End Group Stability of Atom Transfer Radical Polymerization (ATRP)-Synthesized Poly(N-isopropylacrylamide): Perspectives for Diblock Copolymer Synthesis



Abstract

1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Characterization
2.3. AtomTransfer Radical Polymerization (ATRP) of N-isopropylacrylamide (NIPAAm)
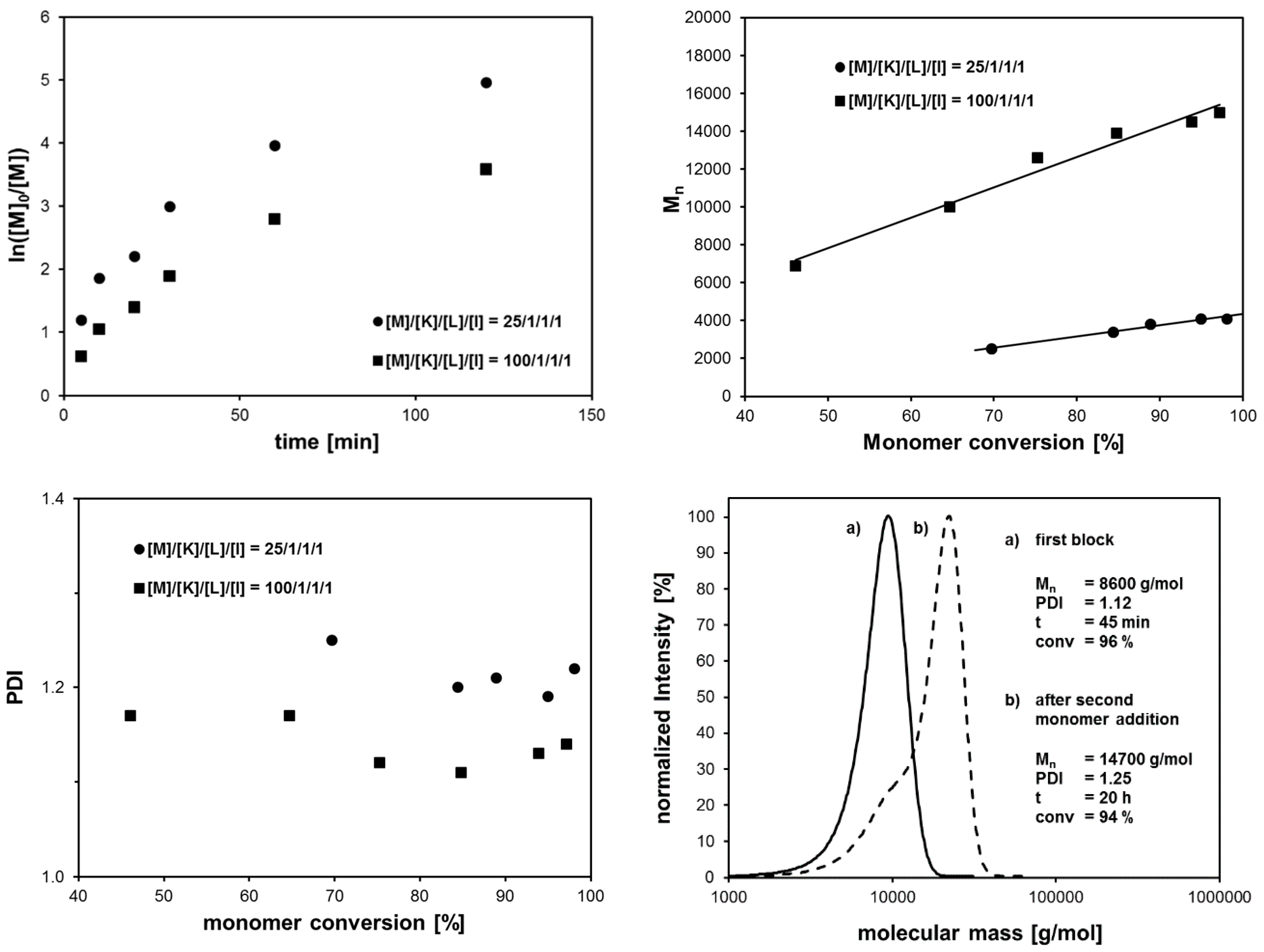
2.4. Blocking Experiments via Sequential Monomer Addition
2.5. Synthesis of Poly(solketal acrylate) (PSKA) Macroinitiators
2.6. Synthesis of Poly(solketal acrylate-b-N-isopropylacrylamide) Block Copolymers
2.7. Synthesis of Poly(2,3-dihydroxypropyl acrylate-b-N-isopropylacrylamide) Block Copolymers
3. Discussion
3.1. End Group Stability
3.2. Perspectives for the Synthesis of Diblock Copolymers with a PNIPAAm Block Using ATRP
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Wang, J.-S.; Matyjaszewski, K. Controlled/“living” radical polymerization. atom transfer radical polymerization in the presence of transition-metal complexes. J. Am. Chem. Soc. 1995, 117, 5614–5615. [Google Scholar] [CrossRef]
- Kato, M.; Kamigaito, M.; Sawamoto, M.; Higashimura, T. Polymerization of Methyl Methacrylate with the Carbon Tetrachloride/Dichlorotris-(triphenylphosphine)ruthenium(II)/Methylaluminum Bis(2,6-di-tert-butylphenoxide) Initiating System: Possibility of Living Radical Polymerization. Macromolecules 1995, 28, 1721–1723. [Google Scholar] [CrossRef]
- Barner, L.; Davis, T.P.; Stenzel, M.H.; Barner-Kowollik, C. Complex Macromolecular Architectures by Reversible Addition Fragmentation Chain Transfer Chemistry: Theory and Practice. Macromol. Rapid Commun. 2007, 28, 539–559. [Google Scholar] [CrossRef]
- Matyjaszewski, K.; Spanswick, J. Controlled/living radical polymerization. Mater. Today 2005, 8, 26–33. [Google Scholar] [CrossRef]
- Braunecker, W.A.; Matyjaszewski, K. Controlled/living radical polymerization: Features, developments, and perspectives. Progr. Polym. Sci. 2007, 32, 93–146. [Google Scholar] [CrossRef]
- Wolf, F.F.; Friedemann, N.; Frey, H. Poly(lactide)-block-Poly(HEMA) Block Copolymers: An Orthogonal One-Pot Combination of ROP and ATRP, Using a Bifunctional Initiator. Macromolecules 2009, 42, 5622–5628. [Google Scholar] [CrossRef]
- Spasojević, M.; Vorenkamp, J.; Jansen, M.; de Vos, P.; Schouten, A.J. Synthesis and Phase Behavior of Poly(N-isopropylacrylamide)-b-Poly(l-Lysine Hydrochloride) and Poly(N-Isopropylacrylamide-co-Acrylamide)-b-Poly(l-Lysine Hydrochloride). Materials 2014, 7, 5305–5326. [Google Scholar] [CrossRef]
- Kipping, M.; Krahl, F.; Döring, A.; Adler, H.-J.P.; Kuckling, D. Synthesis and characterization of particles consisting of a biodegradable poly(l-lactide) core and a functional hydrophilic shell. Eur. Polym. J. 2010, 46, 313–323. [Google Scholar] [CrossRef]
- Yu, Y.C.; Cho, H.S.; Yu, W.-R.; Youk, J.H. One-step synthesis of poly(2-oxazoline)-based amphiphilic block copolymers using a dual initiator for RAFT polymerization and CROP. Polymer 2014, 55, 5986–5990. [Google Scholar] [CrossRef]
- Berges, C.; Javakhishvili, I.; Hvilsted, S.; Sánchez, C.; Alcalá, R. Photoresponsive Azopolyester-PMMA Block Copolymers Obtained by Combination of ATRP, Polycondensation, and “Click” Chemistry. Macromol. Chem. Phys. 2012, 213, 2299–2310. [Google Scholar] [CrossRef]
- Siegwart, D.J.; Oh, J.K.; Matyjaszewski, K. ATRP in the design of functional materials for biomedical applications. Progr. Polym. Sci. 2012, 37, 18–37. [Google Scholar] [CrossRef] [PubMed]
- Ran, J.; Wu, L.; Zhang, Z.; Xu, T. Atom transfer radical polymerization (ATRP): A versatile and forceful tool for functional membranes. Progr. Polym. Sci. 2014, 39, 124–144. [Google Scholar] [CrossRef]
- Hui, C.M.; Pietrasik, J.; Schmitt, M.; Mahoney, C.; Choi, J.; Bockstaller, M.R.; Matyjaszewski, K. Surface-Initiated Polymerization as an Enabling Tool for Multifunctional (Nano-)Engineered Hybrid Materials. Chem. Mater. 2014, 26, 745–762. [Google Scholar] [CrossRef]
- Neugebauer, D. Two decades of molecular brushes by ATRP. Polymer 2015, 72, 413–421. [Google Scholar] [CrossRef]
- Wycisk, A.; Döring, A.; Schneider, M.; Schönhoff, M.; Kuckling, D. Synthesis of β-cyclodextrin-Based Star Block Copolymers with Thermo-Responsive Behavior. Polymers 2015, 7, 921–938. [Google Scholar] [CrossRef]
- Charles, L. MALDI of synthetic polymers with labile end-groups. Mass Spectr. Rev. 2014, 33, 523–543. [Google Scholar] [CrossRef] [PubMed]
- Aseyev, V.; Tenhu, H.; Winnik, F.M. Non-ionic Thermoresponsive Polymers in Water. In Self Organized Nanostructures of Amphiphilic Block Copolymers; Müller, A.H.E., Borisov, O., Ballauff, M., Aseyev, V., Eds.; Springer: Berlin, Germany; London, UK, 2011; pp. 29–89. ISBN 978-3-642-22296-2. [Google Scholar]
- Döring, A.; Birnbaum, W.; Kuckling, D. Responsive hydrogels—Structurally and dimensionally optimized smart frameworks for applications in catalysis, micro-system technology and material science. Chem. Soc. Rev. 2013, 42, 7391–7420. [Google Scholar] [CrossRef]
- Kuckling, D.; Doering, A.; Krahl, F.; Arndt, K.-F. Stimuli-Responsive Polymer Systems. In Polymer Science: A Comprehensive Reference; Matyjaszewski, K., Möller, M., Eds.; Elsevier: Amsterdam, The Netherlands, 2012; pp. 377–413. ISBN 9780080878621. [Google Scholar]
- Rademacher, J.T.; Baum, M.; Pallack, M.E.; Brittain, W.J.; Simonsick, W.J. Atom Transfer Radical Polymerization of N,N-Dimethylacrylamide. Macromolecules 2000, 33, 284–288. [Google Scholar] [CrossRef]
- Masci, G.; Giacomelli, L.; Crescenzi, V. Atom Transfer Radical Polymerization of N-Isopropylacrylamide. Macromol. Rapid Commun. 2004, 25, 559–564. [Google Scholar] [CrossRef]
- Bontempo, D.; Li, R.C.; Ly, T.; Brubaker, C.E.; Maynard, H.D. One-step synthesis of low polydispersity, biotinylated poly(N-isopropylacrylamide) by ATRP. Chem. Commun. 2005, 4702–4704. [Google Scholar] [CrossRef]
- Millard, P.-E.; Mougin, N.C.; Böker, A.; Müller, A.H.E. Controlling the Fast ATRP of N-Isopropylacrylamide in Water. In Controlled: Progress in ATRP; Matyjaszewski, K., Ed.; American Chemical Society: Washington DC, USA, 2009; pp. 127–137. ISBN 0-8412-6995-5. [Google Scholar]
- Xia, Y.; Yin, X.; Burke, N.A.D.; Stöver, H.D.H. Thermal Response of Narrow-Disperse Poly(N-isopropylacrylamide) Prepared by Atom Transfer Radical Polymerization. Macromolecules 2005, 38, 5937–5943. [Google Scholar] [CrossRef]
- Fischer, H. The Persistent Radical Effect: A Principle for Selective Radical Reactions and Living Radical Polymerizations. Chem. Rev. 2001, 101, 3581–3610. [Google Scholar] [CrossRef]
- Percec, V.; Guliashvili, T.; Ladislaw, J.S.; Wistrand, A.; Stjerndahl, A.; Sienkowska, M.J.; Monteiro, M.J.; Sahoo, S. Ultrafast synthesis of ultrahigh molar mass polymers by metal-catalyzed living radical polymerization of acrylates, methacrylates, and vinyl chloride mediated by SET at 25 degrees C. J. Am. Chem. Soc. 2006, 128, 14156–14165. [Google Scholar] [CrossRef] [PubMed]
- Alsubaie, F.; Anastasaki, A.; Wilson, P.; Haddleton, D.M. Sequence-controlled multi-block copolymerization of acrylamides via aqueous SET-LRP at 0 °C. Polym. Chem. 2015, 6, 406–417. [Google Scholar] [CrossRef]
- Matyjaszewski, K.; Tsarevsky, N.V.; Braunecker, W.A.; Dong, H.; Huang, J.; Jakubowski, W.; Kwak, Y.; Nicolay, R.; Tang, W.; Yoon, J.A. Role of Cu 0 in Controlled/“Living” Radical Polymerization. Macromolecules 2007, 40, 7795–7806. [Google Scholar] [CrossRef]
- Birnbaum, W.; Kuckling, D. Synthesis of α-biotinyl poly(ethylene glycol-b-N-isopropylacrylamide) block copolymers with different fluorescent dyes at the ω-side. Polym. Chem. 2012, 3, 2039. [Google Scholar] [CrossRef]
- D’hooge, D.R.; Vachaudez, M.; Stadler, F.J.; Reyniers, M.-F.; Coulembier, O.; Bailly, C.; Dubois, P.; Marin, G.B. Assessment of end-group functionality in atom transfer radical polymerization of N-isopropylacrylamide. Eur. Polym. J. 2013, 49, 2344–2355. [Google Scholar] [CrossRef]
- Ashaduzzaman, M.; Kai, S.; Uemura, S.; Kunitake, M. Synthesis and Aqueous Dispersion Properties of Triarm Poly(NIPAAm-b-HEAAm) Diblock Copolymers. Chem. Lett. 2011, 40, 165–167. [Google Scholar] [CrossRef]
- Couthouis, J.; Keul, H.; Möller, M. MALDI-TOF Analysis of Halogen Telechelic Poly(methyl methacrylate)s and Poly(methyl acrylate)s Prepared by Atom Transfer Radical Polymerization (ATRP) or Single Electron Transfer-Living Radical Polymerization (SET-LRP). Macromol. Chem. Phys. 2015, 216, 1791–1800. [Google Scholar] [CrossRef]
- Altintas, O.; Josse, T.; de Winter, J.; Matsumoto, N.M.; Gerbaux, P.; Wilhelm, M.; Barner-Kowollik, C. Ready access to end-functional polystyrenes via a combination of ARGET ATRP and thiol–ene chemistry. Polym. Chem. 2015, 6, 6931–6935. [Google Scholar] [CrossRef]
- Altintas, O.; Josse, T.; Abbasi, M.; de Winter, J.; Trouillet, V.; Gerbaux, P.; Wilhelm, M.; Barner-Kowollik, C. ATRP-based polymers with modular ligation points under thermal and thermomechanical stress. Polym. Chem. 2015, 6, 2854–2868. [Google Scholar] [CrossRef]
- Cai-Yuan, P.; Lei, T.; De-Cheng, W. Synthesis and characterizations of the four-armed amphiphilic block copolymer S[poly(2,3-dihydroxypropyl acrylate)-block-poly(methyl acrylate)]4. J. Polym. Sci. A Polym. Chem. 2001, 39, 3062–3072. [Google Scholar] [CrossRef]
- Ciampolini, M.; Nardi, N. Five-Coordinated High-Spin Complexes of Bivalent Cobalt, Nickel, and Copper with Tris(2-dimethylaminoethyl)amine. Inorg. Chem. 1966, 5, 41–44. [Google Scholar] [CrossRef]
- Ganachaud, F.; Monteiro, M.J.; Gilbert, R.G.; Dourges, M.-A.; Thang, S.H.; Rizzardo, E. Molecular Weight Characterization of Poly(N-isopropylacrylamide) Prepared by Living Free-Radical Polymerization. Macromolecules 2000, 33, 6738–6745. [Google Scholar] [CrossRef]
- Guillaneuf, Y.; Castignolles, P. Using apparent molecular weight from SEC in controlled/living polymerization and kinetics of polymerization. J. Polym. Sci. A Polym. Chem. 2008, 46, 897–911. [Google Scholar] [CrossRef]
- Montaudo, G.; Lattimer, R. Mass Spectrometry of Polymers; CRC Press: Boca Raton, FL, USA, 2002; ISBN 9780849331275. [Google Scholar]
- Gruendling, T.; Weidner, S.; Falkenhagen, J.; Barner-Kowollik, C. Mass spectrometry in polymer chemistry: A state-of-the-art up-date. Polym. Chem. 2010, 1, 599. [Google Scholar] [CrossRef]
- Xu, J.; Liu, S. Synthesis of well-defined 7-arm and 21-arm poly(N-isopropylacrylamide) star polymers with β-cyclodextrin cores via click chemistry and their thermal phase transition behavior in aqueous solution. J. Polym. Sci. A Polym. Chem. 2009, 47, 404–419. [Google Scholar] [CrossRef]
- Liu, Z.; Liao, Q.; Yang, D.; Gao, Y.; Luo, X.; Lei, Z.; Li, H. Well-defined poly(N-isopropylacrylamide) with a bifunctional end-group: Synthesis, characterization, and thermoresponsive properties. Des. Monomers Polym. 2013, 16, 465–474. [Google Scholar] [CrossRef]
- Brandt, H.; Ehmann, T.; Otto, M. Toward prediction: Using chemometrics for the optimization of sample preparation in MALDI-TOF MS of synthetic polymers. Anal. Chem. 2010, 82, 8169–8175. [Google Scholar] [CrossRef]
- Byrd, H.C.M.; McEwen, C.N. The Limitations of MALDI-TOF Mass Spectrometry in the Analysis of Wide Polydisperse Polymers. Anal. Chem. 2000, 72, 4568–4576. [Google Scholar] [CrossRef]
- Wetzel, S.J.; Guttman, C.M.; Girard, J.E. The influence of matrix and laser energy on the molecular mass distribution of synthetic polymers obtained by MALDI-TOF-MS. Int. J. Mass Spectrom. 2004, 238, 215–225. [Google Scholar] [CrossRef]
- Ladavière, C.; Lacroix-Desmazes, P.; Delolme, F. First Systematic MALDI/ESI Mass Spectrometry Comparison to Characterize Polystyrene Synthesized by Different Controlled Radical Polymerizations. Macromolecules 2009, 42, 70–84. [Google Scholar] [CrossRef]
- Song, J.; Grün, C.H.; Heeren, R.M.A.; Janssen, H.-G.; van den Brink, O.F. High-Resolution Ion Mobility Spectrometry-Mass Spectrometry on Poly(methyl methacrylate). Angew. Chem. 2010, 122, 10366–10369. [Google Scholar] [CrossRef]
- Schilli, C.; Lanzendörfer, M.G.; Müller, A.H.E. Benzyl and Cumyl Dithiocarbamates as Chain Transfer Agents in the RAFT Polymerization of N-Isopropylacrylamide. In Situ FT-NIR and MALDI−TOF MS Investigation. Macromolecules 2002, 35, 6819–6827. [Google Scholar] [CrossRef]
- Raffa, P.; Wever, D.A.Z.; Picchioni, F.; Broekhuis, A.A. Polymeric Surfactants: Synthesis, Properties, and Links to Applications. Chem. Rev. 2015, 115, 8504–8563. [Google Scholar] [CrossRef] [PubMed]
- Mai, Y.; Eisenberg, A. Self-assembly of block copolymers. Chem. Soc. Rev. 2012, 41, 5969–5985. [Google Scholar] [CrossRef]
- Lutz, J.-F. Solution self-assembly of tailor-made macromolecular building blocks prepared by controlled radical polymerization techniques. Polym. Int. 2006, 55, 979–993. [Google Scholar] [CrossRef]
- Hadjichristidis, N.; Pispas, S.; Floudas, G. Block Copolymers: Synthetic Strategies, Physical Properties, and Applications; Hadjichristidis, N., Pispas, S., Floudas, G., Eds.; Wiley: New York, NY, USA, 2003; ISBN 047139436X. [Google Scholar]
- Keddie, D.J. A guide to the synthesis of block copolymers using reversible-addition fragmentation chain transfer (RAFT) polymerization. Chem. Soc. Rev. 2014, 43, 496–505. [Google Scholar] [CrossRef]
- Binder, W.H.; Sachsenhofer, R. ‘Click’ Chemistry in Polymer and Materials Science. Macromol. Rapid Commun. 2007, 28, 15–54. [Google Scholar] [CrossRef]
- Lowe, A.B. Thiol–ene “click” reactions and recent applications in polymer and materials synthesis: A first update. Polym. Chem. 2014, 5, 4820–4870. [Google Scholar] [CrossRef]
- Sarkar, B.; Alexandridis, P. Block copolymer–nanoparticle composites: Structure, functional properties, and processing. Progr. Polym. Sci. 2015, 40, 33–62. [Google Scholar] [CrossRef]
- Ge, Z.; Liu, S. Functional block copolymer assemblies responsive to tumor and intracellular microenvironments for site-specific drug delivery and enhanced imaging performance. Chem. Soc. Rev. 2013, 42, 7289–7325. [Google Scholar] [CrossRef] [PubMed]
- Nakabayashi, K.; Mori, H. Donor-Acceptor Block Copolymers: Synthesis and Solar Cell Applications. Materials 2014, 7, 3274–3290. [Google Scholar] [CrossRef] [PubMed]
- Schattling, P.; Jochum, F.D.; Theato, P. Multi-stimuli responsive polymers—The all-in-one talents. Polym. Chem. 2014, 5, 25–36. [Google Scholar] [CrossRef]
- Hu, Y.; Darcos, V.; Monge, S.; Li, S. Synthesis and self-assembling of poly(N-isopropylacrylamide-block-poly(l-lactide)-block-poly(N-isopropylacrylamide) triblock copolymers prepared by combination of ring-opening polymerization and atom transfer radical polymerization. J. Polym. Sci. A Polym. Chem. 2013, 51, 3274–3283. [Google Scholar] [CrossRef]
- Glassman, M.J.; Olsen, B.D. Structure and Mechanical Response of Protein Hydrogels Reinforced by Block Copolymer Self-Assembly. Soft Matter 2013, 9, 6814–6823. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.J.; Kang, S.M.; Kong, B.; Kim, W.-J.; Paik, H.-j.; Choi, H.; Choi, I.S. Formation of Thermoresponsive Gold Nanoparticle/PNIPAAm Hybrids by Surface-Initiated, Atom Transfer Radical Polymerization in Aqueous Media. Macromol. Chem. Phys. 2005, 206, 1941–1946. [Google Scholar] [CrossRef]
- Kotsuchibashi, Y.; Kuboshima, Y.; Yamamoto, K.; Aoyagi, T. Synthesis and characterization of double thermo-responsive block copolymer consisting N-isopropylacrylamide by atom transfer radical polymerization. J. Polym. Sci. A Polym. Chem. 2008, 46, 6142–6150. [Google Scholar] [CrossRef]
- Huang, C.-J.; Chang, F.-C. Polypeptide Diblock Copolymers: Syntheses and Properties of Poly(N-isopropylacrylamide)-b-Polylysine. Macromolecules 2008, 41, 7041–7052. [Google Scholar] [CrossRef]
- Coullerez, G.; Carlmark, A.; Malmström, E.; Jonsson, M. Understanding Copper-Based Atom-Transfer Radical Polymerization in Aqueous Media. J. Phys. Chem. A 2004, 108, 7129–7131. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Solvent | Initiator | [M]/[I]/[CuI]/[CuII]/Me6Tren |
|---|---|---|---|
| A | Water/DMF (v/v 1:1) | MCP | 50/1/1/-/1 |
| B | DMSO | MCP | 50/1/1.6/0.4/2 |
| C | ACN | MCP | 50/1/1/-/1 |
| Method | Aspired Monomer Conversion at the Point of Termination | Termination | Solvent for Polymer Work-Up |
|---|---|---|---|
| 1 | 100% | freezing/O2 | THF |
| 2 | 100% | freezing/O2 | chloroform |
| 3 | 50% a | freezing/O2 | THF |
| 4 | 100% | addition of CuCl2 | THF |
| Sample a | Reaction Time [h] | Monomer Conversion b [%] | Mn,SEC c [g/mol] | PDI c |
|---|---|---|---|---|
| A1 | 0.75 | 91 | 7910 | 1.13 |
| A2 | 0.75 | 94 | 8750 | 1.17 |
| A3 | 0.08 | 52 | 13,490 | 1.14 |
| A4 | 0.75 | 93 | 8750 | 1.15 |
| B1 | 16 | 70 | 10,060 | 1.11 |
| B2 | 16 | 69 | 8870 | 1.11 |
| B3 | 6 | 54 | 8740 | 1.11 |
| B4 | 16 | 74 | 9750 | 1.12 |
| C1 | 16 | 38 | 2670 | 1.19 |
| C2 | 16 | 53 | 7900 | 1.13 |
| C3 | 16 | 50 | 8040 | 1.14 |
| C4 | 16 | 44 | 4190 | 1.17 |
| END Groups of the PNIPAAm Chain (Main Series) | |||||
|---|---|---|---|---|---|
| Method | 1 | 2 | 3 | 4 | |
| System | |||||
| A | α 2-MP ω1 olefin ω2 pyrrolidinone | α 2-MP ω pyrrolidinone | α n.a. a ω n.a. a | α 2-MP ω1 olefin ω2 pyrrolidinone | |
| B | α 2-MP ω olefin | α 2-MP ω1 olefin ω2 pyrrolidinone | α 2-MP ω1 olefin ω2 pyrrolidinone | α 2-MP ω1 olefin ω2 pyrrolidinone | |
| C | α 2-MP ω1 olefin ω2 pyrrolidinone | α 2-MP ω olefin | α 2-MP ω olefin | α 2-MP ω olefin | |
| Sample | Solvent | [M]/[C]/[L]/[I] | θ | Time | Conv. | Mn,SEC a | PDI a |
|---|---|---|---|---|---|---|---|
| [°C] | [h] | [%] | [g/moL] | ||||
| PN17 | iPrOH/H2O (v/v 5:1) | 50/1/1/1 | 0 | 19 | 99 | 8200 | 1.11 |
| PN18 | iPrOH/H2O (v/v 5:1) | 100/1/1/1 | 0 | 22 | 91 | 12,400 | 1.13 |
| PN19 | iPrOH/H2O (v/v 5:1) | 50/1/1/1 | 30 | 21 | 70 | 7300 | 1.20 |
| Sample | ATRP Reaction Mixture | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| PNIPAAm | PNIPAAm-b-PDMAAm | ||||||||
| [M1]/[C]/[L]/[I1] | t [h] | Conv. (NIPAAm) a [%] | [M2]/[C]/[L]/[I2] | t [h] | Conv. (NIPAAm) a [%] | Conv. (DMAAm) a [%] | Mn,GPC b [g/moL] | PDI b | |
| BC1 | 50/1/1/1 | 1 | 98.2 | 50/1/1/1 | 15 | 98.7 | 68.3 | 13,100 | 1.21 |
| BC2 | 50/1/1/1 | 1 | 98.0 | 50/2/2/1 | 18 | 98.9 | 82.2 | 12,900 | 1.26 |
| BC3 | 50/1/1/1 | 1 | 96.5 | 100/2/2/1 | 18 | 97.7 | 73.8 | 16,100 | 1.39 |
| BC4 | 25/1/1/1 | 0.5 | 95.0 | 100/2/2/2 | 18 | 96.6 | 71.3 | 14,900 | 1.47 |
| Sample | ATRP Reaction Mixture | PNIPAAm-b-PDMAAm Block Copolymers after Centrifugation | ||||
|---|---|---|---|---|---|---|
| Targeted Absolute Block Length Ration | X a,b | m:n a (NMR, abs.) | Mn,NMR a [g/mol] | Mn,GPC c [g/mol] | PDI c | |
| BC1 | 50:50 | 0.25 | 44:47 | 9700 | 13,400 | 1.21 |
| BC2 | 50:50 | 0.45 | 43:54 | 10,200 | 13,900 | 1.24 |
| BC3 | 50:100 | 0.60 | 47:106 | 15,800 | 17,600 | 1.35 |
| BC4 | 25:100 | 0.40 | 24:106 | 13,200 | 16,500 | 1.42 |
| Sample | PSKA | |||
|---|---|---|---|---|
| [M]/[C]/[L]/[I] | Conv. a | Mn,SEC b | PDI b | |
| [%] | [g/moL] | |||
| MI1 | 50/1/1/1 | 89 | 6100 | 1.16 |
| Sample | m:n a | Mn,SEC b | PDI b |
|---|---|---|---|
| (rel.) | [g/moL] | ||
| BC5 | 1.0:2.3 | 20,700 | 1.28 |
| BC6 | 1.0:1.2 | 23,900 | 1.24 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herberg, A.; Yu, X.; Kuckling, D. End Group Stability of Atom Transfer Radical Polymerization (ATRP)-Synthesized Poly(N-isopropylacrylamide): Perspectives for Diblock Copolymer Synthesis. Polymers 2019, 11, 678. https://doi.org/10.3390/polym11040678
Herberg A, Yu X, Kuckling D. End Group Stability of Atom Transfer Radical Polymerization (ATRP)-Synthesized Poly(N-isopropylacrylamide): Perspectives for Diblock Copolymer Synthesis. Polymers. 2019; 11(4):678. https://doi.org/10.3390/polym11040678
Chicago/Turabian StyleHerberg, Artjom, Xiaoqian Yu, and Dirk Kuckling. 2019. "End Group Stability of Atom Transfer Radical Polymerization (ATRP)-Synthesized Poly(N-isopropylacrylamide): Perspectives for Diblock Copolymer Synthesis" Polymers 11, no. 4: 678. https://doi.org/10.3390/polym11040678
APA StyleHerberg, A., Yu, X., & Kuckling, D. (2019). End Group Stability of Atom Transfer Radical Polymerization (ATRP)-Synthesized Poly(N-isopropylacrylamide): Perspectives for Diblock Copolymer Synthesis. Polymers, 11(4), 678. https://doi.org/10.3390/polym11040678