Highly Porous Organic Polymers for Hydrogen Fuel Storage


Abstract
1. Introduction
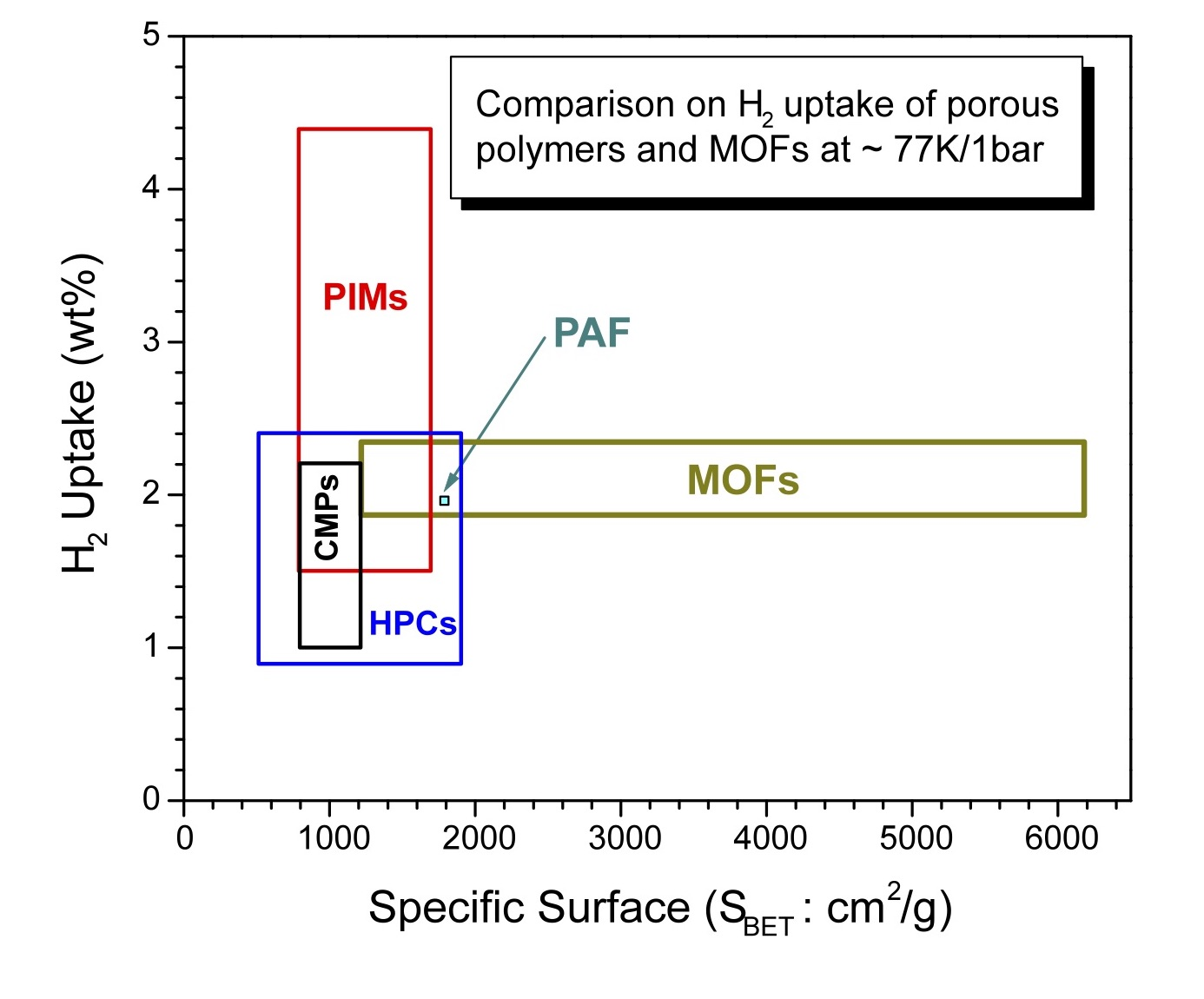
2. Theoretical Considerations
3. Highly Porous Organic Polymers for H2 Storage
3.1. Hypercrosslinked Polymers (HCPs)
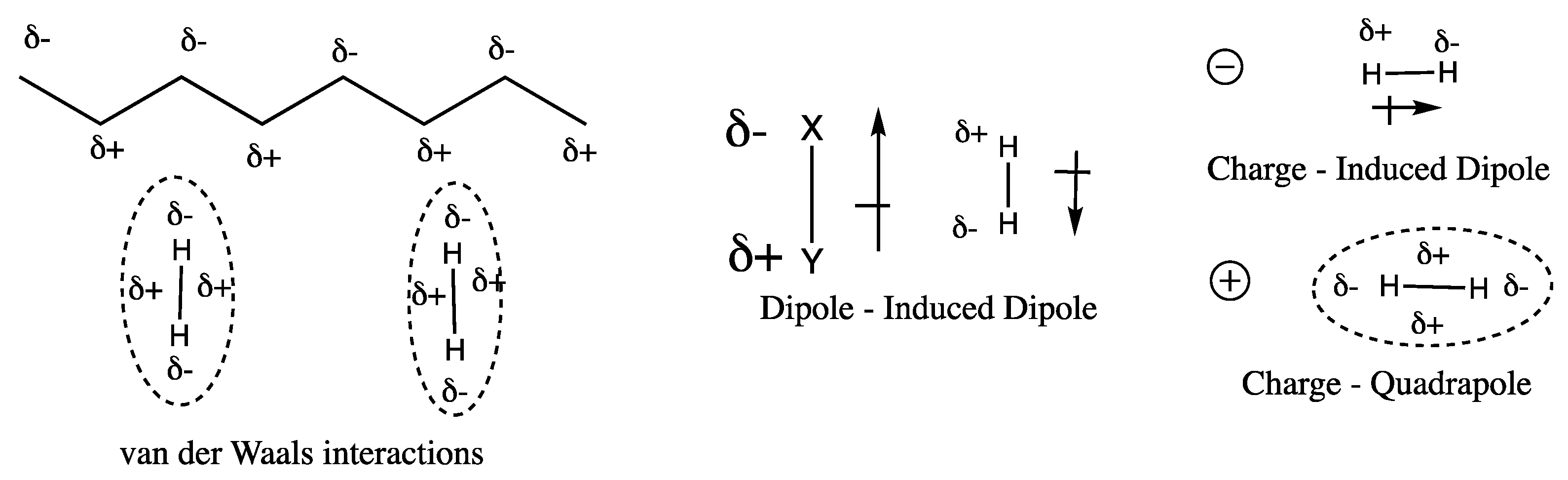
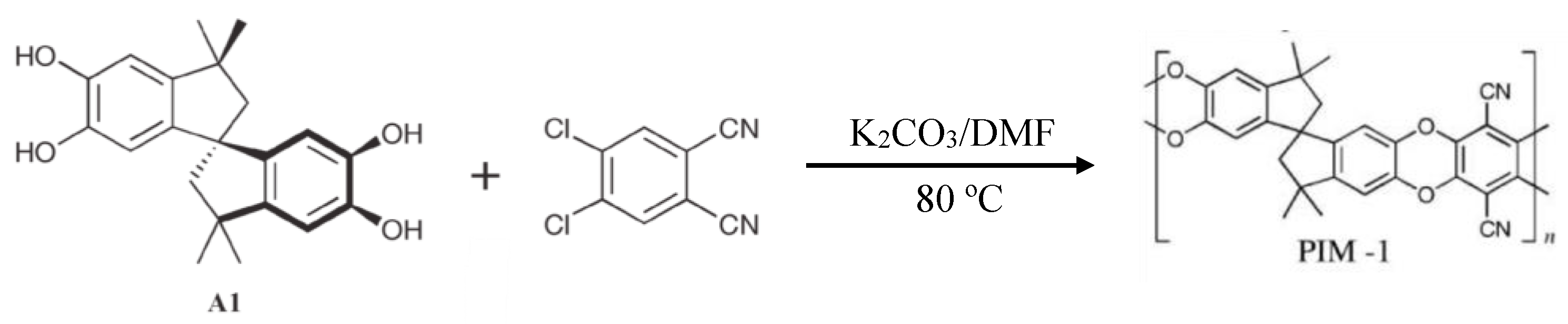
3.2. Polymers of Intrinsic Microporosity (PIMs)
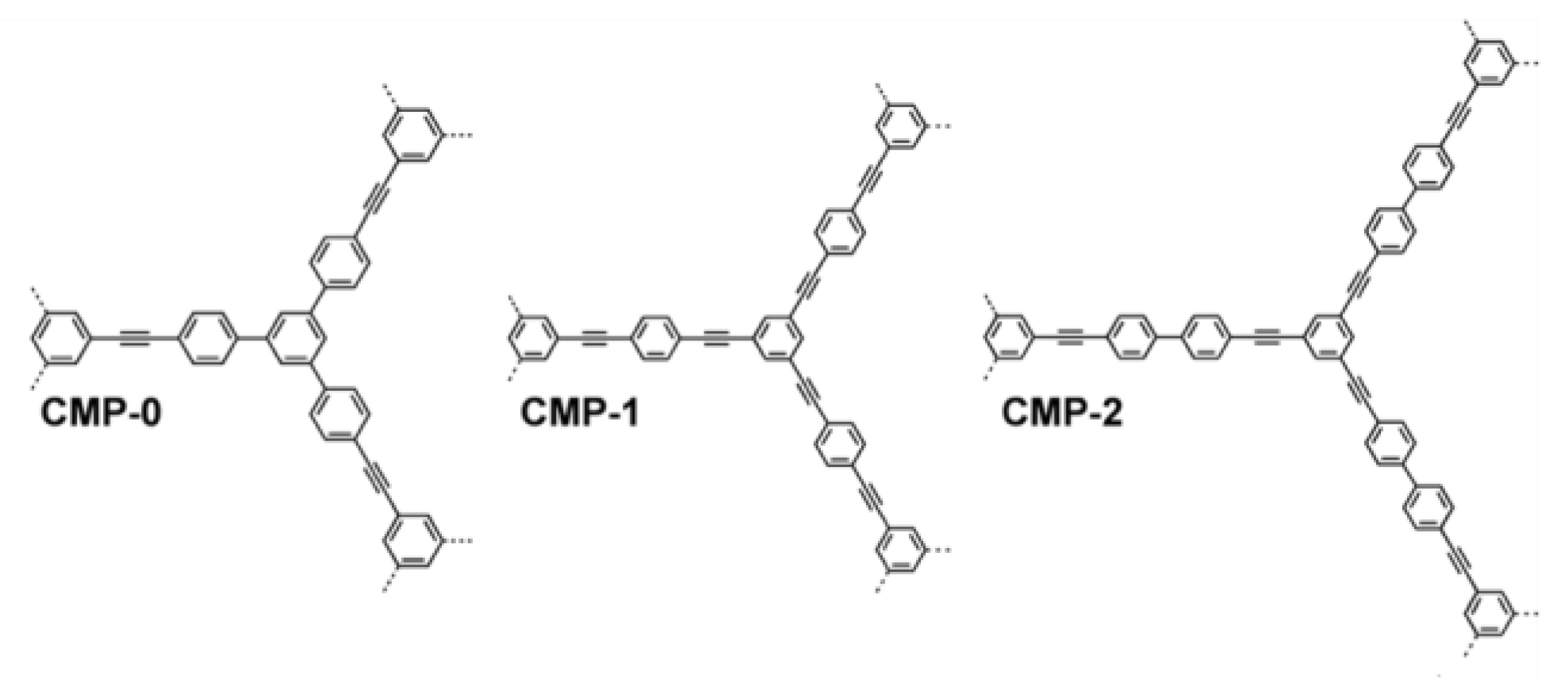
3.3. Conjugated Microporous Polymers (CMPs)
3.4. Porous Aromatic Frameworks (PAFs)
3.5. Other Porous Polymers
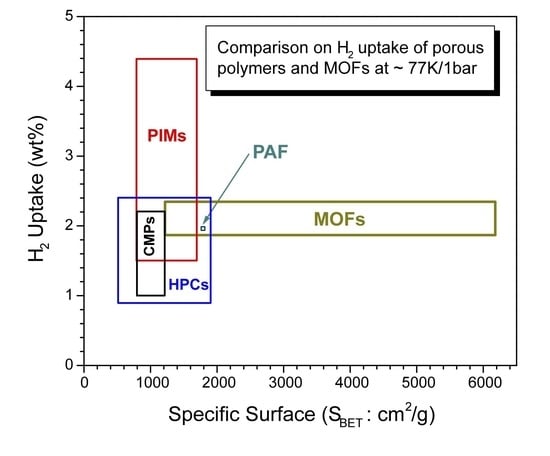
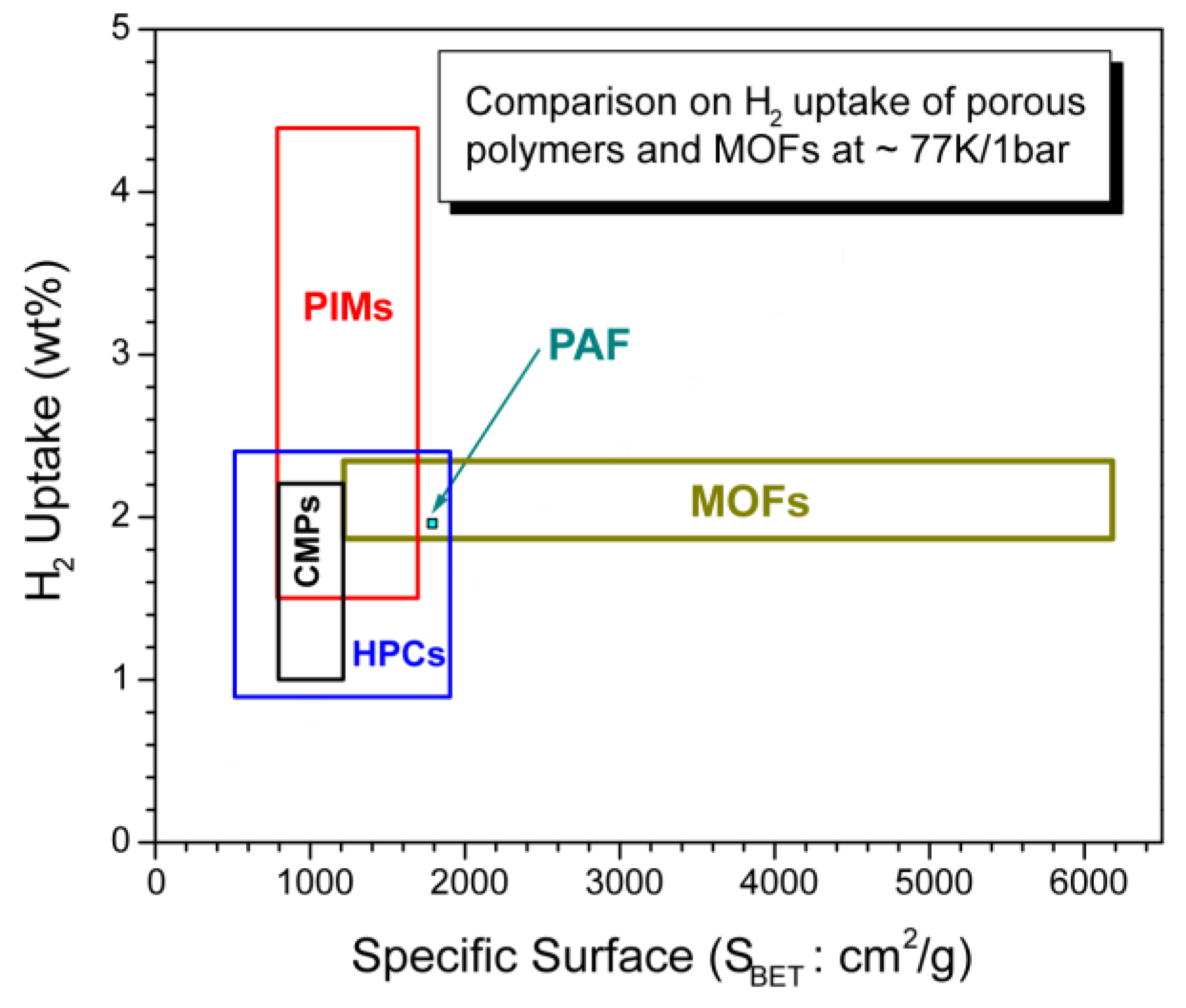
3.6. Brief comparison of highly porous organic polymers with MOFs
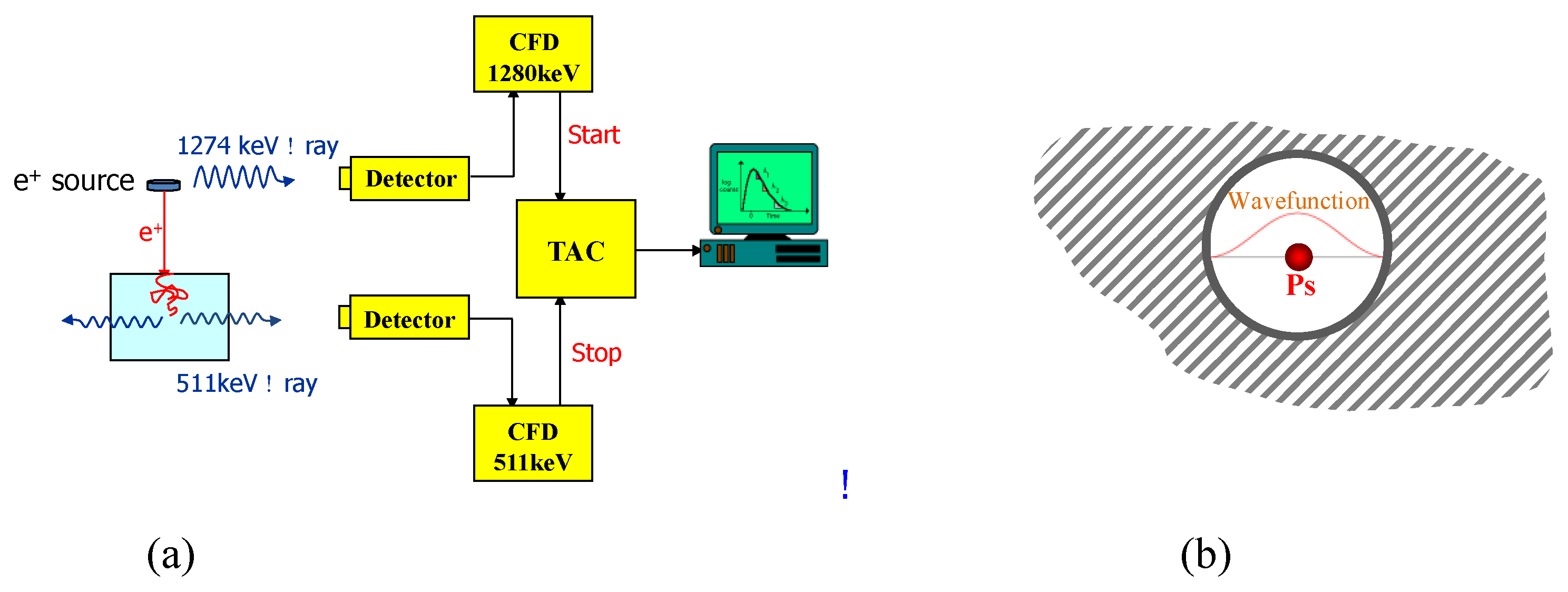
4. Characterization of Porosity in Highly Porous Organic Polymers
5. Conclusions and Prospective Future
Author Contributions
Funding
Conflicts of Interest
References
- Van den Berg, A.W.C.; Areán, C.O. Materials for Hydrogen Storage: Current Research Trends and Perspectives. Chem. Commun. 2008, 668–681. [Google Scholar] [CrossRef]
- Schlapbach, L.; Zutell, A. Hydrogen Storage Materials for Mobile Applications. Nature 2001, 414, 353–358. [Google Scholar] [CrossRef]
- Jena, P. Materials for Hydrogen Storage: Past, Present, and Future. J. Phys. Chem. Lett. 2011, 2, 206–211. [Google Scholar] [CrossRef]
- Stetson, N.T. Hydrogen Storage—2010 Annual Merit Review and Peer Evaluation Meeting. 2010. Available online: http://www.hydrogen.energy.gov/pdfs/review10/st00a_stetson_2010_o_web.pdf (accessed on 31 January 2019).
- Satyapal, S.; Petrovic, J.; Read, C.; Thomas, G.; Ordaz, G. The U.S. Department of Energy’s National Hydrogen Storage Project: Progress towards Meeting Hydrogen-Powered Vehicle Requirements. Catal. Today 2007, 120, 246–256. [Google Scholar] [CrossRef]
- 2015 Hydrogen Storage. Available online: https://www.energy.gov/sites/prod/files/2015/05/f22/fcto_myrdd_storage.pdf (accessed on 31 January 2019).
- Target Explanation Document: Hydrogen Storage for Light-Duty Fuel Cell Vehicles 2017. Available online: https://www.energy.gov/sites/prod/files/2017/05/f34/fcto_targets_onboard_hydro_storage_explanation.pdf (accessed on 31 January 2019).
- Sakintuna, B.; Lamari-Darkrim, F.; Hirscher, M.; Dogan, B. Metal hydride materials for solid hydrogen storage: A review. Int. J. Hydrogen Energy 2007, 32, 1121–1240. [Google Scholar] [CrossRef]
- Amica, G.; Larochette, P.A.; Gennari, F.C. Hydrogen storage properties of LiNH2–LiH system with MgH2, CaH2 and TiH2 added. Int. J. Hydrogen Energy 2015, 40, 9335–9346. [Google Scholar] [CrossRef]
- Xiong, R.; Sang, G.; Zhang, G.; Yan, X.; Li, P.; Yao, Y.; Luo, D.; Chen, C.A.; Tang, T. Evolution of the active species and catalytic mechanism of Ti doped NaAlH4 for hydrogen storage. Int. J. Hydrogen Energy 2017, 42, 6088–6095. [Google Scholar] [CrossRef]
- Petit, J.F.; Miele, P.; Demirci, U.B. Ammonia borane H3NBH3 for solid-state chemical hydrogen storage: Different samples with different thermal behaviors. Int. J. Hydrogen Energy 2016, 41, 15462–15470. [Google Scholar] [CrossRef]
- Zubizarreta, L.; Arenillas, A.; Pis, J.J. Carbon materials for H2 storage. Int. J. Hydrogen Energy 2009, 34, 4575–4581. [Google Scholar] [CrossRef]
- Gogotsi, Y.; Dash, R.K.; Yushin, G.; Yildirim, T.; Laudisio, G.; Fischer, J.E. Tailoring of Nanoscale Porosity in Carbide-Derived Carbons for Hydrogen Storage. J. Am. Chem. Soc. 2005, 127, 16006–16007. [Google Scholar] [CrossRef] [PubMed]
- Benard, P.; Chahine, R.; Chandonia, P.A.; Cossement, D.; Dorval-Douville, G.; Lafi, L.; Lachance, P.; Paggiaro, R.; Poirier, E. Comparison of hydrogen adsorption on nanoporous materials. J. Alloys Compd. 2007, 446, 380–384. [Google Scholar] [CrossRef]
- Patchkovskii, S.; Tse, J.S.; Yurchenko, S.N.; Zhechkov, L.; Heine, T.; Seifert, G. Graphene Nanostructures as Tunable Storage Media for Molecular Hydrogen. Proc. Natl. Acad. Sci. USA 2005, 102, 10439–10444. [Google Scholar] [CrossRef] [PubMed]
- Jordá-Beneyto, M.; Suárez-García, F.; Lozano-Castelló, D.; Cazorla-Amorós, D.; Linares-Solano, A. Hydrogen storage on chemically activated carbons and carbon nanomaterials at high pressures. Carbon 2007, 45, 293–303. [Google Scholar] [CrossRef]
- Yürüm, Y.; Taralp, A.; Veziroglu, T.N. Storage of Hydrogen in Nanostructured Carbon Materials. Int. J. Hydrogen Energy 2009, 34, 3784–3798. [Google Scholar] [CrossRef]
- Dalebrook, A.F.; Gan, W.; Grasemann, M.; Moret, S.; Laurenczy, G. Hydrogen Storage: Beyond Conventional Methods. Chem. Commun. 2013, 49, 8735–8751. [Google Scholar] [CrossRef]
- Broom, D.P.; Webb, C.J.; Fanourgakis, G.S.; Froudakis, G.E.; Trikalitis, P.N.; Hirscher, M. Concepts for Improving Hydrogen Storage in Nanoporous Materials. Int. J. Hydrogen Energy 2019, 44, 7768–7779. [Google Scholar] [CrossRef]
- Bhatia, S.K.; Myers, A.L. Optimum conditions for adsorptive storage. Langmuir 2006, 22, 1688–1700. [Google Scholar] [CrossRef]
- Rosi, N.L.; Eckert, J.; Eddaoudi, M.; Vodak, D.T.; Kim, J.; O’keeffe, M.; Yaghi, O.M. Hydrogen storage in microporous metal-organic frameworks. Science 2003, 300, 1127–1129. [Google Scholar] [CrossRef]
- Sagara, T.; Klassen, J.; Ganz, E. Computational study of hydrogen binding by metal-organic framework-5. J. Chem. Phys. 2004, 121, 12543–12547. [Google Scholar] [CrossRef]
- Allendorf, M.D.; Hulvey, Z.; Gennett, T.; Ahmed, A.; Autrey, T.; Camp, J.; Cho, E.S.; Furukawa, H.; Haranczyk, M.; Head-Gordon, M.; et al. An assessment of strategies for the development of solid-state adsorbents for vehicular hydrogen storage. Energy Environ. Sci. 2018, 11, 2784–2812. [Google Scholar] [CrossRef]
- Heine, T.; Zhechkov, L.; Seifert, G. Hydrogen storage by physisorption on nanostructured graphite platelets. Phys. Chem. Chem. Phys. 2004, 6, 980–984. [Google Scholar] [CrossRef]
- Otero Areán, C.; Manoilova, O.V.; Bonelli, B.; Rodríguez Delgado, M.; Turnes Palomino, G.; Garrone, E. Thermodynamics of Hydrogen Adsorption on the Zeolite Li-ZSM-5. Chem. Phys. Lett. 2003, 370, 631–635. [Google Scholar] [CrossRef]
- Garberoglio, G.; Skoulidas, A.I.; Johnson, J.K. Adsorption of Gases in Metal Organic Materials: Comparison of Simulations and Experiments. J. Phys. Chem. B 2005, 109, 13094–13103. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Johnson, J.K. Molecular simulation of hydrogen adsorption in single-walled carbon nanotubes and idealized carbon slit pores. J. Chem. Phys. 1990, 110, 577–586. [Google Scholar] [CrossRef]
- Cabria, I.; Lopez, M.J.; Alonso, J.A. Simulation of the hydrogen storage in nanoporous carbons with different pore shapes. Int. J. Hydrogen Energy 2011, 36, 10748–10759. [Google Scholar] [CrossRef]
- Rzepka, M.; Lamp, P.; de la Casa-Lillo, M.A. Physisorption of Hydrogen on Microporous Carbon and Carbon Nanotubes. J. Phys. Chem. B 1998, 102, 10894–10898. [Google Scholar] [CrossRef]
- Wang, Q.; Challa, S.R.; Sholl, D.S.; Johnson, J.K. Quantum sieving in carbon nanotubes and zeolites. Phys. Rev. Lett. 1999, 82, 956–959. [Google Scholar] [CrossRef]
- Cai, J.; Xing, Y.; Zhao, X. Quantum sieving: Feasibility and challenges for the separation of hydrogen isotopes in nanoporous materials. RSC Adv. 2012, 2, 8579–8586. [Google Scholar] [CrossRef]
- Oh, H.; Hirscher, M. Quantum sieving for separation of hydrogen isotopes using MOFs. Eur. J. Inorg. Chem. 2016, 27, 4278–4289. [Google Scholar] [CrossRef]
- Gallego, N.C.; He, L.; Saha, D.; Contescu, C.I.; Melnichenko, Y.B. Hydrogen confinement in carbon nanopores: Extreme densification at ambient temperature. J. Am. Chem. Soc. 2011, 133, 13794–13797. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.S.; Bein, T. Covalent Organic Frameworks: Structures, Synthesis, and Applications. Adv. Funct. Mater. 2018, 28, 1705553. [Google Scholar] [CrossRef]
- Lochan, R.C.; Head-Gordon, M. Computational studies of molecular hydrogen binding affinities: The role of dispersion forces, electrostatics, and orbital interactions. Phys. Chem. Chem. Phys. 2006, 8, 1357–1370. [Google Scholar] [CrossRef]
- Gonzalez, A.A.; Zhang, K.; Nolan, S.P.; Lopez de la Vega, R.L.; Mukerjee, S.L.; Hoff, C.D.; Kubas, G.J. Thermodynamic and Kinetic Studies of the Complexes W(CO)3(PCy3)2(L) (L = H2, N2, NCCH3, Pyridine, P(OMe)3, CO). Organometallics 1988, 7, 2429–2435. [Google Scholar] [CrossRef]
- Sevilla, M.; Fuertes, A.B.; Mokaya, R. High Density Hydrogen Storage in Superactivated Carbons from Hydrothermally Carbonized Renewable Organic Materials. Energy Environ. Sci. 2011, 4, 1400–1410. [Google Scholar] [CrossRef]
- Rossetti, I.; Ramis, G.; Gallo, A.; Michele, A.D. Hydrogen Storage over Metal-Doped Activated Carbon. Int. J. Hydrogen Energy 2015, 40, 7609–7616. [Google Scholar] [CrossRef]
- Kuchta, B.; Firlej, L.; Mohammadhosseini, A.; Boulet, P.; Beckner, M.; Romanos, J.; Pfeifer, P. Hypothetical High-Surface-Area Carbons with Exceptional Hydrogen Storage Capacities: Open Carbon Frameworks. J. Am. Chem. Soc. 2012, 134, 15130–15137. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.M.; Park, K.S.; Choi, Y.C.; Park, Y.S.; Bok, J.M.; Bae, D.J.; Nahm, K.S.; Choi, Y.G.; Yu, S.C.; Kim, N.; et al. Hydrogen Adsorption and Storage in Carbon Nanotubes. Synth. Met. 2000, 113, 209–216. [Google Scholar] [CrossRef]
- Li, F.; Zhao, J.; Chen, Z. Carbon-Based Nanomaterials for H2 Storage. In Carbon Nanomaterials for Advanced Energy Systems; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2015; pp. 407–437. [Google Scholar]
- Li, G.; Kobayashi, H.; Taylor, J.M.; Ikeda, R.; Kubota, Y.; Kato, K.; Takata, M.; Yamamoto, T.; Toh, S.; Matsumura, S.; et al. Hydrogen Storage in Pd Nanocrystals Covered with a Metal–Organic Framework. Nat. Mater. 2014, 13, 802. [Google Scholar] [CrossRef]
- Furukawa, H.; Miller, M.A.; Yaghi, O.M. Independent Verification of the Saturation Hydrogen Uptake in MOF-177 and Establishment of a Benchmark for Hydrogen Adsorption in Metal–Organic Frameworks. J. Mater. Chem. 2007, 17, 3197–3204. [Google Scholar] [CrossRef]
- Wong-Foy, A.G.; Matzger, A.J.; Yaghi, O.M. Exceptional H2 saturation uptake in microporous metal−organic frameworks. J. Am. Chem. Soc. 2006, 128, 3494–3495. [Google Scholar] [CrossRef]
- Feng, X.; Ding, X.; Jiang, D. Covalent organic frameworks. Chem. Soc. Rev. 2012, 41, 6010–6022. [Google Scholar] [CrossRef]
- Pramudya, Y.; Mendoza-Cortes, J.L. Design principles for high H2 storage using chelation of abundant transition metals in covalent organic frameworks for 0–700 bar at 298 K. J. Am. Chem. Soc. 2016, 138, 15204–15213. [Google Scholar] [CrossRef]
- Mendoza-Cortes, J.L.; Goddard, W.A.; Furukawa, H.; Yaghi, O.M. A Covalent Organic Framework That Exceeds the DOE 2015 Volumetric Target for H2 Uptake at 298 K. J. Phys. Chem. Lett. 2012, 3, 2671–2675. [Google Scholar] [CrossRef]
- Klontzas, E.; Tylianakis, E.; Froudakis, G.E. Hydrogen Storage in Lithium-Functionalized 3-D Covalent-Organic Framework Materials. J. Phys. Chem. C 2009, 113, 21253–21257. [Google Scholar] [CrossRef]
- Gao, F.; Sun, J.T.; Meng, S. “H2 Sponge”: Pressure as a Means for Reversible High-Capacity Hydrogen Storage in Nanoporous Ca-Intercalated Covalent Organic Frameworks. Nanoscale 2015, 7, 6319–6324. [Google Scholar] [CrossRef]
- Liu, J.; Zou, R.; Zhao, Y. Recent Developments in Porous Materials for H2 and CH4 Storage. Tetrahedron Lett. 2016, 57, 4873–4881. [Google Scholar] [CrossRef]
- Yuan, D.; Zhao, D.; Sun, D.; Zhou, H. An Isoreticular Series of Metal–Organic Frameworks with Dendritic Hexacarboxylate Ligands and Exceptionally High Gas-Uptake Capacity. Angew. Chem. Int. Ed. 2010, 49, 5357–5361. [Google Scholar] [CrossRef] [PubMed]
- Veverka, P.; Jeřábek, K. Mechanism of Hypercrosslinking of Chloromethylated Styrene–Divinylbenzene Copolymers. React. Funct. Polym. 1999, 41, 21–25. [Google Scholar] [CrossRef]
- Germain, J.; Hradil, J.; Fréchet, J.M.J.; Svec, F. High Surface Area Nanoporous Polymers for Reversible Hydrogen Storage. Chem. Mater. 2006, 18, 4430–4435. [Google Scholar] [CrossRef]
- Germain, J.; Fréchet, J.M.J.; Svec, F. Nanoporous Polymers for Hydrogen Storage. Small 2009, 5, 1098–1111. [Google Scholar] [CrossRef]
- Fontanals, N.; Marcé, R.M.; Cormack, P.A.G.; Sherrington, D.C.; Borrull, F. Monodisperse, Hypercrosslinked Polymer Microspheres as Tailor-Made Sorbents for Highly Efficient Solid-Phase Extractions of Polar Pollutants from Water Samples. J. Chromatogr. A 2008, 1191, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Guiochon, G.; Gritti, F. Shell particles, trials, tribulations and triumphs. J. Chrom. A 2011, 1218, 1915–1938. [Google Scholar] [CrossRef] [PubMed]
- Germain, J.; Fréchet, J.M.J.; Svec, F. Hypercrosslinked Polyanilines with Nanoporous Structure and High Surface Area: Potential Adsorbents for Hydrogen Storage. J. Mater. Chem. 2007, 17, 4989–4997. [Google Scholar] [CrossRef]
- Germain, J.; Fréchet, J.M.J.; Svec, F. Nanoporous, Hypercrosslinked Polypyrroles: Effect of Crosslinking Moiety on Pore Size and Selective Gas Adsorption. Chem. Commun. Camb. Engl. 2009, 12, 1526–1528. [Google Scholar] [CrossRef]
- Wood, C.D.; Tan, B.; Trewin, A.; Niu, H.; Bradshaw, D.; Rosseinsky, M.J.; Khimyak, Y.Z.; Campbell, N.L.; Kirk, R.; Stöckel, E.; et al. Hydrogen Storage in Microporous Hypercrosslinked Organic Polymer Networks. Chem. Mater. 2007, 19, 2034–2048. [Google Scholar] [CrossRef]
- Gagnon-Thibault, É.; Cossement, D.; Guillet-Nicolas, R.; Masoumifard, N.; Bénard, P.; Kleitz, F.; Chahine, R.; Morin, J.F. Nanoporous Ferrocene-Based Cross-Linked Polymers and Their Hydrogen Sorption Properties. Microporous Mesoporous Mater. 2014, 188, 182–189. [Google Scholar] [CrossRef]
- Li, G.; Liu, Q.; Xia, B.; Huang, J.; Li, S.; Guan, Y.; Zhou, H.; Liao, B.; Zhou, Z.; Liu, B. Synthesis of Stable Metal-Containing Porous Organic Polymers for Gas Storage. Eur. Polym. J. 2017, 91, 242–247. [Google Scholar] [CrossRef]
- Shu, G.; Zhang, C.; Li, Y.; Jiang, J.X.; Wang, X.; Li, H.; Wang, F. Hypercrosslinked silole-containing microporous organic polymers with N-functionalized pore surfaces for gas storage and separation. J. Appl. Polym. Sci. 2018, 135, 45907. [Google Scholar] [CrossRef]
- Fu, S.; Yao, J.; Yang, Z.; Sun, H.; Liu, W. Silane-Based Hyper-Cross-Linked Porous Polymers and Their Applications in Gas Storage and Water Treatment. J. Mater. Sci. 2018, 53, 10469–10478. [Google Scholar] [CrossRef]
- Gao, H.; Ding, L.; Bai, H.; Liu, A.; Li, S.; Li, L. Pitch-based hyper-cross-linked polymers with high performance for gas adsorption. J. Mater. Chem. A 2016, 4, 16490–16498. [Google Scholar] [CrossRef]
- Li, B.; Huang, X.; Liang, L.; Tan, B. Synthesis of uniform microporous polymer nanoparticles and their applications for hydrogen storage. J. Mater. Chem. 2010, 20, 7444–7450. [Google Scholar] [CrossRef]
- Pan, L.; Chen, Q.; Zhu, J.H.; Yu, J.G.; He, Y.J.; Han, B.H. Hypercrosslinked porous polycarbazoles via one-step oxidative coupling reaction and Friedel–Crafts alkylation. Polym. Chem. 2015, 6, 2478–2487. [Google Scholar] [CrossRef]
- Yang, X.; Yu, M.; Zhao, Y.; Zhang, C.; Wang, X.; Jiang, J.X. Hypercrosslinked microporous polymers based on carbazole for gas storage and separation. RSC Adv. 2014, 4, 61051–61055. [Google Scholar] [CrossRef]
- Gao, H.; Ding, L.; Li, W.; Ma, G.; Bai, H.; Li, L. Hyper-cross-linked organic microporous polymers based on alternating copolymerization of bismaleimide. ACS Macro Lett. 2016, 5, 377–381. [Google Scholar] [CrossRef]
- Liu, G.; Wang, Y.; Shen, C.; Ju, Z.; Yuan, D. A facile synthesis of microporous organic polymers for efficient gas storage and separation. J. Mater. Chem. A 2015, 3, 3051–3058. [Google Scholar] [CrossRef]
- Li, B.; Gong, R.; Wang, W.; Huang, X.; Zhang, W.; Li, H.; Hu, C.; Tan, B. A new strategy to microporous polymers: Knitting rigid aromatic building blocks by external cross-linker. Macromolecules 2011, 44, 2410–2414. [Google Scholar] [CrossRef]
- Schwab, M.G.; Lennert, A.; Pahnke, J.; Jonschker, G.; Koch, M.; Senkovska, I.; Rehahn, M.; Kaskel, S. Nanoporous copolymer networks through multiple Friedel–Crafts-alkylation—Studies on hydrogen and methane storage. J. Mater. Chem. 2011, 21, 2131–2135. [Google Scholar] [CrossRef]
- Dawson, R.; Stevens, L.A.; Drage, T.C.; Snape, C.E.; Smith, M.W.; Adams, D.J.; Cooper, A.I. Impact of water coadsorption for carbon dioxide capture in microporous polymer sorbents. J. Am. Chem. Soc. 2012, 134, 10741–10744. [Google Scholar] [CrossRef]
- Wang, T.; Zhao, Y.C.; Zhang, L.M.; Cui, Y.; Zhang, C.S.; Han, B.H. Novel approach to hydroxy-group-containing porous organic polymers from bisphenol A. Beilstein J. Org. Chem. 2017, 13, 2131–2137. [Google Scholar] [CrossRef]
- Chen, S.; Liu, J.; Li, Z.; Wang, H.; Wang, X.; Xu, Y. Hydrogen storage properties of highly cross-linked polymers derived from chlorinated polypropylene and polyethylenimine. Int. J. Hydrogen Energy 2017, 42, 23028–23034. [Google Scholar] [CrossRef]
- McKeown, N.B.; Budd, P.M. Polymers of Intrinsic Microporosity (PIMs): Organic Materials for Membrane Separations, Heterogeneous Catalysis and Hydrogen Storage. Chem. Soc. Rev. 2006, 35, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Rochat, S.; Polak-Kraśna, K.; Tian, M.; Mays, T.J.; Bowen, C.R.; Burrows, A.D. Assessment of the Long-Term Stability of the Polymer of Intrinsic Microporosity PIM-1 for Hydrogen Storage Applications. State Art Mater. Hydrogen Energy 2019, 44, 332–337. [Google Scholar] [CrossRef]
- McKeown, N.B.; Gahnem, B.; Msayib, K.J.; Budd, P.M.; Tattershall, C.E.; Mahmood, K.; Tan, S.; Book, D.; Langmi, H.W.; Walton, A. Towards Polymer-based Hydrogen Storage Materials: Engineering Ultramicroporous Cavities within Polymers of Intrinsic Microporosity. Angew. Chem. Int. Ed. 2006, 45, 1804–1807. [Google Scholar] [CrossRef] [PubMed]
- McKeown, N.B.; Budd, P.M.; Book, D. Microporous Polymers as Potential Hydrogen Storage Materials. Macromol. Rapid Commun. 2007, 28, 995–1002. [Google Scholar] [CrossRef]
- Budd, P.M.; Butler, A.; Selbie, J.; Mahmood, K.; McKeown, N.B.; Ghanem, B.; Msayib, K.; Book, D.; Walton, A. The Potential of Organic Polymer-Based Hydrogen Storage Materials. Phys. Chem. Chem. Phys. 2007, 9, 1802–1808. [Google Scholar] [CrossRef]
- Ghanem, B.S.; Hashem, M.; Harris, K.D.; Msayib, K.J.; Xu, M.; Budd, P.M.; Chaukura, N.; Book, D.; Tedds, S.; Walton, A.; et al. Triptycene-Based Polymers of Intrinsic Microporosity: Organic Materials That Can Be Tailored for Gas Adsorption. Macromolecules 2010, 43, 5287–5294. [Google Scholar] [CrossRef]
- Bera, R.; Mondal, S.; Das, N. Triptycene Based Microporous Polymers (TMPs): Efficient Small Gas (H2 and CO2) Storage and High CO2/N2 Selectivity. Microporous Mesoporous Mater. 2018, 257, 253–261. [Google Scholar] [CrossRef]
- Makhseed, S.; Ibrahim, F.; Samuel, J. Phthalimide Based Polymers of Intrinsic Microporosity. Polymer 2012, 53, 2964–2972. [Google Scholar] [CrossRef]
- Weber, J.; Antonietti, M.; Tomas, A. Microporous Networks of High-Performance Polymers: Elastic Deformations and Gas Sorption Properties. Macromolecules 2008, 41, 2880–2885. [Google Scholar] [CrossRef]
- Ramimoghadam, D.; Gray, E.M.; Webb, C.J. Review of Polymers of Intrinsic Microporosity for Hydrogen Storage Applications. Int. J. Hydrogen Energy 2016, 41, 16944–16965. [Google Scholar] [CrossRef]
- Cooper, A.I. Conjugated Microporous Polymers. Adv. Mater. 2009, 21, 1291–1295. [Google Scholar] [CrossRef]
- Jiang, J.X.; Su, F.; Niu, H.; Wood, C.D.; Campbell, N.L.; Khimyak, Y.Z.; Cooper, A.I. Conjugated microporous poly(phenylene butadiynylene)s. Chem. Commun. 2008, 486–488. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Dorney, B.; White, D.; Kirklin, S.; Zapol, P.; Yu, L.; Liu, D.J. Microporous polyphenylenes with tunable pore size for hydrogen storage. Chem. Commun. 2010, 46, 4547–4549. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Luo, M.; Wang, T.; Wang, J.X.; Zhou, D.; Han, Y.; Zhang, C.S.; Yan, C.G.; Han, B.H. Porous Organic Polymers Based on Propeller-like Hexaphenylbenzene Building Units. Macromolecules 2011, 44, 5573–5577. [Google Scholar] [CrossRef]
- Chen, Q.; Wang, J.X.; Yang, F.; Zhou, D.; Bian, N.; Zhang, X.J.; Yan, C.G.; Han, B.H. Tetraphenylethylene-Based Fluorescent Porous Organic Polymers: Preparation, Gas Sorption Properties and Photoluminescence Properties. J. Mater. Chem. 2011, 21, 13554–13560. [Google Scholar] [CrossRef]
- Suresh, V.M.; Bonakala, S.; Roy, S.; Balasubramanian, S.; Maji, T.K. Synthesis, characterization, and modeling of a functional conjugated microporous polymer: CO2 storage and light harvesting. J. Phys. Chem. C 2014, 118, 24369–24376. [Google Scholar] [CrossRef]
- Qiao, S.; Du, Z.; Yang, R. Design and Synthesis of Novel Carbazole–Spacer–Carbazole Type Conjugated Microporous Networks for Gas Storage and Separation. J. Mater. Chem. A 2014, 2, 1877–1885. [Google Scholar] [CrossRef]
- Bandyopadhyay, S.; Anil, A.G.; James, A.; Patra, A. Multifunctional Porous Organic Polymers: Tuning of Porosity, CO2, and H2 Storage and Visible-Light-Driven Photocatalysis. ACS Appl. Mater. Interfaces 2016, 8, 27669–27678. [Google Scholar] [CrossRef] [PubMed]
- Gu, C.; Bao, Y.; Huang, W.; Liu, D.; Yang, R. Four Simple Structure Carbazole-Based Conjugated Microporous Polymers with Different Soft Connected Chains. Macromol. Chem. Phys. 2016, 217, 748–756. [Google Scholar] [CrossRef]
- Rabbani, M.G.; Sekizkardes, A.K.; El-Kadri, O.M.; Kaafarani, B.R.; El-Kaderi, H.M. Pyrene-Directed Growth of Nanoporous Benzimidazole-Linked Nanofibers and Their Application to Selective CO2 Capture and Separation. J. Mater. Chem. 2012, 22, 25409–25417. [Google Scholar] [CrossRef]
- Bhunia, A.; Vasylyeva, V.; Janiak, C. From a Supramolecular Tetranitrile to a Porous Covalent Triazine-Based Framework with High Gas Uptake Capacities. Chem. Commun. 2013, 49, 3961–3963. [Google Scholar] [CrossRef]
- Kassab, R.M.; Jackson, K.T.; El-Kadri, O.M.; El-Kaderi, H.M. Nickel-Catalyzed Synthesis of Nanoporous Organic Frameworks and Their Potential Use in Gas Storage Applications. Res. Chem. Intermed. 2011, 37, 747–757. [Google Scholar] [CrossRef]
- Li, A.; Lu, R.F.; Wang, Y.; Wang, X.; Han, K.L.; Deng, W.Q. Lithium-doped Conjugated Microporous Polymers for Reversible Hydrogen Storage. Angew. Chem. 2010, 122, 3402–3405. [Google Scholar] [CrossRef]
- Xu, D.; Sun, L.; Li, G.; Shang, J.; Yang, R.X.; Deng, W.Q. Methyllithium-Doped Naphthyl-Containing Conjugated Microporous Polymer with Enhanced Hydrogen Storage Performance. Chem. Eur. J. 2016, 22, 7944–7949. [Google Scholar] [CrossRef] [PubMed]
- Reich, T.E.; Jackson, K.T.; Li, S.; Jena, P.; El-Kaderi, H.M. Synthesis and Characterization of Highly Porous Borazine-Linked Polymers and Their Performance in Hydrogen Storage Application. J. Mater. Chem. 2011, 21, 10629–10632. [Google Scholar] [CrossRef]
- Liao, Y.; Cheng, Z.; Zuo, W.; Thomas, A.; Fall, C.F. Nitrogen-Rich Conjugated Microporous Polymers: Facile Synthesis, Efficient Gas Storage, and Heterogeneous Catalysis. ACS Appl. Mater. 2017, 9, 38390–38400. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Li, Z.; Zhang, F.; Zhuang, X.; Zeng, Z.; Wei, J. New nitrogen-rich azo-bridged porphyrin-conjugated microporous networks for high performance of gas capture and storage. RSC Adv. 2016, 6, 30048–30055. [Google Scholar] [CrossRef]
- Sun, C.J.; Zhao, X.Q.; Wang, P.F.; Wang, H.; Han, B.H. Thiophene-based conjugated microporous polymers: Synthesis, characterization and efficient gas storage. Sci. China Chem. 2017, 60, 1067–1074. [Google Scholar] [CrossRef]
- Ben, T.; Ren, H.; Ma, S.; Cao, D.; Lan, J.; Jing, X.; Wang, W.; Xu, J.; Deng, F.; Simmons, J.M.; et al. Targeted Synthesis of a Porous Aromatic Framework with High Stability and Exceptionally High Surface Area. Angew. Chem. Int. Ed. 2009, 48, 9457–9460. [Google Scholar] [CrossRef]
- Lu, W.; Yuan, D.; Zhao, D.; Schilling, C.I.; Plietzsch, O.; Muller, T.; Bräse, S.; Guenther, J.; Blümel, J.; Krishna, R.; et al. Porous Polymer Networks: Synthesis, Porosity, and Applications in Gas Storage/Separation. Chem. Mater. 2010, 22, 5964–5972. [Google Scholar] [CrossRef]
- Yuan, D.; Lu, W.; Zhao, D.; Zhou, H.-C. Highly Stable Porous Polymer Networks with Exceptionally High Gas-uptake Capacities. Adv. Mater. 2011, 23, 3723–3725. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Wang, R.; Yang, H.; Wang, W.; Cai, W.; Li, Q. Ultrahigh hydrogen storage capacity of novel porous aromatic frameworks. J. Mater. Chem. A 2015, 3, 10724–10729. [Google Scholar] [CrossRef]
- Huang, L.; Yang, X.; Cao, D. From inorganic to organic strategy to design porous aromatic frameworks for high-capacity gas storage. J. Phys. Chem. C 2015, 119, 3260–3267. [Google Scholar] [CrossRef]
- Konstas, K.; Taylor, J.W.; Thornton, A.W.; Doherty, C.M.; Lim, W.X.; Bastow, T.J.; Kennedy, D.F.; Wood, C.D.; Cox, B.J.; Hill, J.M.; et al. Lithiated Porous Aromatic Frameworks with Exceptional Gas Storage Capacity. Angew. Chem. 2012, 124, 6743–6746. [Google Scholar] [CrossRef]
- Ma, H.; Ren, H.; Zou, X.; Sun, F.; Yan, Z.; Cai, K.; Wang, D.; Zhu, G. Novel Lithium-Loaded Porous Aromatic Framework for Efficient CO2 and H2 Uptake. J. Mater. Chem. A 2013, 1, 752–758. [Google Scholar] [CrossRef]
- Ahmed, A.; Thornton, A.W.; Konstas, K.; Kannam, S.K.; Babarao, R.; Todd, B.D.; Hill, A.J.; Hill, M.R. Strategies toward Enhanced Low-Pressure Volumetric Hydrogen Storage in Nanoporous Cryoadsorbents. Langmuir 2013, 29, 15689–15697. [Google Scholar] [CrossRef]
- Wu, X.; Li, L.; Peng, L.; Wang, Y.; Cai, W. Effect of Coordinatively Unsaturated Metal Sites in Porous Aromatic Frameworks on Hydrogen Storage Capacity. Acta Phys. Chim. Sinca 2017, 34, 286–295. [Google Scholar]
- Demirocak, D.E.; Ram, M.K.; Srinivasan, S.S.; Goswami, D.Y.; Stefanakos, E.K. A Novel Nitrogen Rich Porous Aromatic Framework for Hydrogen and Carbon Dioxide Storage. J. Mater. Chem. A 2013, 1, 13800–13806. [Google Scholar] [CrossRef]
- Rochat, S.; Polak-Kraśna, K.; Tian, M.; Holyfield, L.T.; Mays, T.J.; Bowen, C.R.; Burrows, A.D. Hydrogen storage in polymer-based processable microporous composites. J. Mater. Chem. A 2017, 5, 18752–18761. [Google Scholar] [CrossRef]
- Lyu, H.; Zhang, Q.; Wang, Y.; Duan, J. Unified Meso-Pores and Dense Cu2+ Sites in Porous Coordination Polymers for Highly Efficient Gas Storage and Separation. Dalton Trans. 2018, 47, 4424–4427. [Google Scholar] [CrossRef]
- Kovalev, A.I.; Mart’yanova, E.S.; Khotina, I.A.; Klemenkova, Z.S.; Blinnikova, Z.K.; Volchkova, E.V.; Loginova, T.P.; Ponomarev, I.I. Polyphenylene Gels. Polym. Sci. Ser. B 2018, 60, 675–679. [Google Scholar] [CrossRef]
- Ahmed, S.D.; El-Hiti, A.G.; Yousif, E.; Hameed, S.A.; Abdalla, M. New Eco-Friendly Phosphorus Organic Polymers as Gas Storage Media. Polymers 2017, 9, 336. [Google Scholar] [CrossRef]
- Sing, K.S.W.; Williams, R.T. Physisorption Hysteresis Loops and the Characterization of Nanoporous Materials. Adsorpt. Sci. Technol. 2004, 22, 773–782. [Google Scholar] [CrossRef]
- Rouquerol, J.; Avnir, D.; Fairbridge, C.W.; Everett, D.H.; Haynes, J.H.; Pernicone, N.; Ramsay, J.D.F.; Sing, K.S.W.; Unger, K.K. Recommendations for the Characterization of Porous Solids. Pure Appl. Chem. 1994, 66, 1739–1758. [Google Scholar] [CrossRef]
- Horváth, G.; Kawazoe, K. Method for the Calculation of Effective Pore Size Distribution in Molecular Sieve Carbon. J. Chem. Eng. Jpn. 1983, 16, 470–475. [Google Scholar] [CrossRef]
- Cheng, L.S. Improved Horvath—Kawazoe Equations Including Spherical Pore Models for Calculating Micropore Size Distribution. Chem. Eng. Sci. 1994, 49, 2599–2609. [Google Scholar] [CrossRef]
- Dubinin, M.M. Fundamentals of the Theory of Adsorption in Micropores of Carbon Adsorbents: Characteristics of Their Adsorption Properties and Microporous Structures. Carbon 1989, 27, 457–467. [Google Scholar] [CrossRef]
- Dubinin, M.M. Chemistry and Physics of Carbon; Walker, L.P., Ed.; Marcel Dekker: New York, NY, USA, 1966; Volume 2, p. 51. [Google Scholar]
- Tarazona, P. Solid-Fluid Transition and Interfaces with Density Functional Approaches. Surf. Sci. 1995, 331, 989–994. [Google Scholar] [CrossRef]
- Zhechkov, L.; Heine, T.; Patchkovskii, S.; Seifert, G.; Duarte, H.A. An Efficient a Posteriori Treatment for Dispersion Interaction in Density-Functional-Based Tight Binding. J. Chem. Theory Comput. 2005, 1, 841–847. [Google Scholar] [CrossRef] [PubMed]
- Kolmogorov, A.N.; Crespi, V.H. Registry-Dependent Interlayer Potential for Graphitic Systems. Phys. Rev. B 2005, 71, 235415. [Google Scholar] [CrossRef]
- Breck, D. Zeolite Molecular Sieves: Structure, Chemistry, and Use; Krieger Pub Co.: Malabar, FL, USA, 1984. [Google Scholar]
- Lozano-Castelló, D.; Cazorla-Amoros, D.; Linares-Solano, A. Usefulness of CO2 Adsorption at 273 K for the Characterization of Porous Carbons. Carbon 2004, 42, 1233–1242. [Google Scholar] [CrossRef]
- Zhang, C.; Babonneau, F.; Bonhomme, C.; Laine, R.M.C.; Soles, L.; Hristov, H.A.; Yee, A.F. Highly Porous Polyhedral Silsesquioxane Polymers. Synthesis and Characterization. J. Am. Chem. Soc. 1998, 120, 8380–8391. [Google Scholar] [CrossRef]
- Zhang, R.; Phalen, R.N.; Cataquis, A.; Desta, M.; Kloesel, M. Study of Highly Porous Polymers for H2 Fuel Storage Using Positron Annihilation Lifetime Spectroscopy. Int. J. Hydrogen Energy 2015, 40, 8732–8741. [Google Scholar] [CrossRef]
- Jean, Y.C. Positron and Positronium Chemistry; World Scientific: River Edge, NJ, USA, 1990. [Google Scholar]
- Urban, C.; McCord, E.F.; Webster, O.W.; Abrams, L.; Long, H.W.; Gaede, H.; Tang, P.; Pines, A. 129Xe NMR Studies of Hyper-Cross-Linked Polyarylcarbinols: Rigid versus Flexible Structures. Chem. Matter 1995, 7, 1325–1332. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, Y.; Li, B.; Tan, B.; Chen, C.F.; Xu, H.B.; Yang, X.L. Triptycene-Based Microporous Polymers: Synthesis and Their Gas Storage Properties. ACS Macro Lett. 2011, 1, 190–193. [Google Scholar] [CrossRef]
- Guo, J.H.; Zhang, H.; Tang, Y.; Cheng, X. Hydrogen Spillover Mechanism on Covalent Organic Frameworks as Investigated by Ab Initio Density Functional Calculation. Phys. Chem. Chem. Phys. 2013, 15, 2873–2881. [Google Scholar] [CrossRef]
- Lachawiec, A.J., Jr.; Yang, R.T. Isotope tracer study of hydrogen spillover on carbon-based adsorbents for hydrogen storage. Langmuir 2008, 24, 6159–6165. [Google Scholar] [CrossRef]
- Suri, M.; Dornfeld, M.; Ganz, E. Calculation of hydrogen storage capacity of metal-organic and covalent-organic frameworks by spillover. J. Chem. Phys. 2009, 131, 174703. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.T.; Wang, Y. Catalyzed hydrogen spillover for hydrogen storage. J. Am. Chem. Soc. 2009, 131, 4224–4226. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Gennett, T. Water-mediated cooperative migration of chemisorbed hydrogen on graphene. Phys. Rev. Lett. 2014, 112, 076101. [Google Scholar] [CrossRef] [PubMed]
- Ghimbeu, C.M.; Zlotea, C.; Gadiou, R.; Cuevas, F.; Leroy, E.; Latroche, M.; Vix-Guterl, C. Understanding the mechanism of hydrogen uptake at low pressure in carbon/palladium nanostructured composites. J. Mater. Chem. 2011, 21, 17765–17775. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year | 2020 | 2025 | Ultimate | |
|---|---|---|---|---|
| Target | ||||
| Gravimetric capacity (wt %) | 4.5% | 5.5% | 6.5% | |
| Volumetric capacity (g/L) | 30 | 40 | 50 | |
| Cost ($/kg H2) | 333 | 300 | 266 | |
Durability/Operability:
| −40/60 −40/85 1500 5/12 90% | −40/60 −40/85 1500 5/12 90% | −40/60 −40/85 1500 5/12 90% | |
Charging/Discharging rate:
| 3–5 0.02 0.004 5 15 0.75 | 3–5 0.02 0.004 5 15 0.75 | 3–5 0.02 0.004 5 15 0.75 | |
| Interaction type (Material – H2) | Energy dependence | Typical values (kJ/mol) |
|---|---|---|
| Charge – H2 quadropole | ∝ 1/r 3 | ~ 3.5 |
| Charge – induced H2 dipole | ∝ 1/r 4 | ~ 6.8 |
| Dipole – induced H2 dipole | ∝ 1/r 5 | ~ 0.6 |
| van der Waals | ∝ 1/r 6 | ~ 5 – 6 |
| Orbital interaction | <vdW radii | ~ 20 – 160 |
| Monomer | Crosslinker | SSA (m2/g) | H2 Uptake | Ref. |
|---|---|---|---|---|
| VBC-DVB | VBC | 1300–1900 | 3 wt % 77 K/15 bar, 1.5 wt % 77 K/1 bar | [52,53] |
| VCB | DVB | 1500 | 1.59 wt % 77 K/1.13 bar | [65] |
| Carbazole bromophenyl- methanol | 1,10-Phenantroline | 1000 | 2.39 wt % 77 K/1 bar | [66] |
| Carbazoles | FDA | 1000–1800 | 1.94 wt % 77 K/1 bar | [67] |
| Carbazoles | DCX | 600–1900 | 0.9–17.7 wt % 77 K/1 bar | [59] |
| Polyanilines | CH2I2 | ~500 | 2.2 wt % 77 K/30 bar | [57] |
| Polypyrroles | CH2I2 | ~20–700 | 0.6–1.6 wt % 77 K/4 bar | [58] |
| Bismaleimides | DVB | 841 | 0.82 wt % 77 K/1 bar | [68] |
| TPB | Cl | 1783 | 1.91 wt % 77 K/1 bar | [69] |
| TPB | FDA | 1059 | 1.58 wt % 77 K/1.13 bar | [70] |
| Benzene | FDA | 1391 | 1.45 wt % 77 K/1.13 bar | [70] |
| Fluorene | BCMBP | 1700 | 1.63 wt % 77 K/1 bar | [71] |
| TPE | FDA | 1980 | 1.76 wt % 77 K/1 bar | [72] |
| CMPs | SBET (m2/g) | Pore diamter (nm) | Micropore volume (cm3/g) | H2 uptake |
|---|---|---|---|---|
| POP-1 | 1031 | 0.77 | 0.378 | 2.78 wt % 77K/60bar |
| POP-2 | 1013 | 0.74 | 0.341 | 2.71 wt % 77K/60bar |
| POP-3 | 1246 | 0.88 | 0.448 | 3.07 wt % 77K/60bar |
| POP-4 | 1033 | 0.81 | 0.402 | 2.35 wt % 77K/60bar |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cousins, K.; Zhang, R. Highly Porous Organic Polymers for Hydrogen Fuel Storage. Polymers 2019, 11, 690. https://doi.org/10.3390/polym11040690
Cousins K, Zhang R. Highly Porous Organic Polymers for Hydrogen Fuel Storage. Polymers. 2019; 11(4):690. https://doi.org/10.3390/polym11040690
Chicago/Turabian StyleCousins, Kimberley, and Renwu Zhang. 2019. "Highly Porous Organic Polymers for Hydrogen Fuel Storage" Polymers 11, no. 4: 690. https://doi.org/10.3390/polym11040690
APA StyleCousins, K., & Zhang, R. (2019). Highly Porous Organic Polymers for Hydrogen Fuel Storage. Polymers, 11(4), 690. https://doi.org/10.3390/polym11040690