Ionic Porous Organic Polymers Based on Functionalized Tetraarylborates

 , and
, and
Abstract

1. Introduction
2. Materials and Methods
2.1. General Comments
2.2. Synthesis
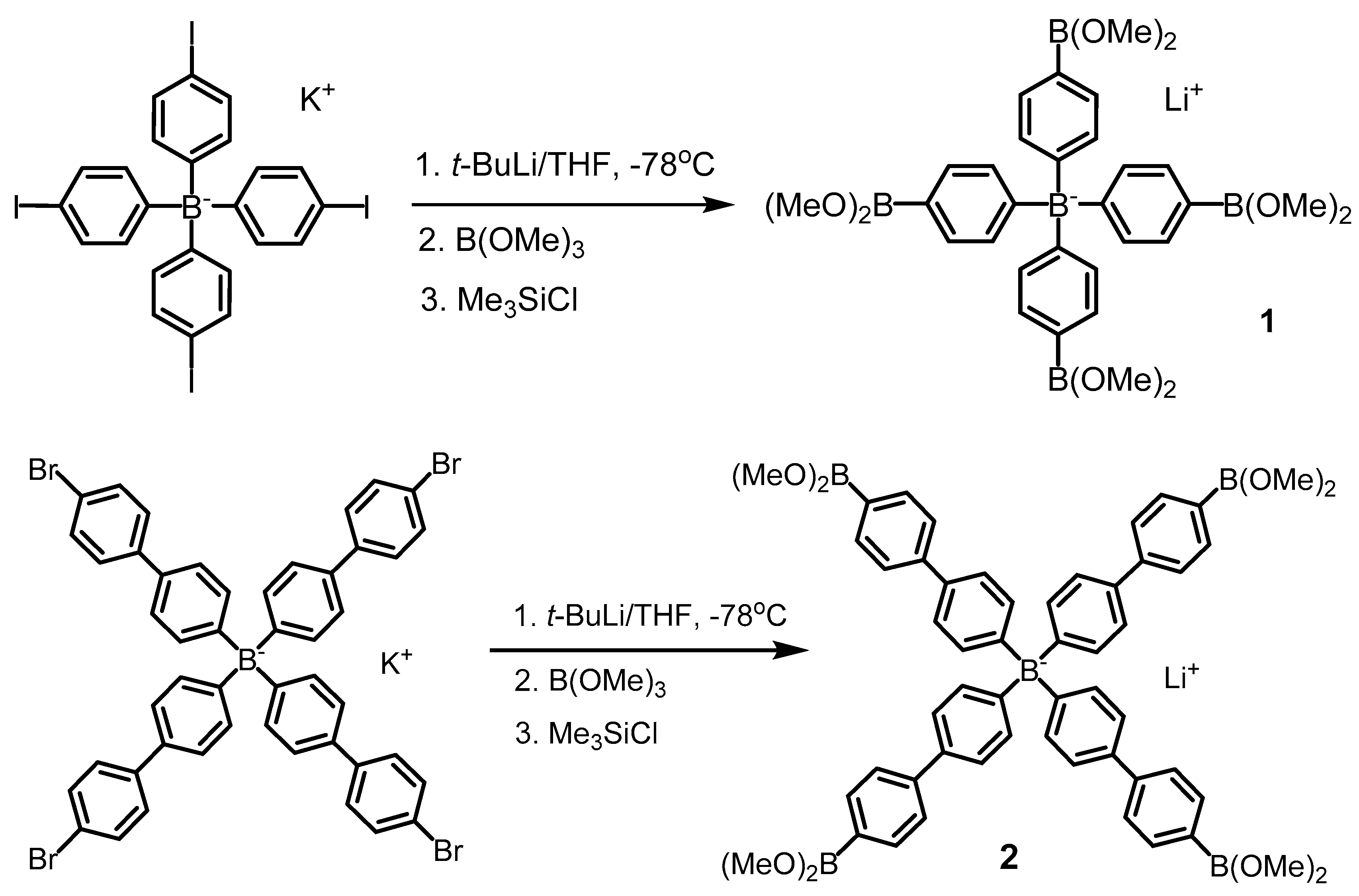
2.2.1. Lithium tetrakis[4-(dimethoxyboryl)phenyl]borate THF solvate (1)
2.2.2. Lithium tetrakis[4′-(dimethoxyboryl)-4-biphenylyl]borate THF solvate (2)
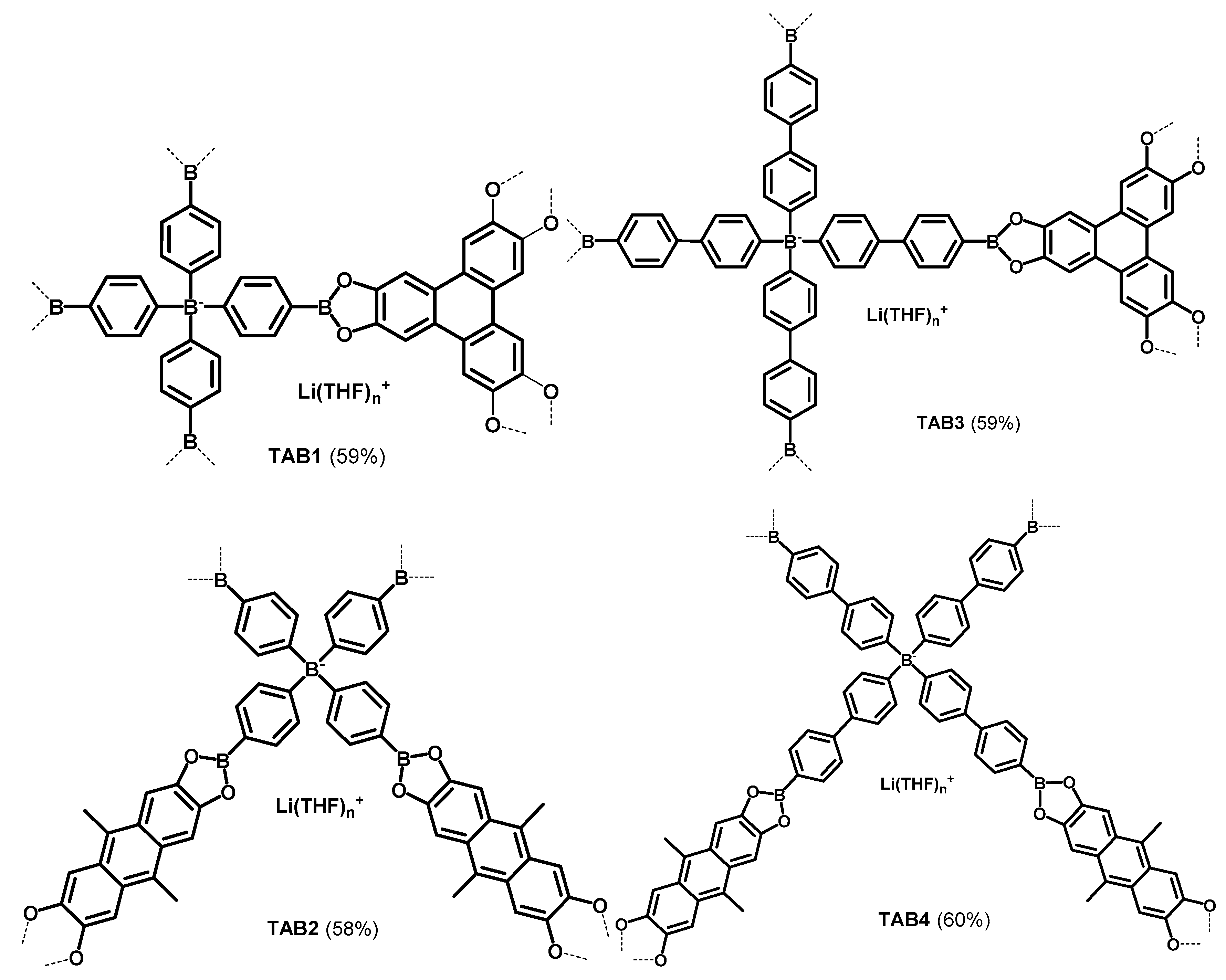
2.2.3. Porous materials TAB1–4
2.3. Physicochemical Characterization
2.3.1. Thermogravimetric analysis
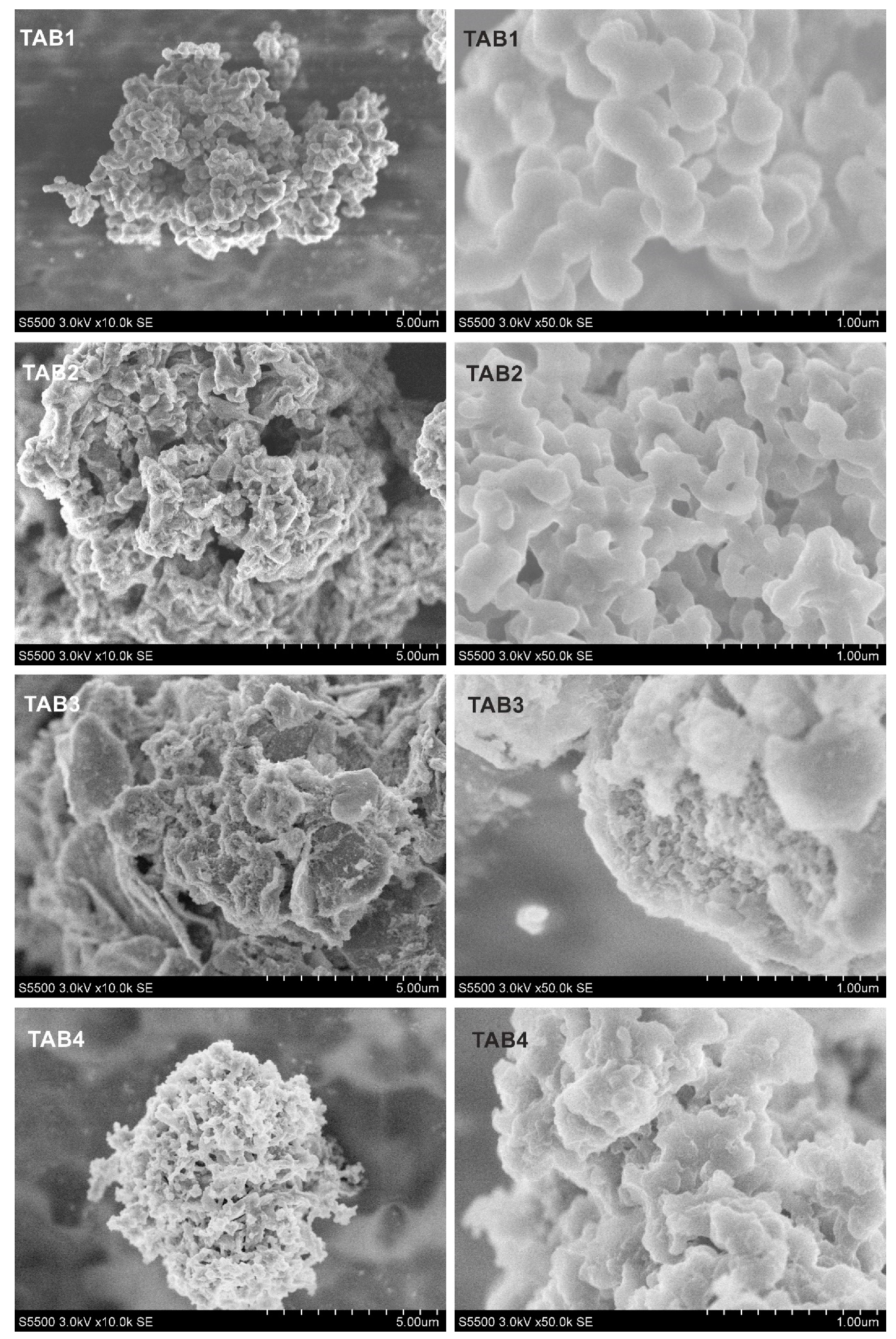
2.3.2. Scanning electron microscopy studies
2.3.3. Powder X-ray diffraction measurements
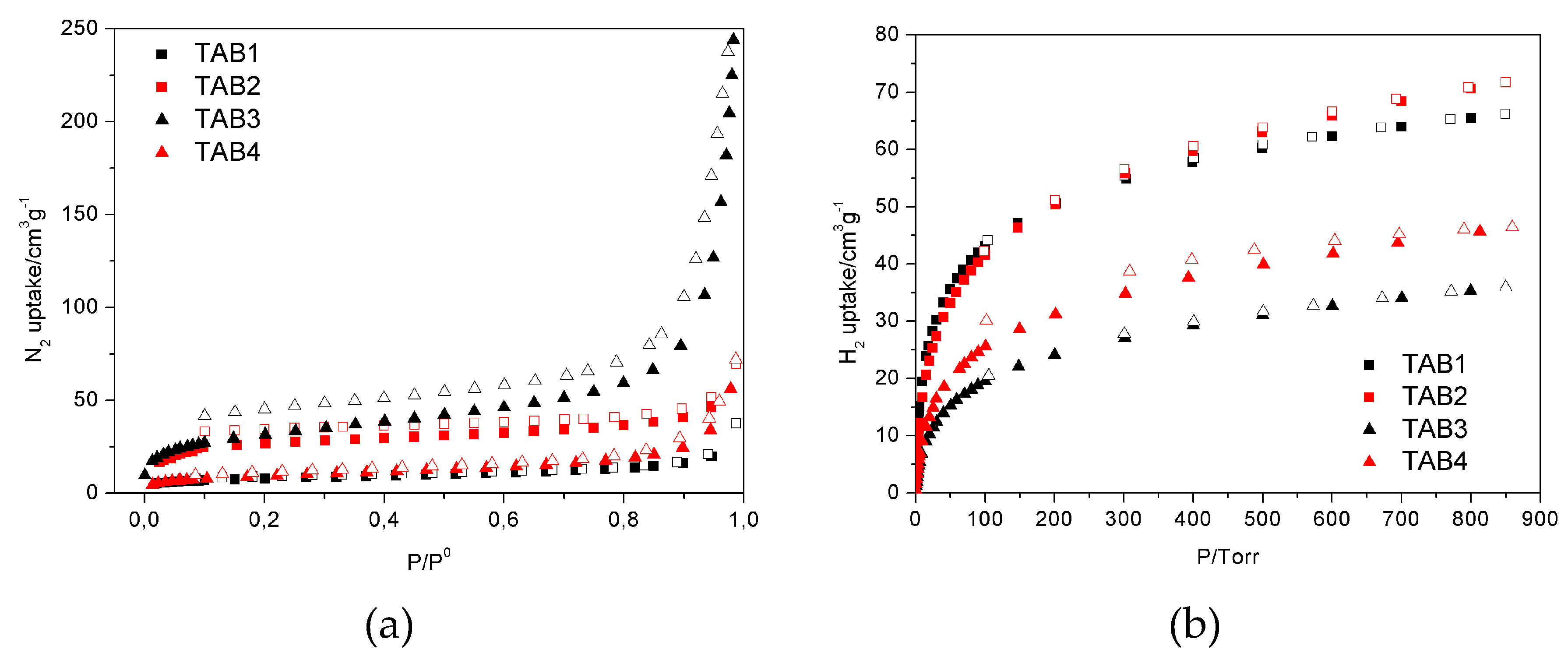
2.3.4. Sorption measurements
3. Results
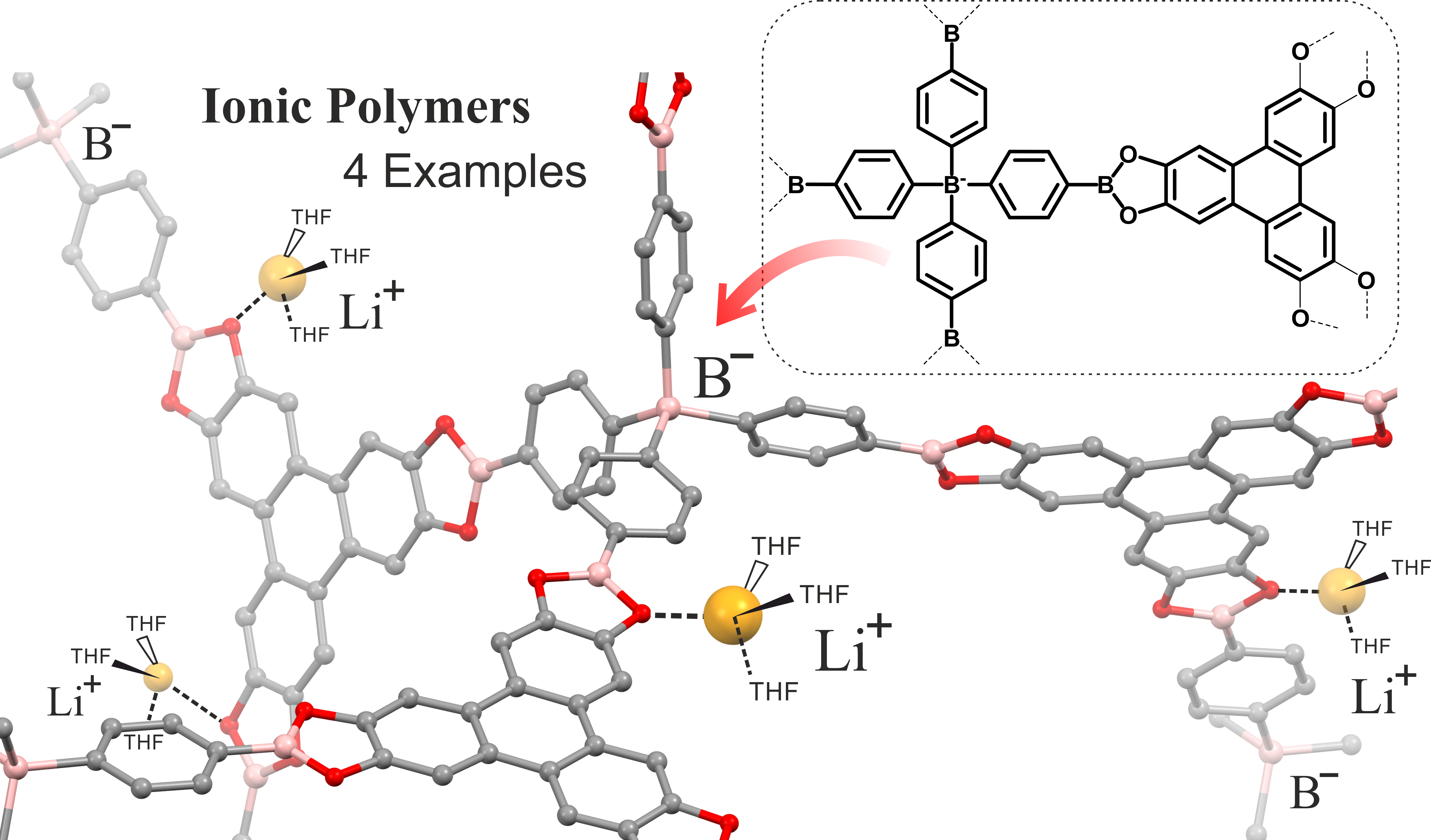
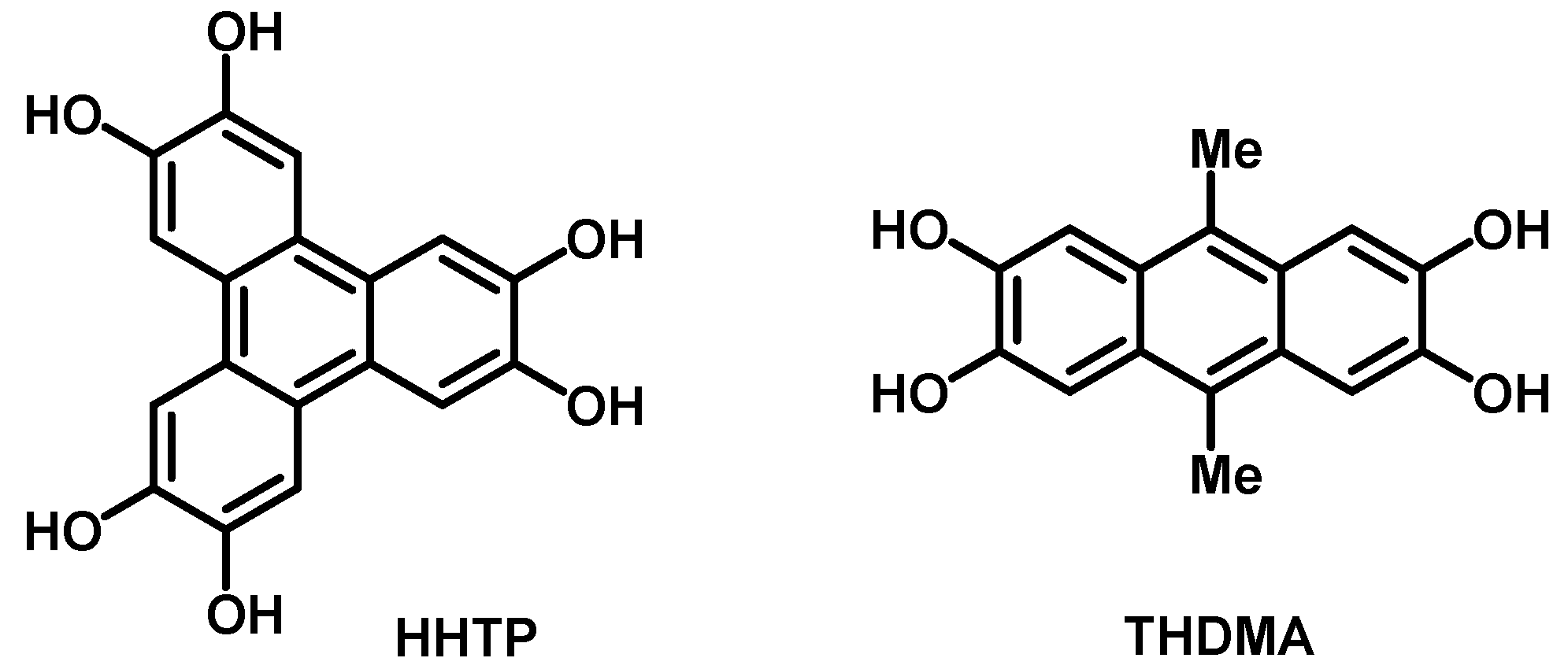
3.1. Synthesis
3.2. Morphology and Structural Studies
3.3. Sorption Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sun, J.-K.; Antonietti, M.; Yuan, J. Nanoporous ionic organic networks: from synthesis to materials applications. Chem. Soc. Rev. 2016, 45, 6627–6656. [Google Scholar] [CrossRef] [PubMed]
- El-Kaderi, H.M.; Hunt, J.R.; Mendoza-Cortés, J.L.; Côté, A.P.; Taylor, R.E.; O’Keeffe, M.; Yaghi, O.M. Designed synthesis of 3D Covalent Organic Frameworks. Science 2007, 316, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Malek, N.; Maris, T.; Simard, M.; Wuest, J.D. Molecular tectonics. Selective exchange of cations in porous anionic hydrogen-bonded networks built from derivatives of tetraphenylborate. J. Am. Chem. Soc. 2005, 127, 5910–5916. [Google Scholar] [CrossRef] [PubMed]
- Qiu, F.; Zhao, W.; Han, S.; Zhuang, X.; Lin, H.; Zhang, F. Recent advances in boron-containing conjugated porous polymers. Polymers 2016, 8, 191. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Yang, H.; Whiteley, J.M.; Wan, S.; Jin, Y.; Lee, S.-H.; Zhang, W. Ionic Covalent Organic Frameworks with spiroborate linkage. Angew. Chem. Int. Ed. 2016, 55, 1737–1741. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Yuan, Y.; Tian, Y.; Zhang, D.; Zhu, G. Highly efficient enrichment of volatile iodine by charged Porous Aromatic Frameworks with three sorption sites. Angew. Chem. Int. Ed. 2015, 54, 12733–12737. [Google Scholar] [CrossRef] [PubMed]
- Türp, D.; Wagner, M.; Enkelmann, V.; Müllen, K. Synthesis of nanometer-sized, rigid, and hydrophobic anions. Angew. Chem. Int. Ed. 2011, 50, 4962–4965. [Google Scholar] [CrossRef]
- Cui, C.; Bonder, E.M.; Jäkle, F. Weakly coordinating amphiphilic organoborate block copolymers. J. Am. Chem. Soc. 2010, 132, 1810–1812. [Google Scholar] [CrossRef]
- Fischer, S.; Schmidt, J.; Strauch, P.; Thomas, A. An anionic microporous polymer network prepared by the polymerization of weakly coordinating anions. Angew. Chem. Int. Ed. 2013, 52, 12174–12178. [Google Scholar] [CrossRef]
- Van Humbeck, J.F.; Aubrey, M.L.; Alsbaiee, A.; Ameloot, R.; Coates, G.W.; Dichtel, W.R.; Long, J.R. Tetraarylborate polymer networks as single-ion conducting solid electrolytes. Chem. Sci. 2015, 6, 5499–5505. [Google Scholar] [CrossRef]
- Zhang, P.; Jiang, X.; Wan, S.; Dai, S. Charged porous polymers using a solid C-O cross-coupling reaction. Chem. Eur. J. 2015, 21, 12866–12870. [Google Scholar] [CrossRef] [PubMed]
- Thapa, S.; Hettiarachchi, E.; Dickie, D.A.; Rubasinghege, G.; Qin, Y. A charge-separated diamondoid Metal–Organic Framework. Chem. Commun. 2018, 54, 12654–12657. [Google Scholar] [CrossRef] [PubMed]
- Tomaszewski, P.; Wiszniewski, M.; Serwatowski, J.; Woźniak, K.; Durka, K.; Luliński, S. Synthesis of tetraarylborates via tetralithio intermediates and the effect of polar functional groups and cations on their crystal structures. Dalton Trans. 2018, 47, 16627–16637. [Google Scholar] [CrossRef] [PubMed]
- Gontarczyk, K.; Bury, W.; Serwatowski, J.; Wieciński, P.; Woźniak, K.; Durka, K.; Luliński, S. Hybrid triazine-boron two-dimensional Covalent Organic Frameworks: synthesis, characterization, and DFT approach to layer interaction energies. ACS Appl. Mater. Interfaces 2017, 9, 31129–31141. [Google Scholar] [CrossRef] [PubMed]
- Rouquerol, J.; Llewellyn, P.; Rouquerol, F. Is the BET equation applicable to microporous adsorbents? Stud. Surf. Sci. Catal. 1986, 160, 49–56. [Google Scholar]
- Côté, A.P.; Benin, A.I.; Ockwig, N.W.; O’Keeffe, M.; Matzger, A.J.; Yaghi, O.M. Porous, crystalline, Covalent Organic Frameworks. Science 2005, 310, 1166–1170. [Google Scholar] [CrossRef] [PubMed]
- Luliński, S.; Smętek, J.; Durka, K.; Serwatowski, J. Tandem synthesis of 5,10-dihydroboranthrenes via elusive ortho-lithiated phenyl boronates. Eur. J. Org. Chem. 2013, 8315–8322. [Google Scholar] [CrossRef]
- Penner, G.H.; Hutzal, J.A. Lithium-6 CP/MAS standard. Magn. Res. Chem. 1997, 35, 222–226. [Google Scholar] [CrossRef]
- Martínez Casado, F.J.; Ramos Riesco, M.; Redondo, M.I.; Choquesillo-Lazarte, D.; López-Andrés, S.; Rodríguez Cheda, J.A. Anhydrous lithium acetate polymorphs and its hydrates: three-dimensional coordination polymers. Cryst. Growth Des. 2011, 11, 1021–1032. [Google Scholar] [CrossRef]
- Smith, M.K.; Northrop, B.H. Vibrational properties of boroxine anhydride and boronate ester materials: model systems for the diagnostic characterization of Covalent Organic Frameworks. Chem. Mater. 2014, 26, 3781–3795. [Google Scholar] [CrossRef]
- Rambo, B.M.; Lavigne, J.J. Defining self-assembling linear oligo(dioxaborole)s. Chem. Mater. 2007, 19, 3732–3739. [Google Scholar] [CrossRef]
- Ismail, A.F.; Khulbe, K.; Matsuura, T. Gas Separation Membranes, Polymeric and Inorganic; Springer: Cham, Switzerland, 2015. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sorption parameter | TAB1 | TAB2 | TAB3 | TAB4 |
|---|---|---|---|---|
| SBET/m2 g−1 | 29.0 | 114.9 | 114.7 | 33.7 |
| Vp/cm3 g−1 | 0.058 | 0.107 | 0.402 | 0.111 |
| N2@77 K/cm3 g−1 STP 1 | 37.4 | 69.4 | 259.7 | 72.0 |
| H2@77 K/cm3 g−1 STP 2 | 66.1 | 71.7 | 35.9 | 46.4 |
| CO2@273 K | 33.7 | 32.1 | 14.8 | 19.7 |
| CH4@273 K | 9.6 | 9.1 | 4.0 | 5.7 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tomaszewski, P.; Wiszniewski, M.; Gontarczyk, K.; Wieciński, P.; Durka, K.; Luliński, S. Ionic Porous Organic Polymers Based on Functionalized Tetraarylborates. Polymers 2019, 11, 1070. https://doi.org/10.3390/polym11061070
Tomaszewski P, Wiszniewski M, Gontarczyk K, Wieciński P, Durka K, Luliński S. Ionic Porous Organic Polymers Based on Functionalized Tetraarylborates. Polymers. 2019; 11(6):1070. https://doi.org/10.3390/polym11061070
Chicago/Turabian StyleTomaszewski, Patryk, Marcin Wiszniewski, Krzysztof Gontarczyk, Piotr Wieciński, Krzysztof Durka, and Sergiusz Luliński. 2019. "Ionic Porous Organic Polymers Based on Functionalized Tetraarylborates" Polymers 11, no. 6: 1070. https://doi.org/10.3390/polym11061070
APA StyleTomaszewski, P., Wiszniewski, M., Gontarczyk, K., Wieciński, P., Durka, K., & Luliński, S. (2019). Ionic Porous Organic Polymers Based on Functionalized Tetraarylborates. Polymers, 11(6), 1070. https://doi.org/10.3390/polym11061070