Twisting of Fibers Balancing the Gel–Sol Transition in Cellulose Aqueous Suspensions

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
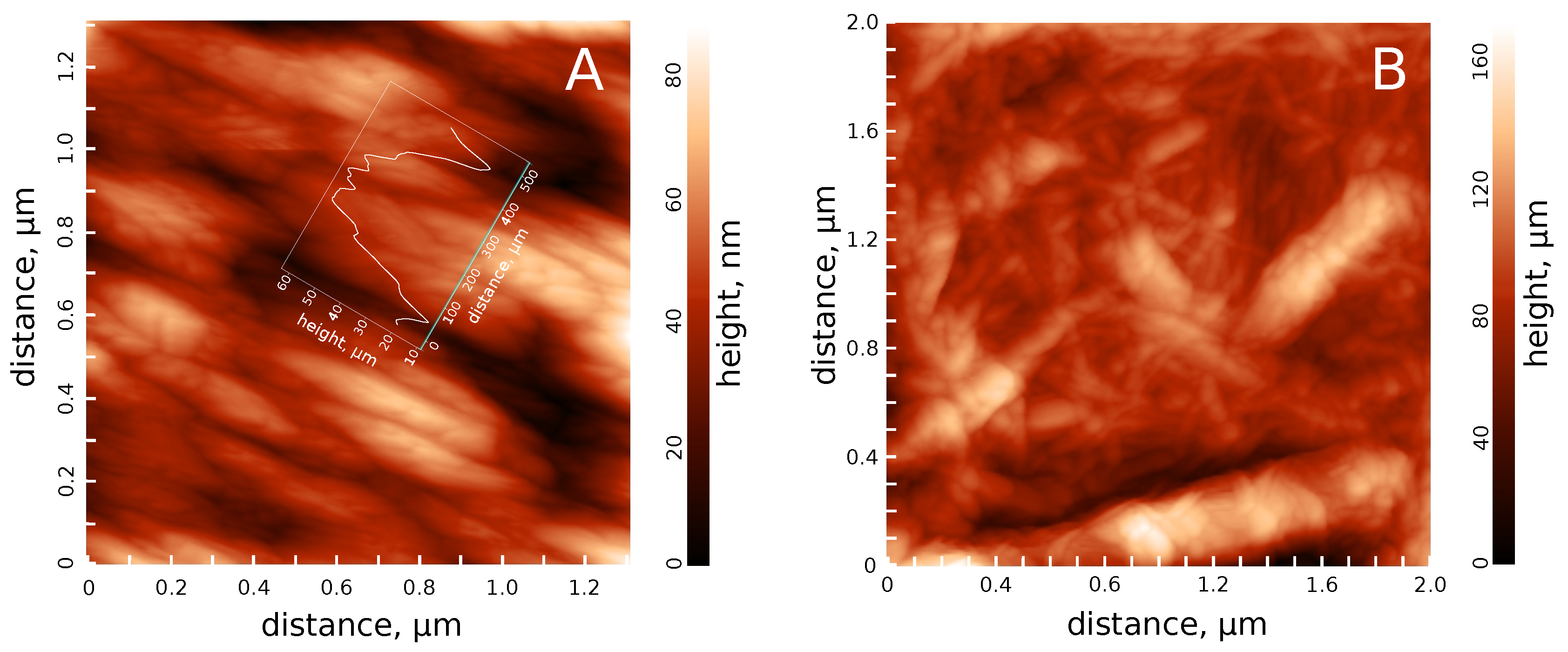
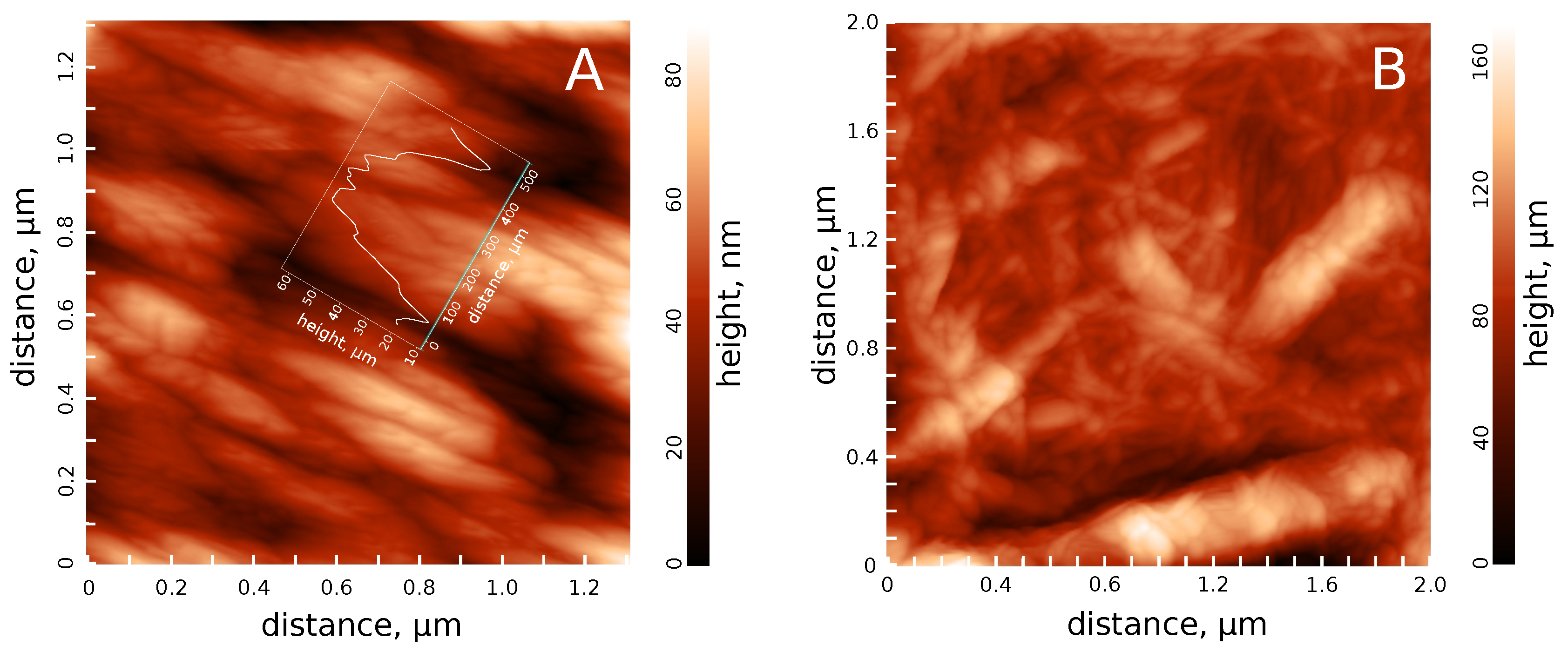
3.1. Hydrosol Sedimentation Stability and Structure
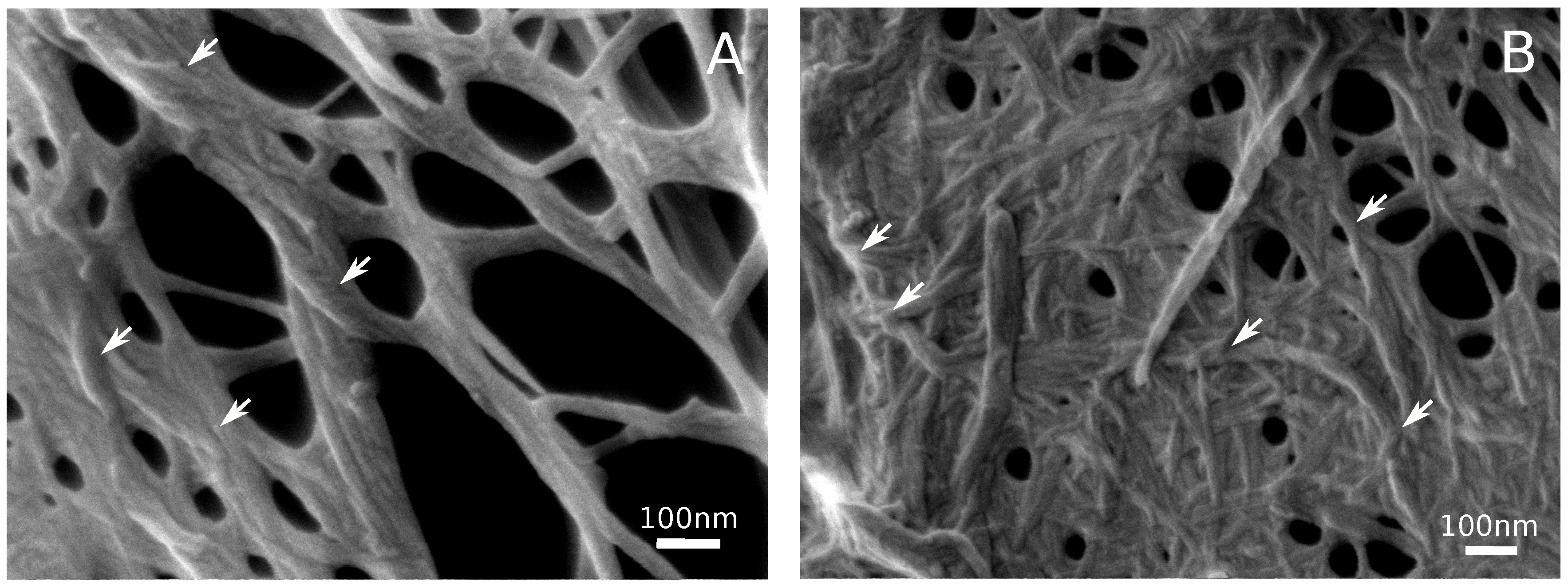
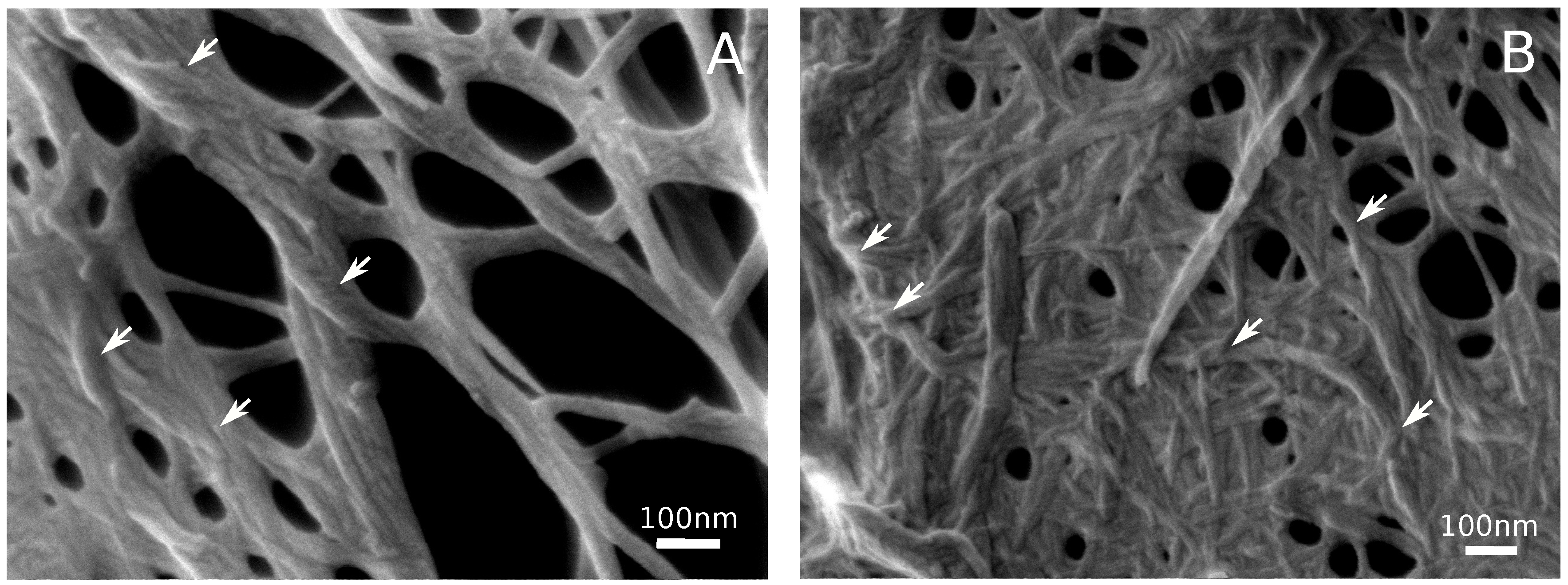
3.2. Hydrogel Structure
3.3. Cellulose Films
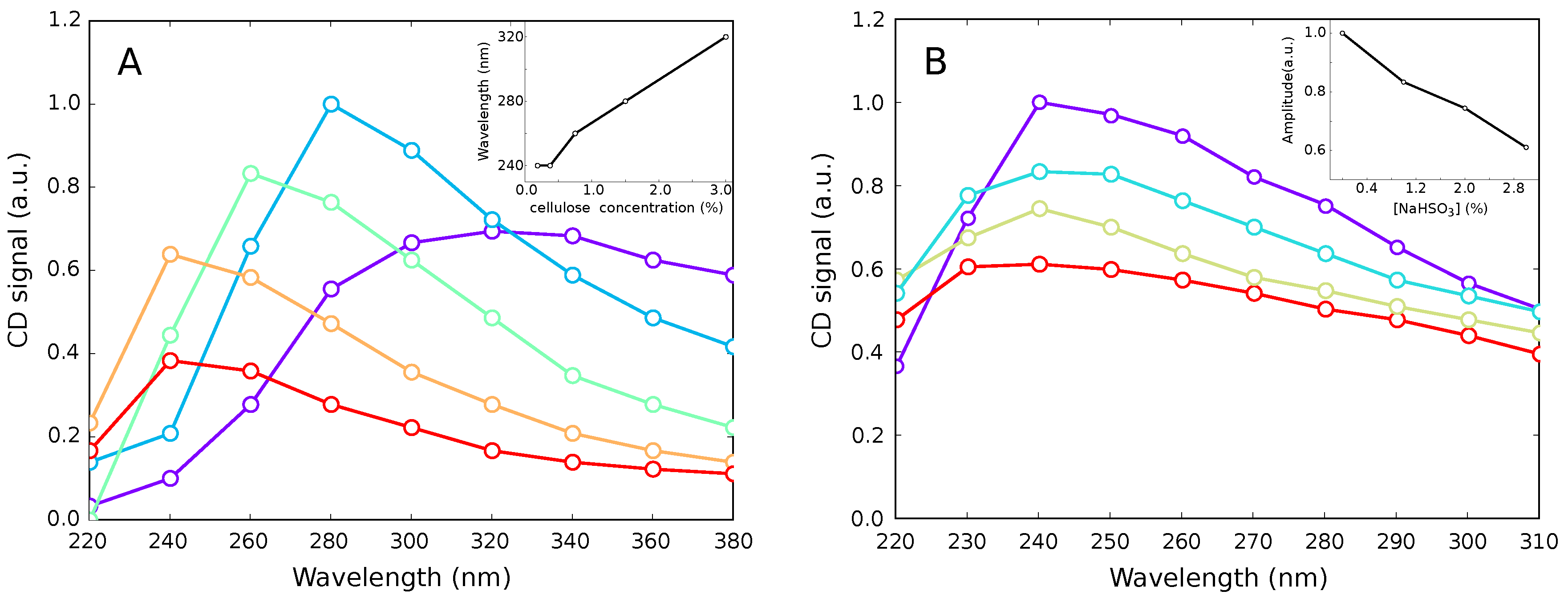
3.4. Optical Activity of Cellulose Hydrosol
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AFM | Atomic Force Microscopy |
| CD | Circular Dichroism |
| SEM | Scanning Electron Microscopy |
References
- Raabe, D.; Sachs, C.; Romano, P. The crustacean Exoskeleton as an Example of a Structurally and Mechanically Graded Biological Nanocomposite Material. Acta Mater. 2005, 53, 4281–4292. [Google Scholar] [CrossRef]
- Korolovych, V.F.; Cherpak, V.; Nepal, D.; Ng, A.; Shaikh, N.R.; Grant, A.; Xiong, R.; Bunning, T.J.; Tsukruk, V.V. Cellulose Nanocrystals with Different Morphologies and Chiral Properties. Polymer 2018, 145, 334–347. [Google Scholar] [CrossRef]
- Nikolsky, S.N.; Zlenko, D.V.; Melnikov, V.P.; Stovbun, S.V. The Fibrils Untwisting Limits the Rate of Cellulose Nitration Process. Cabrb. Polym. 2019, 204, 232–237. [Google Scholar] [CrossRef]
- Jarvis, M. Cellulose Stacks Up. Nature 2003, 426, 611–612. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, J. Excellent Chemical and Material Cellulose from Tunicates: Diversity in Cellulose Production Yield and Chemical and Morphological Structures from Different Tunicate Species. Cellulose 2014, 21, 3427–3441. [Google Scholar] [CrossRef]
- Muhlethaler, K. Ultrastructure and Formation of Plant Cell Walls. Annu. Rev. Plant. Phys. 1967, 18, 1–24. [Google Scholar] [CrossRef]
- Fernandes, A.; Thomas, L.; Altaner, C.; Callow, P.; Forsyth, V.; Apperley, D.; Kennedy, C.; Jarvish, M. Nanostructure of Cellulose Microfibrils in Spruce Wood. Proc. Natl. Acad. Sci. USA 2011, 108, E1195–E1203. [Google Scholar] [CrossRef] [PubMed]
- Usov, I.; Nyström, G.; Adamcik, J.; Handschin, S.; Schültz, C.; Fall, A.; Bergström, L.; Mezzenga, R. Understanding nanocellulose chirality and structure–properties relationship at the single fibril level. Nat. Commun. 2015, 6, 7564. [Google Scholar] [CrossRef]
- Hult, E.; Larsson, P.; Iversen, T. Cellulose Fibril Aggregation—An Inherent Property of Kraft Pulps. Polymer 2001, 42, 3309–3314. [Google Scholar] [CrossRef]
- Newman, R. Estimation of the Lateral Dimensions of Cellulose Crystallites Using 13C NMR Signal Strengths. Solid State Nucl. Magn. Reson. 1999, 15, 21–29. [Google Scholar] [CrossRef]
- Ruben, G.; Bokelman, G. Triple-Stranded, Left-Hand-Twisted Cellulose Microflbril. Carbohydr. Res. 1987, 160, 434–443. [Google Scholar] [CrossRef]
- Hanley, S.J.; Giasson, J.; Revol, J.F.; Gray, D.G. Atomic Force Microscopy of Cellulose Microfibrils: Comparison with Transmission Electron Microscopy. Polymer 1992, 33, 4639–4642. [Google Scholar] [CrossRef]
- Hanley, S.J.; Revol, J.F.; Godbout, L.; Gray, D.G. Atomic Force Microscopy and Transmission Electron Microscopy of Cellulose from Micrasterias Denticulata; Evid. A Chiral Helical Microfibril Twist. Cellulose 1997, 4, 209–220. [Google Scholar] [CrossRef]
- Tseng, P.; Napier, B.; Zhao, S.; Mitropoulos, A.N.; Applegate, M.B.; Marelli, B.; Kaplan, D.L.; Omenetto, F.G. Directed Assembly of Bio-Inspired Hierarchical Materials with Controlled Nanofibrillar Architectures. Nat. Nanotechnol. 2017, 12, 474–480. [Google Scholar] [CrossRef]
- Pääkkö, M.; Ankerfors, M.; Kosonen, H.; Nykänen, A.; Ahola, S.; Österberg, M.; Ruokolainen, J.; Laine, J.; Larsson, P.; Ikkala, O.; Lindström, T. Enzymatic Hydrolysis Combined with Mechanical Shearing and High-Pressure Homogenization for Nanoscale Cellulose Fibrils and Strong Gels. Biomacromolecules 2007, 8, 1934–1941. [Google Scholar] [PubMed]
- Fleming, K.; Gray, D.G.; Matthews, S. Cellulose Crystallites. Chem. Eur. J. 2001, 7, 1831–1835. [Google Scholar] [CrossRef]
- Zhao, Y.; Moser, C.; Lindström, M.E.; Henriksson, G.; Li, J. Cellulose Nanofibers (CNF) from Softwood, Hardwood and Tunicate: Preparation-Structure-Film Performance Interrelation. Appl. Mater. Interfaces 2017, 9, 13508–13519. [Google Scholar] [CrossRef] [PubMed]
- Beck-Candanedo, S.; Roman, M.; Gray, D.G. Effect of Reaction Conditions on the Properties and Behavior of Wood Cellulose Nanocrystal Suspensions. Biomacromolecules 2005, 6, 1048–1054. [Google Scholar]
- Dumanli, A.G.; van der Kooij, H.M.; Kamita, G.; Reisner, E.; Baumberg, J.J.; Steiner, U.; Vignolini, S. Digital Color in Cellulose Nanocrystal Films. Appl. Mater. Interfaces 2014, 6, 12302–12306. [Google Scholar] [CrossRef]
- Fernandes, S.N.; Almeida, P.L.; Monge, N.; Aguirre, L.E.; Reis, D.; de Oliveira, C.L.; Neto, A.M.; Pieranski, P.; Godinho, M.H. Mind the Microgap in Iridescent Cellulose Nanocrystal Films. Adv. Mater. 2016. [Google Scholar] [CrossRef]
- Majoinen, J.; Kontturi, E.; Gray, O.I.D.G. SEM Imaging of Chiral Nematic Films Cast from Cellulose Nanocrystal Suspensions. Cellulose 2012, 19, 1599–1605. [Google Scholar] [CrossRef]
- Kamita, G.; Frka-Petesic, B.; Allard, A.; Dargaud, M.; King, K.; Dumanli, A.G.; Vignolini, S. Biocompatible and Sustainable Optical Strain Sensors for Large-Area Applications. Adv. Opt. Mater. 2016, 4, 1950–1954. [Google Scholar] [CrossRef]
- Honorato-Rios, C.; Kuhnhold, A.; Bruckner, J.R.; Dannert, R. Equilibrium Liquid Crystal Phase Diagrams and Detection of Kinetic Arrest in Cellulose Nanocrystal Suspensions. Front. Matter. 2016, 3, 21. [Google Scholar] [CrossRef]
- Klemm, D.; Kramer, F.; Moritz, S.; Lindström, T.; Ankerfors, M.; Gray, D.; Dorris, A. Nanocelluloses: A New Family of Nature-Based Materials. Angew. Chem. Int. Ed. 2011, 50, 5438–5466. [Google Scholar] [CrossRef]
- Shen, X.; Shamshina, J.L.; Berton, P.; Gurauc, G.; Rogers, R.D. Hydrogels Based on Cellulose and Chitin: Fabrication, Properties, and Applications. Green Chem. 2016, 18, 53–75. [Google Scholar] [CrossRef]
- France, K.J.D.; Hoare, T.; Cranston, E.D. Review of Hydrogels and Aerogels Containing Nanocellulose. Chem. Matter. 2017, 29, 4609–4631. [Google Scholar] [CrossRef]
- Ye, C.; Malak, S.T.; Hu, K.; Wu, W.; Tsukruk, V.V. Cellulose Nanocrystal Microcapsules as Tunable Cages for Nano- and Microparticles. ACS Nano 2015, 9, 10887–10895. [Google Scholar] [CrossRef]
- Sannino, A.; Demitri, C.; Madaghiele, M. Biodegradable Cellulose-based Hydrogels: Design and Applications. Materials 2009, 2, 353–373. [Google Scholar] [CrossRef]
- Lavoine, N.; Desloges, I.; Dufresne, A.; Bras, J. Microfibrillated Cellulose – Its Barrier Properties and Applications in Cellulosic Materials: A review. Carbohydr. Polym. 2012, 90, 735–764. [Google Scholar] [CrossRef]
- Yue, Y.; Zhou, C.; French, A.D.; Xia, G.; Han, G.; Wang, Q.; Wu, Q. Comparative Properties of Cellulose Nano-Crystals from Native and Mercerized Cotton Fibers. Cellulose 2012, 19, 1173–1187. [Google Scholar] [CrossRef]
- Lagerwall, J.P.; Schültz, C.; Salajkova, M.; Noh, J.H.; Park, J.H.; Scalia, G.; Bergström, L. Cellulose Nanocrystal-Based Materials: From Liquid Crystal Self-Assembly and Glass Formation to Multifunctional Thin Films. NPG Asia Mater. 2014, 6, e80. [Google Scholar] [CrossRef]
- Qiu, Y.; Park, K. Environment-Sensitive Hydrogels for Drug Delivery. Adv. Drug. Deliv. Rev. 2001, 52, 321–339. [Google Scholar] [CrossRef]
- Miao, C.; Hamad, W.Y. Cellulose Reinforced Polymer Composites and Nanocomposites: A Critical Review. Cellulose 2013, 20, 2221–2262. [Google Scholar] [CrossRef]
- Sarko, A.; Marchessault, R. Supermolecular Structure of Polysaccharides. J. Polym. Sci. Part C 1969, 28, 317–331. [Google Scholar] [CrossRef]
- O’Sullivan, A.C. Cellulose: The Structure Slowly Unravels. Cellulose 1997, 4, 173–207. [Google Scholar] [CrossRef]
- Mittal, N.; Ansari, F.; Gowda, K.V.; Brouzet, C.; Chen, P.; Larsson, P.T.; Roth, S.V.; Lundell, F.; Wågberg, L.; Kotov, N.A.; et al. Multiscale Control of Nanocellulose Assembly: Transferring Remarkable Nanoscale Fibril Mechanics to Macroscale Fibers. ACS Nano 2018, 12, 6378–6388. [Google Scholar] [CrossRef] [PubMed]
- Gray, D.G. Order and Gelation of Cellulose Nanocrystal Suspensions: An Overview of Some Issues. Philos. Trans. A Math. Phys. Eng. Sci. 2018, 376, 2112. [Google Scholar] [CrossRef] [PubMed]
- Stovbun, S.V.; Skoblin, A.A.; Zlenko, D.V. Self Assembly and Gelation in Solutions of Chiral N-trifluoroacetylated α-aminoalcohols. Chem. Phys. 2018, 508, 34–44. [Google Scholar] [CrossRef]
- Kalia, S.; Dufresne, A.; Cherian, B.M.; Kaith, B.; Avéros, L.; Njuguna, J.; Nassiopoulos, E. Cellulose-Based Bio- and Nanocomposites: A Review. Int. J. Polym. Sci. 2011, 2011, 837875. [Google Scholar] [CrossRef]
- Israelachvili, J.N. Intermolecular and Surface Forces; Academic Press: Cambridge, MA, USA, 2011. [Google Scholar]
- Preston, R. The Organisation of the Cell Wall of the Conifer Tracheid. Philos. Trans. B 1934, 61, 131–174. [Google Scholar]
- Bardage, S.; Donaldson, L.; Tokoh, C.; Daniel, G. Ultrastructure of the Cell Wall of Unbeaten Norway Spruce Pulp Fibre Surfaces. Nord. Pulp Pap. Res. J. 2004, 19, 448–452. [Google Scholar] [CrossRef]
- Liu, D.; Wang, S.; Ma, Z.; Tian, D.; Gu, M.; Lin, F. Structure–Color Mechanism of Iridescent Cellulose Nanocrystal Films. RSC Adv. 2014, 4, 39322–39331. [Google Scholar] [CrossRef]
- Li, M.; Wu, Q.; Song, K.; Lee, S.; Qing, Y.; Wu, Y. Cellulose Nanoparticles: Structure-morphology-rheology Relationships. ACS Sustain. Chem. Eng. 2015, 3, 821–832. [Google Scholar] [CrossRef]
- Meudtner, R.M.; Hecht, S. Helicity Inversion in Responsive Foldamers Induced by Achiral Halide Ion Guests. Angew. Chem. Int. Ed. 2008, 47, 4926–4930. [Google Scholar] [CrossRef]
- Zhao, Z.; Shklyaev, O.; Nili, A.; Mohamed, M.; Kubicki, J.; Crespi, V.; Zhong, L. Cellulose Microfibril Twist, Mechanics, and Implication for Cellulose Biosynthesis. J. Phys. Chem. A. 2013, 117, 2580–2589. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zlenko, D.V.; Nikolsky, S.N.; Vedenkin, A.S.; Politenkova, G.G.; Skoblin, A.A.; Melnikov, V.P.; Mikhaleva, M.G.; Stovbun, S.V. Twisting of Fibers Balancing the Gel–Sol Transition in Cellulose Aqueous Suspensions. Polymers 2019, 11, 873. https://doi.org/10.3390/polym11050873
Zlenko DV, Nikolsky SN, Vedenkin AS, Politenkova GG, Skoblin AA, Melnikov VP, Mikhaleva MG, Stovbun SV. Twisting of Fibers Balancing the Gel–Sol Transition in Cellulose Aqueous Suspensions. Polymers. 2019; 11(5):873. https://doi.org/10.3390/polym11050873
Chicago/Turabian StyleZlenko, Dmitry V., Sergey N. Nikolsky, Alexander S. Vedenkin, Galina G. Politenkova, Aleksey A. Skoblin, Valery P. Melnikov, Maria G. Mikhaleva, and Sergey V. Stovbun. 2019. "Twisting of Fibers Balancing the Gel–Sol Transition in Cellulose Aqueous Suspensions" Polymers 11, no. 5: 873. https://doi.org/10.3390/polym11050873
APA StyleZlenko, D. V., Nikolsky, S. N., Vedenkin, A. S., Politenkova, G. G., Skoblin, A. A., Melnikov, V. P., Mikhaleva, M. G., & Stovbun, S. V. (2019). Twisting of Fibers Balancing the Gel–Sol Transition in Cellulose Aqueous Suspensions. Polymers, 11(5), 873. https://doi.org/10.3390/polym11050873