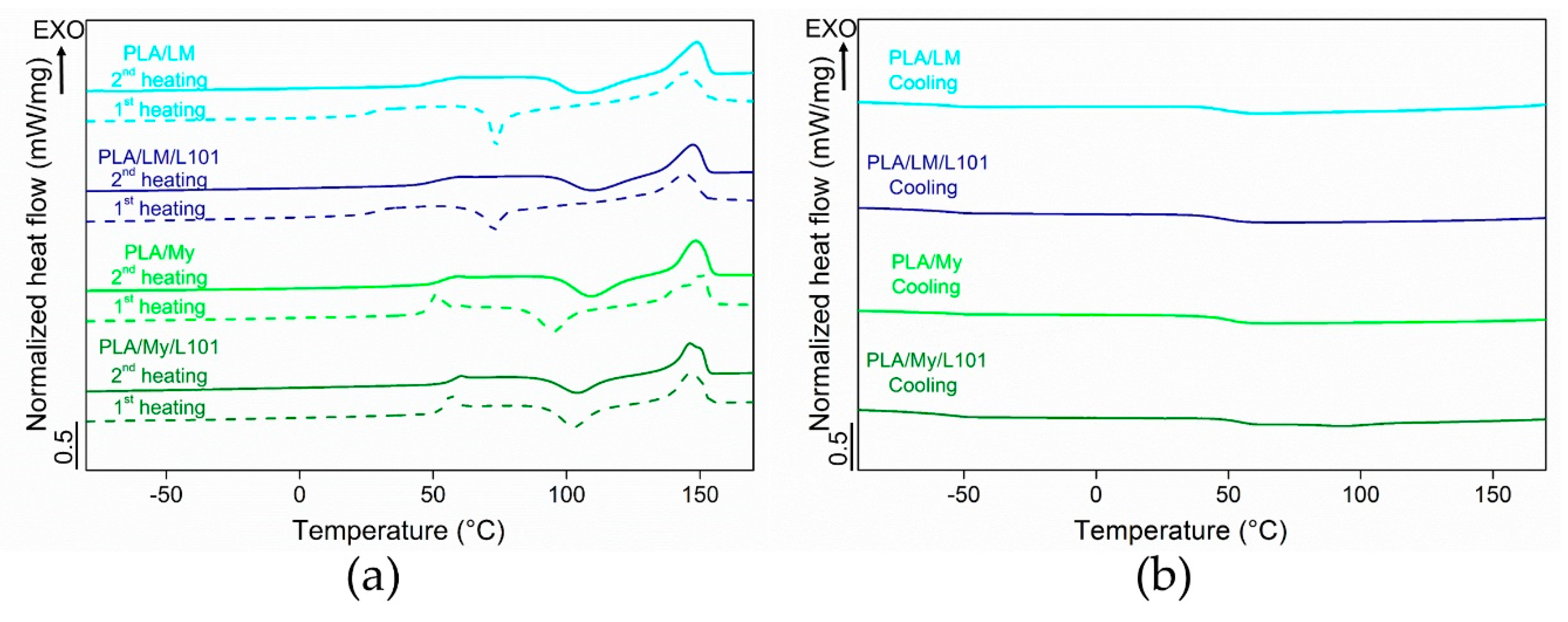
Some representative DSC thermograms of the materials were represented in
Figure 6, while the extracted data was listed in
Table 2. In particular, the thermal properties of PLA-based materials were measured during the first heating stage, the subsequent cooling stage, and the second heating stage. During the cooling stage of all the PLA-based materials, only the glass transition was noted (no crystallization peak). In the case of PLA, the measured
was 54.4 °C ± 0.4 °C. All the plasticized PLAs exhibited a lower
compared to neat PLA. In the case of PLA plasticized with LM, a
of 46.5 ± 1.3 °C was obtained for PLA/LM and a
of 48.1 ± 1.1 °C was obtained for PLA/LM/L101. In the case of PLA plasticized with My, a of 50.5 ± 0.5 °C was obtained for PLA/My and a
of 52.6 ± 0.5 °C was obtained for PLA/My/L101. Therefore, in the case of PLA/LM, the glass transition temperature is quite unaffected when adding L101, while in the case of PLA/My,
was few degrees higher in the case of the reactive blending in comparison with the non-reactive one.
Our results indicate that LM and My, with or without L101, enable to plasticize PLA according to the obtained glass transition temperature, lower for plasticized PLAs compared to neat PLA. The more marked decrease of
results from LM, as this plasticizer presents a better miscibility with PLA compared to My (
Table 1). Extrusion in the presence of L101 conducts to an increase of
for My that can be due to the enhanced My polymerization (
Figure 4c,d). Indeed, the dispersion of My within PLA, and hence the availability of My for increasing the PLA chains’ free volume is supposed to be reduced by the reactive extrusion. In the case of LM, the addition of L101 during the extrusion seems to have no effect on LM polymerization, confirming extrusion force monitoring (
Figure 1), and microstructure observations (
Figure 4a,b and
Figure 5a,b).
During the heating stages, differences between the two families of plasticized PLA were more marked. PLA/LM and PLA/LM/L101 exhibited a strong decrease of
to a value below 30 °C in the first heating stage. Interestingly, the
was in the cooling stage observed at over 40 °C and finally in the second heating stage it was observed at around 50 °C, just 10 °C lower than for PLA having a
of 60.9 ± 0.1 °C. The same behavior was noted concerning the cold-crystallization temperature
. Indeed, a low
was measured at around 70 °C in the first heating stage, while in the second heating stage, the
was higher than 100 °C for both PLA-limonene materials. The melting temperature increased just by a few degrees from the first to the second heating stage for PLA/LM and PLA/LM/L101. It is obvious that the cold-crystallization was characterized by a narrow peak in the first heating stage and by a broad peak in the second heating stage. Overall, the effect of the addition of the radical initiator L101 on the thermal properties of PLA/LM was very low, since all the thermal properties were quite similar for PLA/LM and PLA/LM/L101. This supports the previous interpretation of the extrusion force, where no difference between the non-reactive and the reactive blending was observed in the case of LM (
Figure 1), although the crystallinity increased from PLA/LM to PLA/LM/L101. This increase of crystallinity is probably due to a weak branching/crosslinking level of PLA. Indeed, a low concentration of compact grafting/crosslinking regions may act as crystallization precursor, enhancing PLA crystallization [
32,
33].
In the case of My-plasticized PLA materials, higher glass transition temperatures were observed in the first heating stage compared to the LM-plasticized PLA materials and just an increase by a few degrees was measured in the second heating stage. Furthermore, in the second heating stage, the glass transition temperature of PLA/My equal to 54.3 ± 0.4 °C and of PLA/My/L101 equal to 57.1 ± 0.5 °C were just a few degrees lower than in the case of PLA (
= 60.9 ± 0.1 °C). It is also to be noted that the glass transition occurring in the first heating stage was accompanied by a small endothermal peak, which disappeared in the second heating stage. This additional peak, also observed in the case of neat PLA, was attributed to the melting of a mesophase or to an ageing phenomenon [
34,
35]. For PLA/My blend, the cold-crystallization temperature increased by 13 °C from the first to the second heating stage. In the case of PLA/My/L101 material, just a 3 °C increase was noted between the first and the second heating stage. This stabilization of
was the only relevant finding when comparing the non-reactive blend with the reactive blends of PLA with myrcene. During the first heating stage, both PLA/My and PLA/My/L101 exhibited a double melting peak resulting from two families of crystals, which transformed into a single melting peak during the second heating stage. As a result, the heating step (melting) followed by the cooling step (crystallization) may have homogenized the crystals into one unique family. Furthermore, the crystallinity was slightly higher for the non-reactive blend PLA/My compared to the reactive blend PLA/My/L101 during the first heating stage. Although the weak branching of PLA is supposed to fasten PLA crystallization as for PLA/LM/L101, the presence of L101 is also supposed to accelerate My polymerization. The formation of pMy inclusions may decrease the availability of My to ensure a good chain mobility of PLA and may tend to hinder PLA crystallization at a given step of crystallization. So, pMy is limiting PLA crystallization, despite PLA crystallization is initially accelerated by the presence of the compact grafting/crosslinking regions [
32,
33]. However, during the second heating stage, the opposite result was obtained. It is suggested that My plasticizer in PLA/My has been further polymerized (higher conversion of the monomers) by the first heating stage of DSC measurements, conducting to less available plasticizer for PLA matrix, and hence, less plasticization and chain mobility to attain a high crystallinity. Such a phenomenon is less probable in PLA/My/L101 for which polymerization may reach its limit during the extrusion step.
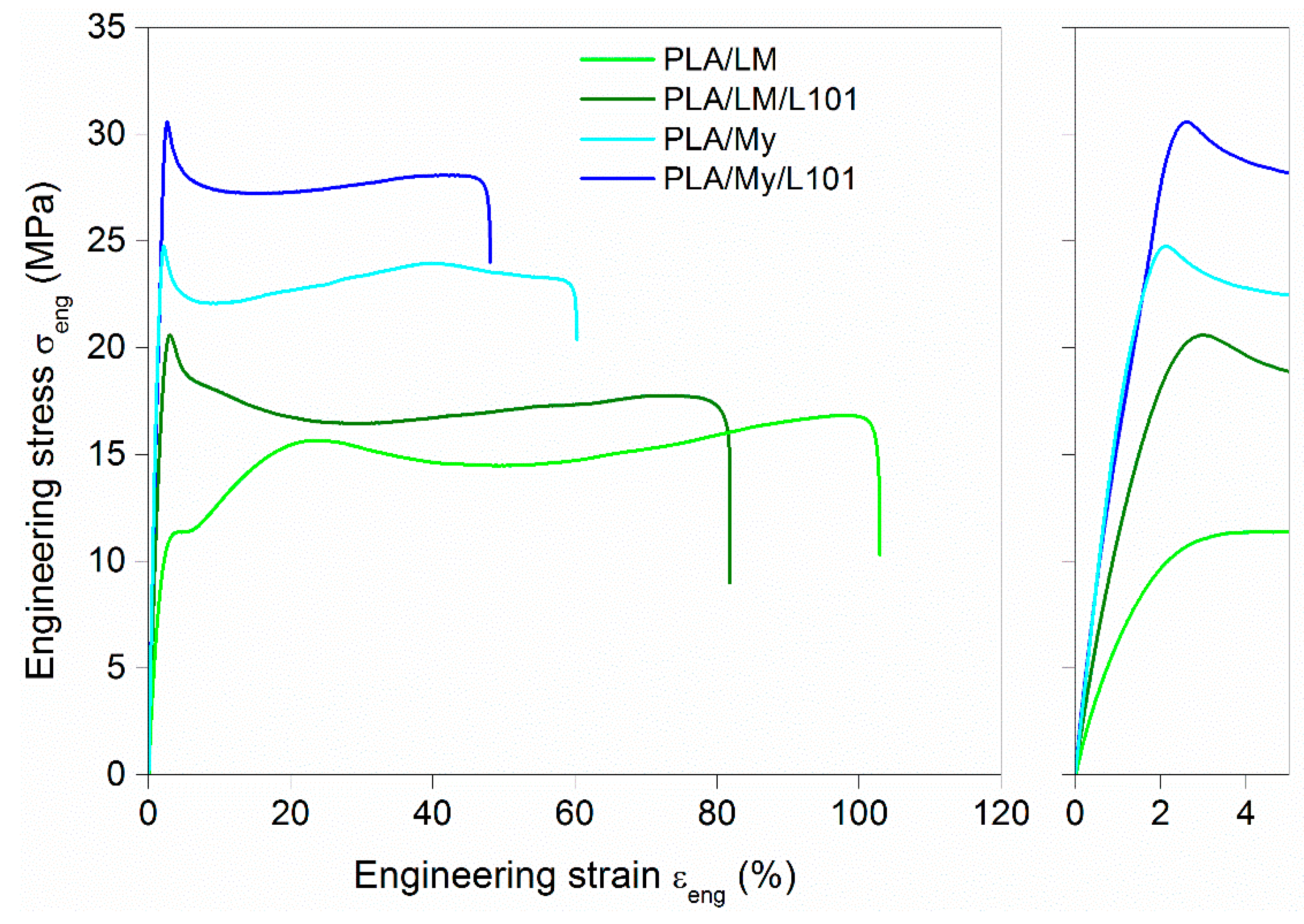
It is now important to investigate the effect of adding My and LM into PLA, with or without radical initiator, on the tensile and impact properties of the PLA-based materials. The engineering stress–engineering strain curves of the materials were plotted in
Figure 7, while the extracted parameters were listed in
Table 3. Compared to neat PLA, the addition of plasticizer, with or without L101, conducted to an important decrease of the elastic modulus E, decrease of the stress at yield
, increase of the ultimate strain
, and decrease of ultimate stress
. The intensity of these variations depended on the plasticization method (physical blending vs. reactive blending) and of the type of plasticizer (My vs. LM). The differences between LM and My were quite marked. Indeed, the ultimate strain of PLA/LM was almost the double of that of PLA/My. In the case of LM, the ultimate strain was similar for both plasticization methods (
≈ 120%), and in the case of My, the ultimate strain was about one quarter lower for the reactive blending (
= 45.0%) compared to the non-reactive blending (
= 62.7%). The tensile modulus for LM-plasticized materials (E = 1.0 GPa for PLA/LM and E = 1.2 GPa for PLA/LM/L101) was lower compared to PLA (E = 2.3 GPa), and E was slightly higher in the case of the use of L101 as shown previously with acryl-PEG [
8]. In the case of My-plasticized materials, E was comprised between that of PLA and that of LM-plasticized materials (E = 1.9 GPa and E = 1.7 GPa for PLA/My and PLA/My/L101, respectively). Contrary to what was observed for LM-plasticized PLA, the tensile modulus of the non-reactive PLA/My blend was slightly higher than that of reactive PLA/My/L101 blend. Concerning impact behavior, the addition of LM or My to PLA improved the impact strength
compared to neat PLA for which
was equal to 2.7 ± 0.1 kJ·m
−2. The highest impact strength,
= 12.5 ± 0.9 kJ·m
−2, was achieved in the case of PLA/My, while for the other cases the impact strength was comprised between 4.9 ± 0.1 kJ·m
−2 and 5.8 ± 0.6 kJ·m
−2.
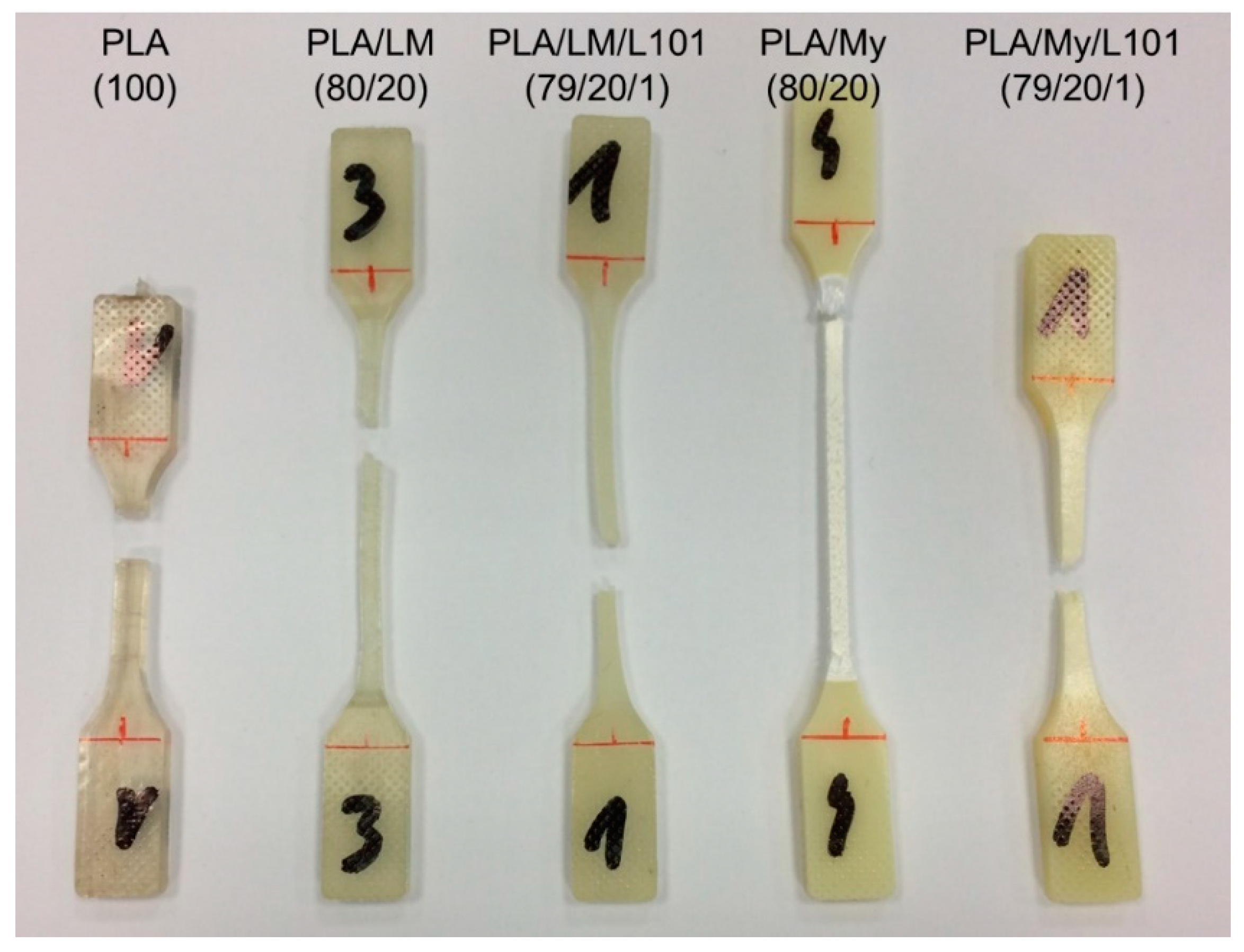
The observation of the tensile specimen after breakage can also provide information about the influence of the PLA plasticization methodology on the mechanical behavior, and in particular, on the deformation heterogeneities. A picture showing the broken tensile specimen of PLA, PLA/LM, PLA/LM/L101, PLA/My, and PLA/My/L101 was shown in
Figure 8. PLA tensile specimen clearly exhibited limited deformation and no particular change of aspect remaining transparent. Concerning the tensile specimen of PLA plasticized with LM, it exhibited important deformation, while the initial transparency and green color were retained. Finally, in the case of PLA plasticized with My, an important whitening of the broken tensile specimen was noted. These specimens were further investigated by µCT to identify more clearly deformation heterogeneities (
Figure 9). In the case of neat PLA (
Figure 9a), the presence of crazes was noted by µCT confirming previous study [
36]. In the case of PLA plasticized with LM, the presence of cracks generated from the surface were noted (
Figure 9b,c). In the case of PLA plasticized with My, other deformation heterogeneities were observed, namely surface delamination (
Figure 9d,e), plasticizer–matrix debonding forming cavities (
Figure 9d,e), and coalescence of these cavities (
Figure 9e).
Obviously, the plasticization of PLA with LM or My, with or without radical initiator, has increased PLA ductility and impact strength. Prior to modification, PLA exhibited a brittle tensile behavior characterized by crazing development to withstand the imposed macroscopic strain, since chain mobility was very low [
35]. The presence of LM or My seems to significantly increase PLA chain mobility since plastic deformation of PLA is active and crazing was no more observed. Nevertheless, some cracks developing from the tensile specimen surface are noted in PLA/LM (
Figure 9b). These deformation heterogeneities may be generated to withstand the imposed strain once the chain orientation mechanisms may not be active anymore. Concerning PLA/My, it exhibits inclusion-matrix debonding forming cavities, as deformation heterogeneities appearing to withstand large strain. It is possible that the propagation and coalescence of such cavities engender the formation of surface debonding as observed in
Figure 9d. The formation of such cavities may also explain the whitening phenomenon of this material as shown in
Figure 8. The higher ultimate strain of PLA/LM compared to PLA/My can be due to the larger size of My plasticizer inclusions compared to LM inclusions (
Figure 4a,c and
Figure 5a,c) and higher miscibility of PLA with LM compared to My (
Table 1). As a result, LM plasticizer-PLA matrix debonding may not be present in PLA/LM (no whitening phenomenon) or appears at a nanometric scale not captured by µCT and does not scattering light. Cavities forming at the micron scale in PLA/My may be generated at a moderate imposed strain and may propagate very fast conducting to an early failure compared to PLA/LM in which nanometric debonding may slowly propagate and/or surface cracks may not be generated yet. In the case of My, the extrusion in the presence of L101 decreases the ultimate strain compared to the extrusion without L101 as the plasticizer inclusion size increases, enhancing cavitation by debonding and reducing ductility (
Figure 9d,e). In the case of LM, the presence of L101 does not influence the elongation at break, confirming that no significant effect of LM structure is induced by L101 during the extrusion except some very local grafting/crosslinking. For the two plasticizers, reactive extrusion conducts to an increase of the stress at yield and the ultimate strength. In the case of LM, this is probably engendered by the increased crystallinity compared to the extrusion without L101 (
Table 2). In the case of My, crystallinity being almost the same between PLA/My and PLA/My/L101, the increase of strength occurring after reactive extrusion may be linked to the increase of
(
Table 2). Indeed, as stated in the DSC section, the formation of pMy makes less My available for the PLA plasticization.
The increase of PLA chain mobility resulting from the addition of plasticizer is reflected by an increase of impact strength in comparison with neat PLA. Nevertheless, a remarkable result has been obtained in the case of PLA/My that exhibits the highest impact strength among plasticized PLA materials. It is thought that this finding is due to the plasticizer inclusion–PLA matrix debonding acting as an additional dissipation mechanisms to chain rearrangement during the impact. This is true as soon as the cavitation produced at the inclusion-matrix interface does not propagate too fast to neighboring cavities. Such a mechanism implies that inclusions are small and well dispersed. This is not the case for PLA/My/L101 where inclusions are quite big (
Figure 4d and
Figure 5d), enhancing cavitation propagation and surface delamination conducting to an earlier material failure compared to PLA/My. It is to be noted that the formation of surface delamination in PLA/My can be responsible for the slow decrease of tensile stress from 40% of tensile strain (
Figure 7).
3.4. Hydrothermal Ageing
Polyester hydrolysis was followed by the number of chain scissions
in mol·g
−1 defined as follows [
37]:
where
represented the number average molecular weight of PLA as a function of ageing time, and
represented the initial number average molecular weight of PLA.
was calculated by SEC in THF at 40 °C based on PS calibration and using the Kuhn–Mark–Houwink equation [
38]:
In addition, since hydrolysis of PLA was expected to destroy ester functions, the concentration of the remaining ester functions
in mol·g
−1 as a function of time could be calculated according to:
with the initial concentration of esters
being proportional to the molar mass of PLA repetitive unit (
) of 72 g·mol
−1:
Assuming that ester hydrolysis kinetic had a first order for each reactant (ester and water) and degenerated in one of the reactants (the water being constant in concentration), one could write:
with k being the kinetic constant of the degenerated order in h
−1 (considering hour as unit of time) such as:
In Equation (14), K corresponded to the hydrolysis kinetic constant of the second order in L·mol
−1·h
−1.
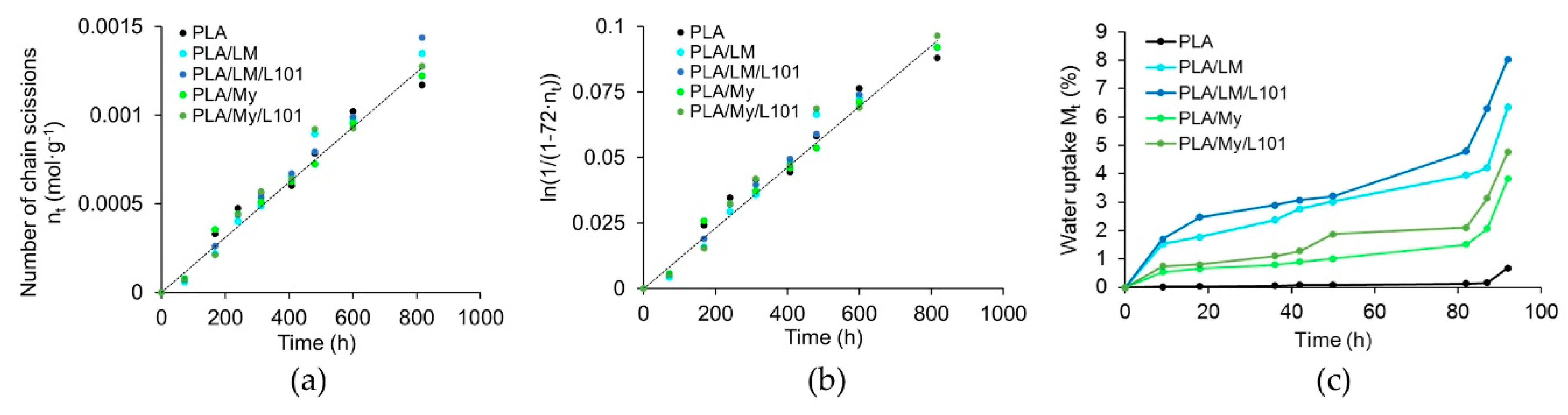
The evolution of the number of chain scission and water uptake of PLA, PLA/LM, PLA/LM/L101, PLA/My, and PLA/My/L101 were represented in
Figure 10. All the materials exhibited similar chain scission number evolutions (
Figure 10a). It was possible to calculate the k constant of PLA at 70 °C that was equal to 1.18·10
−4 h
−1 (coefficient of determination of 0.98), which was in good agreement with the literature (a k value of 1.20·10
−4 h
−1 was found in Reference [
39] for the same hydrothermal ageing temperature). Besides this, K was estimated to be in the range of 1.53·10
−4 L·mol
−1·h
−1 (
Figure 10b). At the early stage of the ageing procedure, the water uptake
of the different blends deferred. Indeed,
increased in the following material order: PLA < PLA/My ≈ PLA/My/L101 < PLA/LM ≈ PLA/LM/L101 (
Figure 10c). After 3 days of ageing, DSC measurements (based on the first heating stage) revealed that the glass transition temperature of PLA decreased by 10%, while that of all the plasticized PLAs increased (+27% for PLA/LM, + 22% for PLA/LM/L101, +8% for PLA/My, and + 6% for PLA/LM/L101). At the same time, the crystallinity of all the materials reached values comprised between 51 wt% and 61 wt%, excepted for PLA/My/L101 that exhibited a crystallinity of 36 wt%. Note that an important increase in water uptake was observed after 80 h of ageing for all the materials. Last, during the ageing, the water of all the plasticized PLA blends was slightly colored, while that of PLA remained colorless.
Obviously, PLA degradation kinetic is not influenced during the hydrothermal ageing by the plasticization as shown by the chain scission number evolutions. This finding suggests that the initial PLA structure is not drastically modified by the presence of L101. Indeed, if PLA would have been significantly branched by the free radical initiator, its stability against ageing would have increased [
30]. In the case of PLA, the decrease of the glass transition temperature and the increase of crystallinity after 3 days of ageing are linked to the chain scission mechanisms providing a higher chain mobility. At the same time, the plasticization markedly influences the water uptake of PLA materials. LM brings more water uptake than My in plasticized PLAs, which can be directly connected to the lower
of PLA/LM blends compared to PLA/My blends (
Table 2). In addition, the increase of
and the slight coloration of the water in the case of plasticized PLA materials indicate the release of the two plasticizers during the hydrothermal ageing, explaining also the water uptake. The higher increase of
in the case of PLA/LM blends compared to PLA/My blends demonstrate that LM is probably more released than My during the ageing. This can be explained by the polymerization of My during the reactive extrusion providing a higher stability compared to LM. Note that the increase of
observed after the hydrothermal ageing of plasticized PLA materials can be related to the increase of
when comparing the first and the second DSC heating stage of the same materials (
Table 2), both cases being due to the release of the plasticizer. The use of L101 has no or a limited effect on the material water uptake during the hydrothermal ageing. Finally, the authors cannot account for the increase of hydrolysis observed after 80 h of ageing, though they believe that this trend can be connected to the increase of pH in the non-buffered media.

 ,
, 











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}