Ethylene-co-norbornene Copolymerization Using a Dual Catalyst System in the Presence of a Chain Transfer Agent

 ,
, 
 ,
, 

Abstract
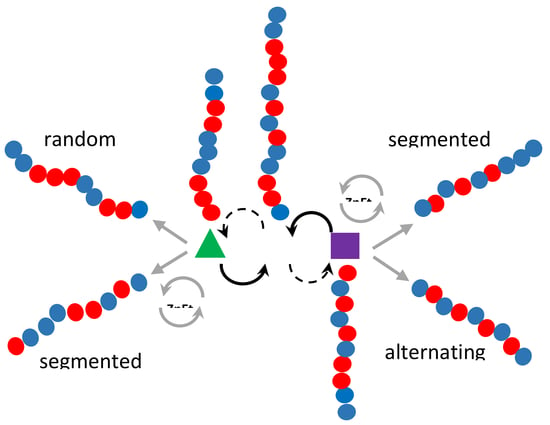
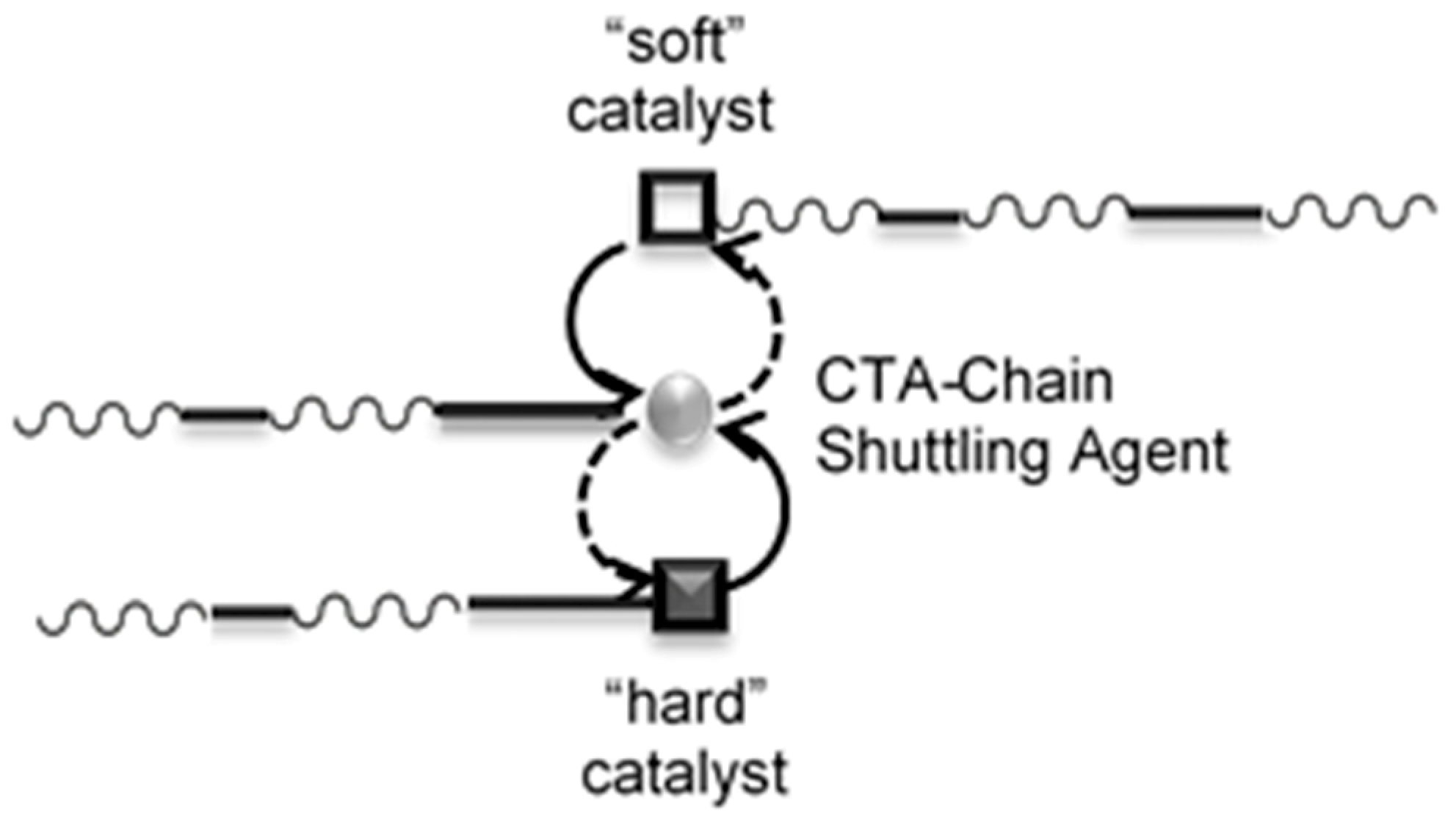
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Analytical Measurements
2.3. Polymerization Reaction
3. Results
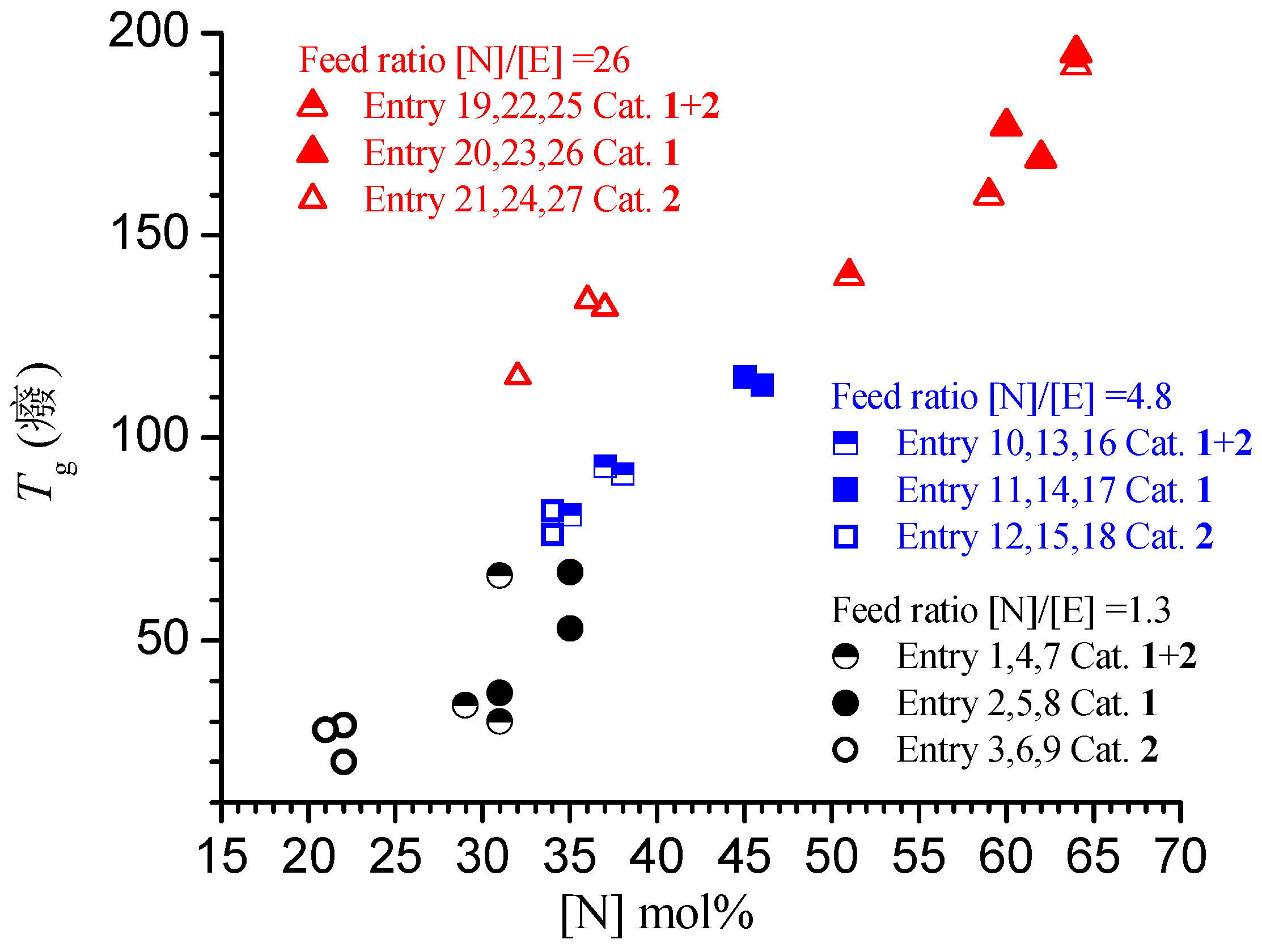
3.1. Synthesis and Characterization of Poly(E-co-N)
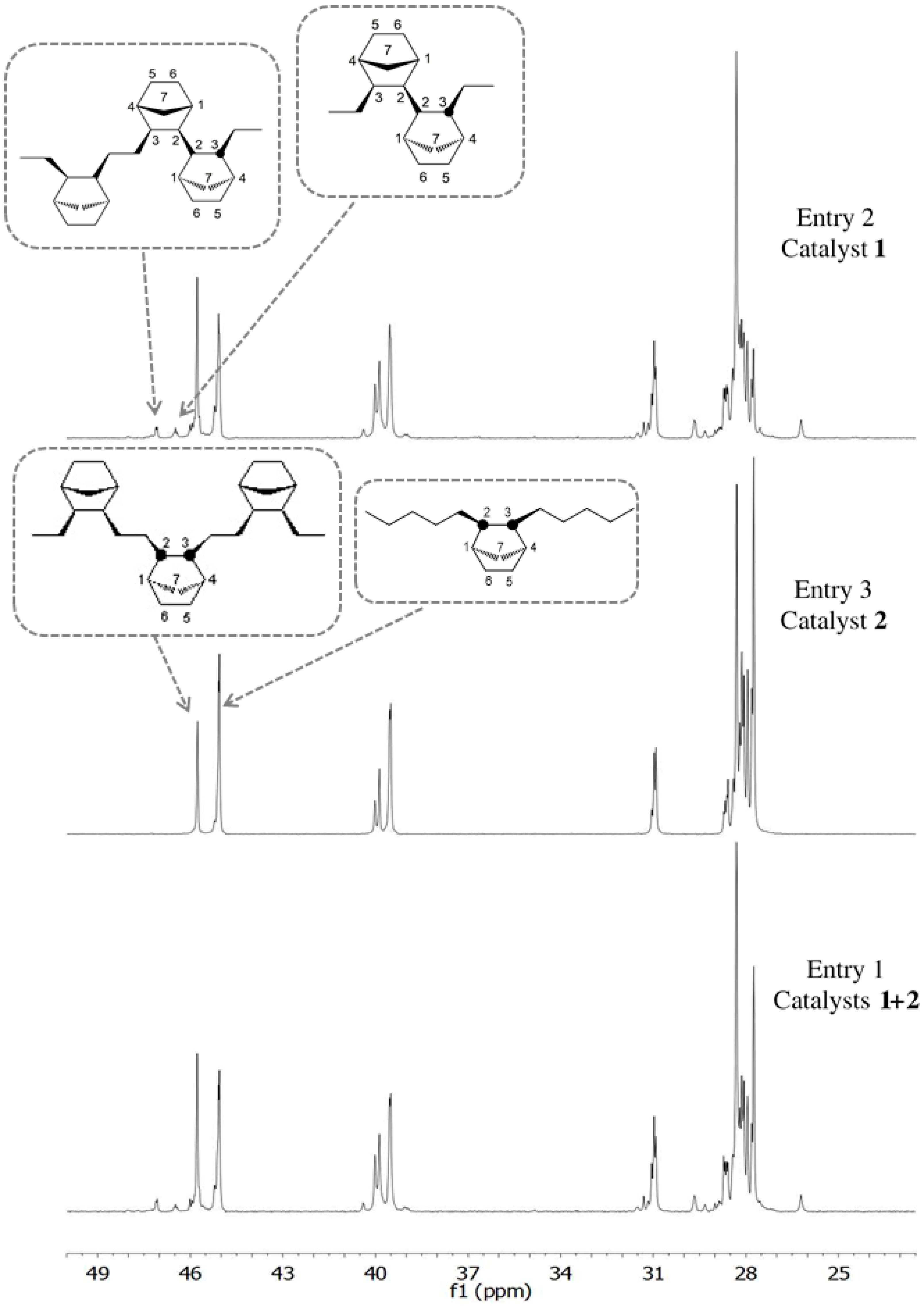
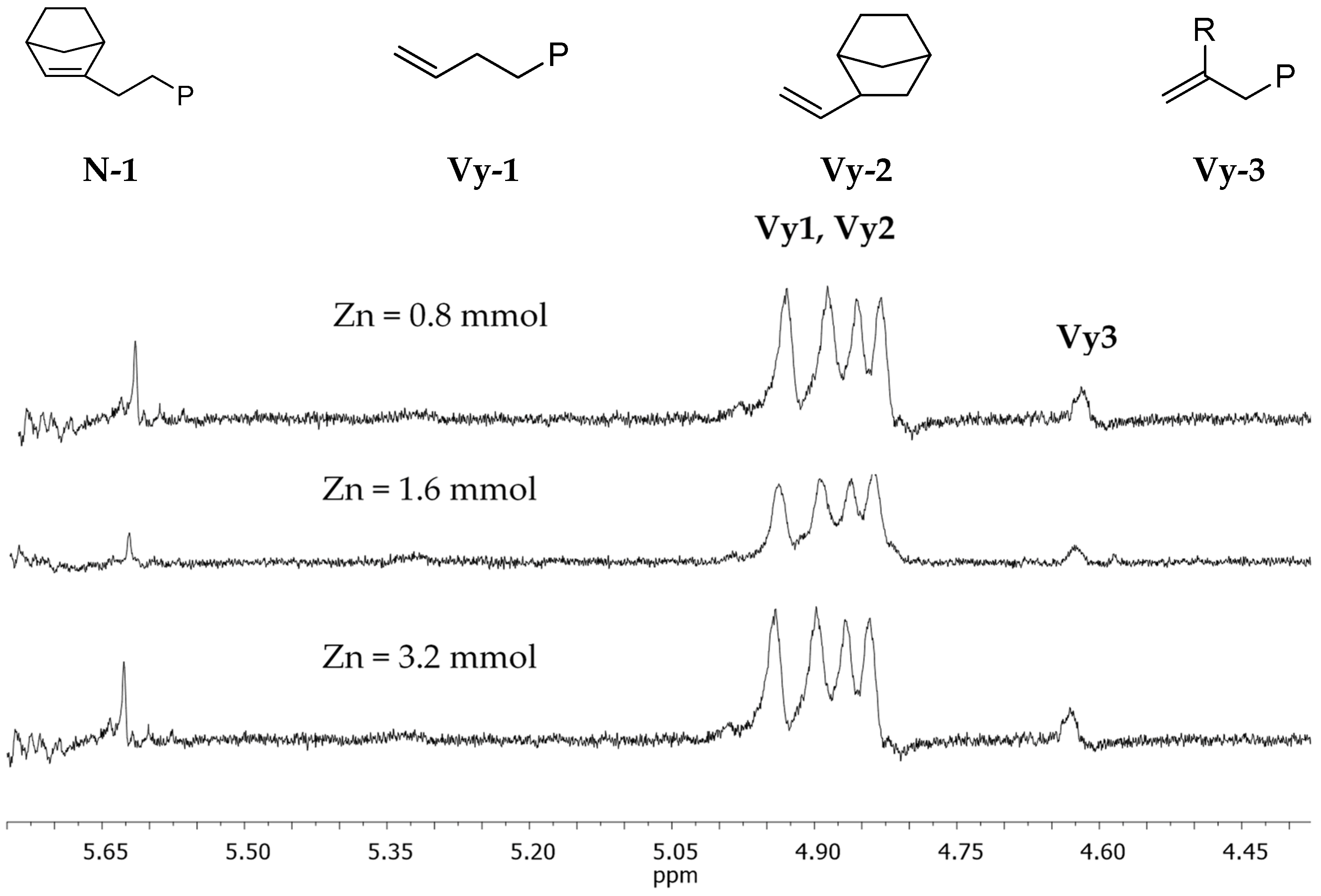
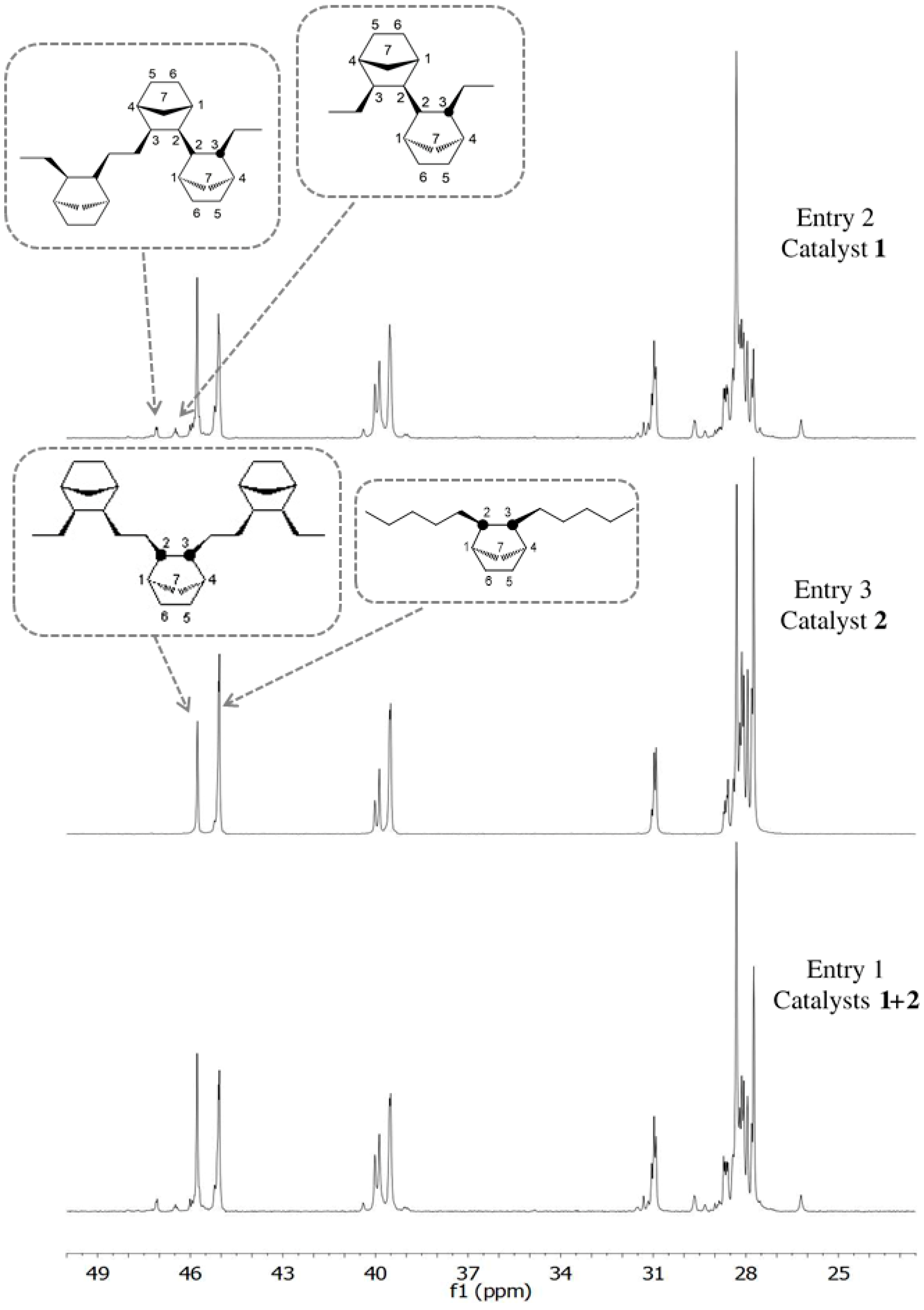
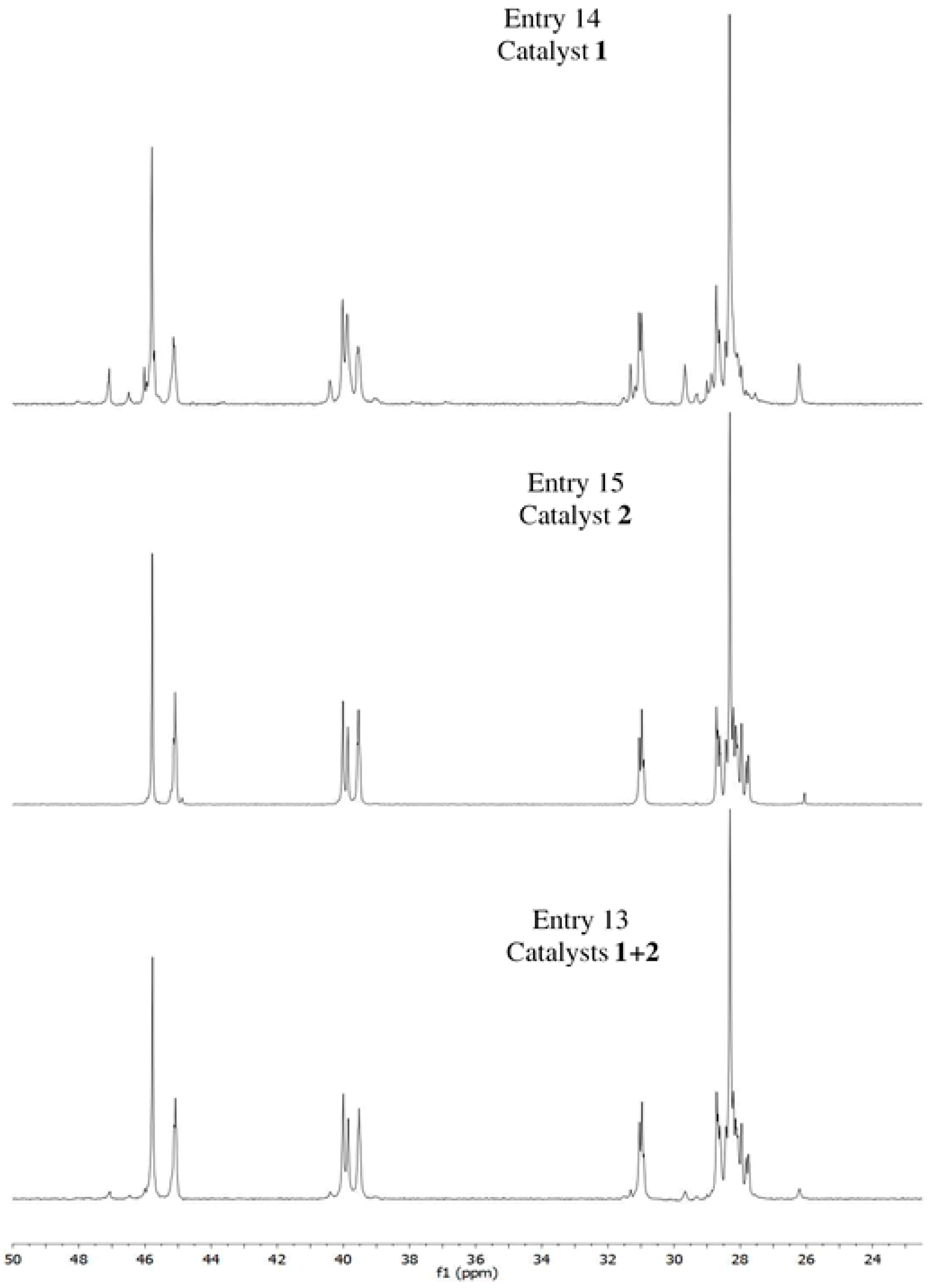
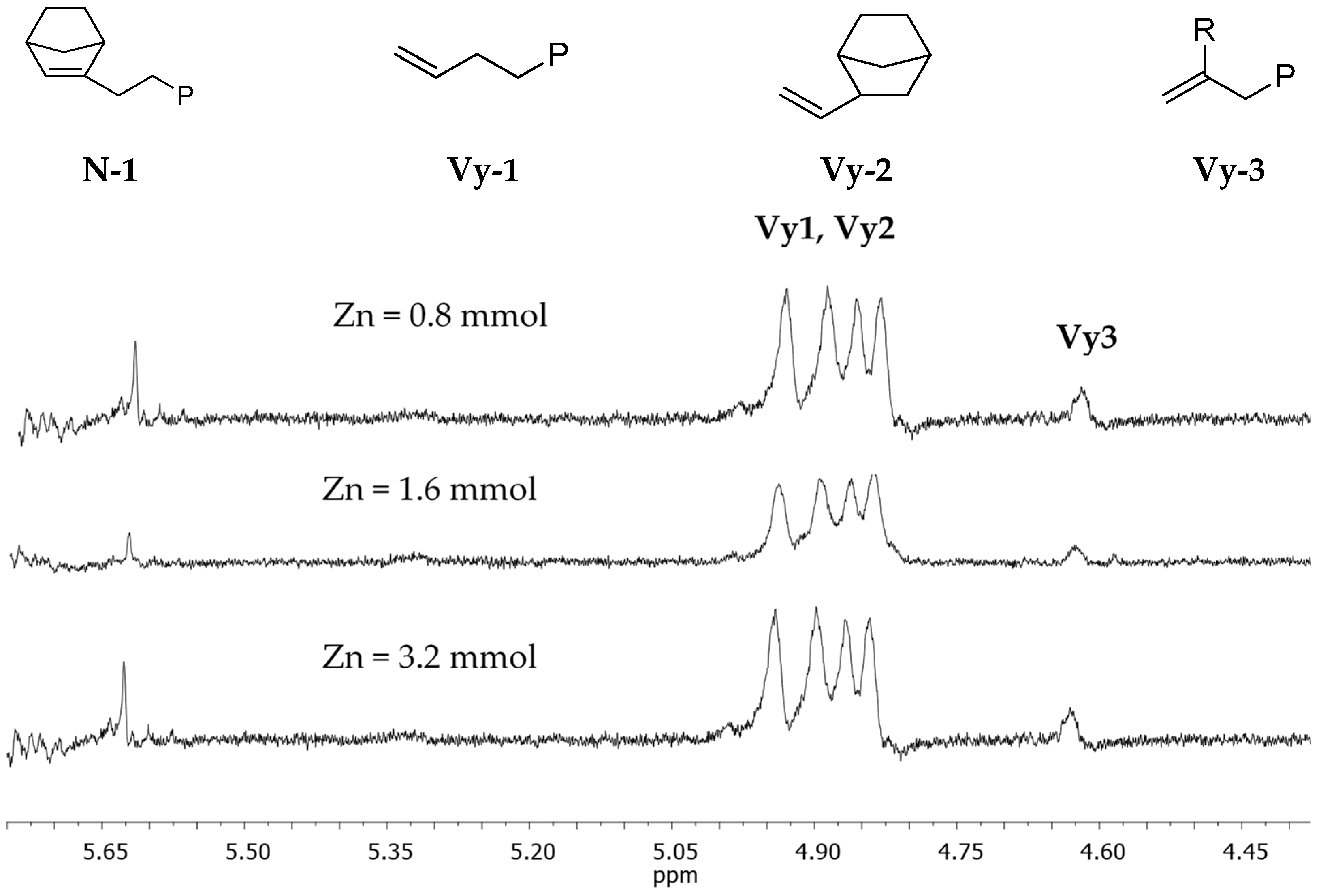
3.2. Chain End Analysis and Number of Chains
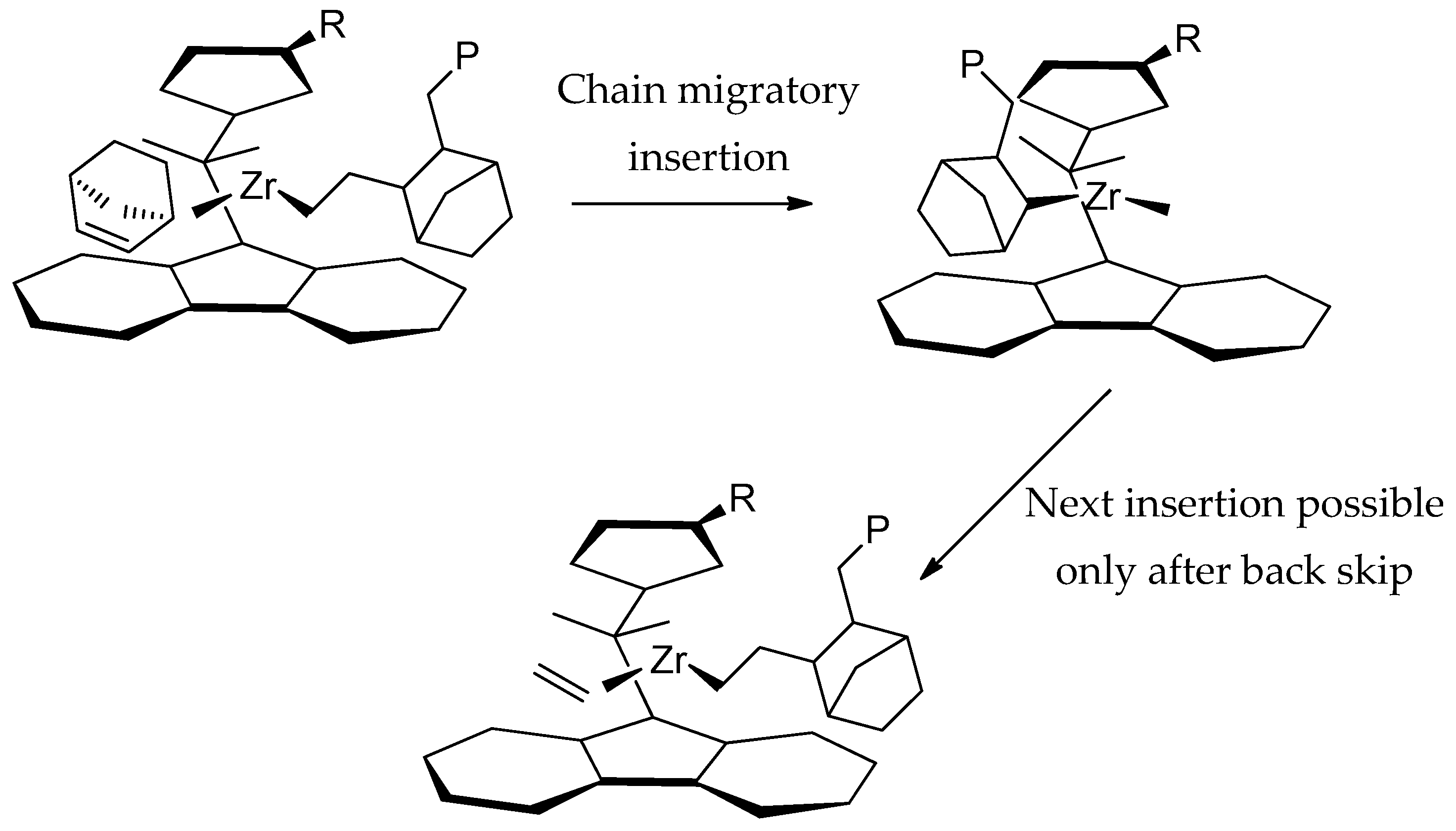
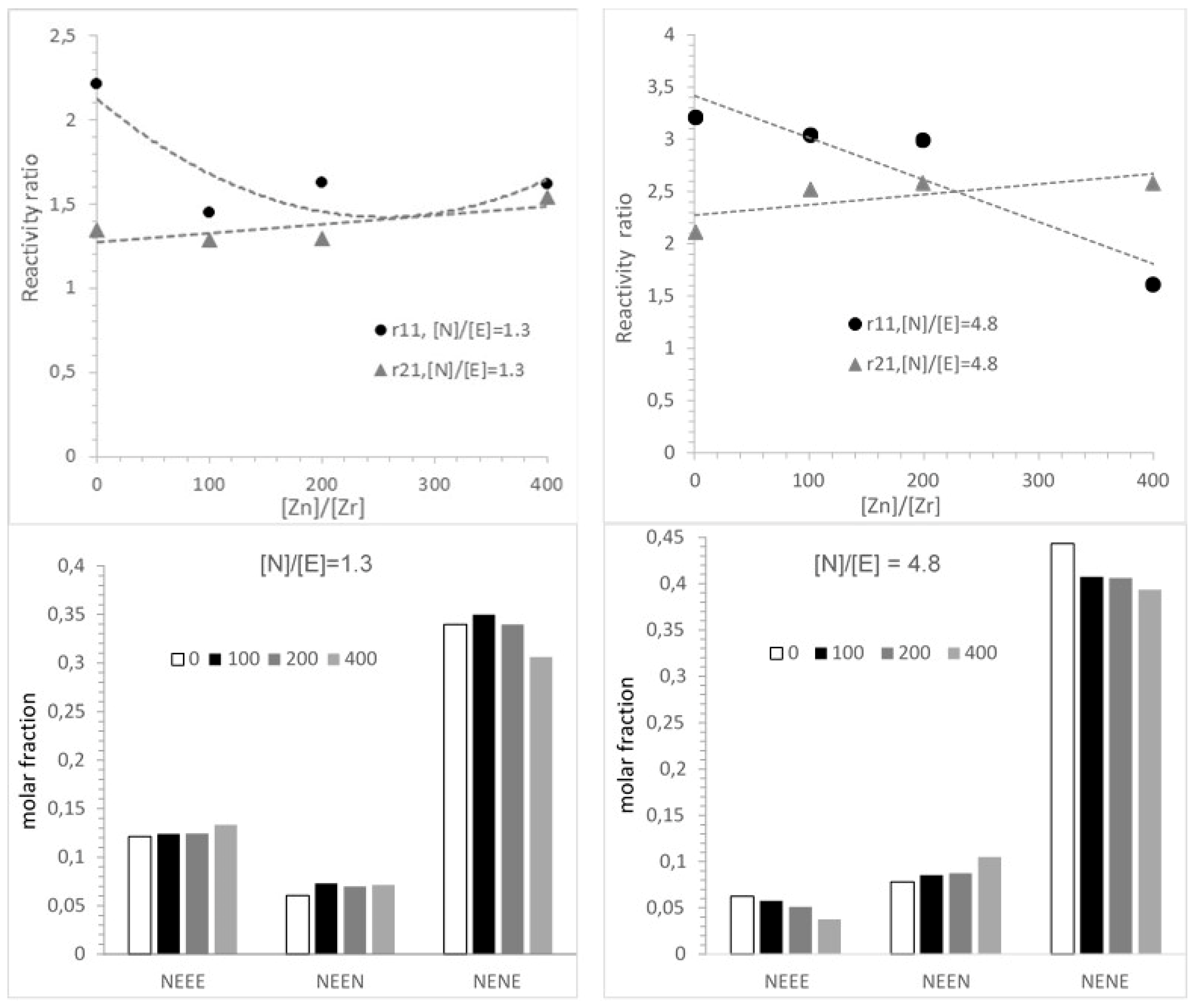
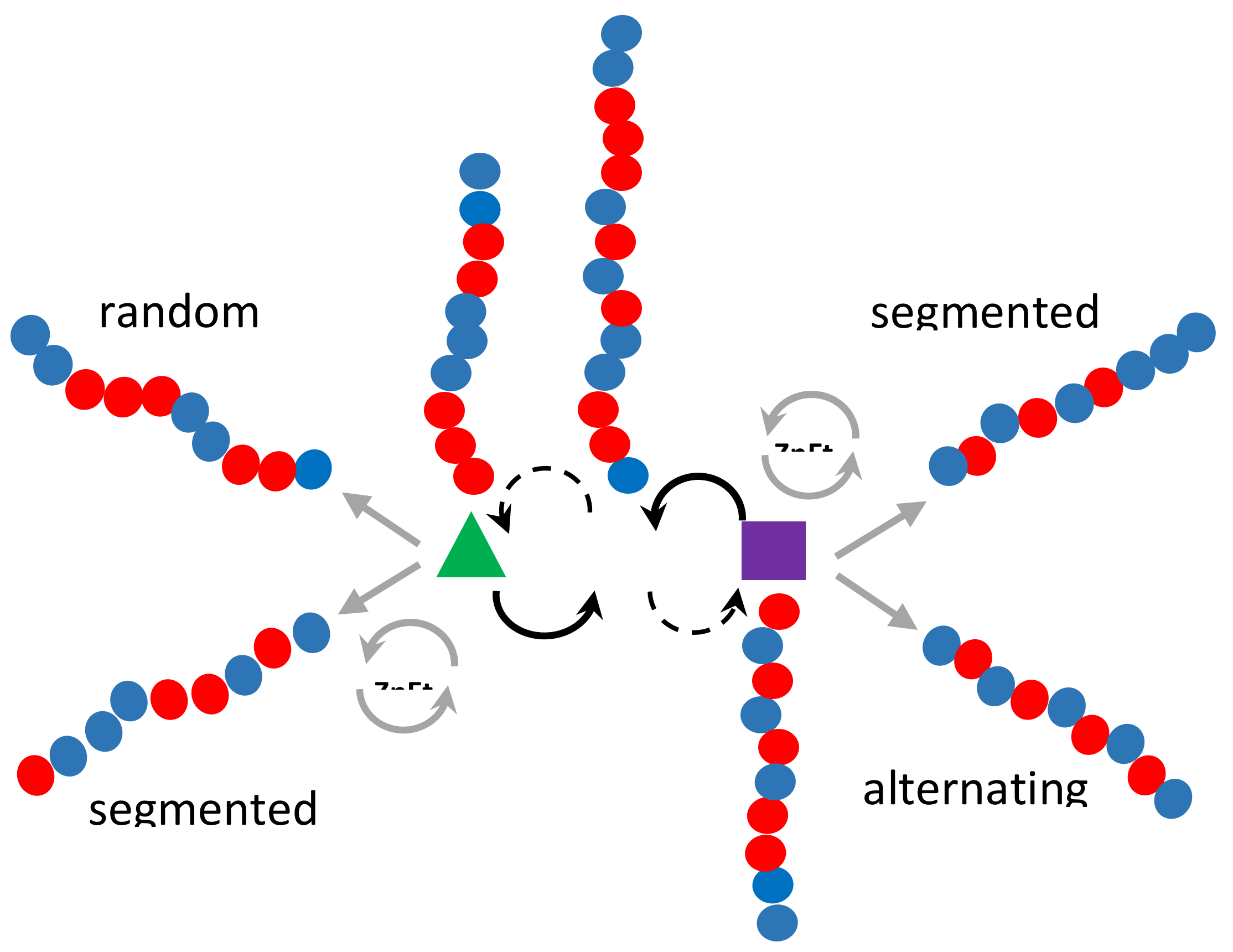
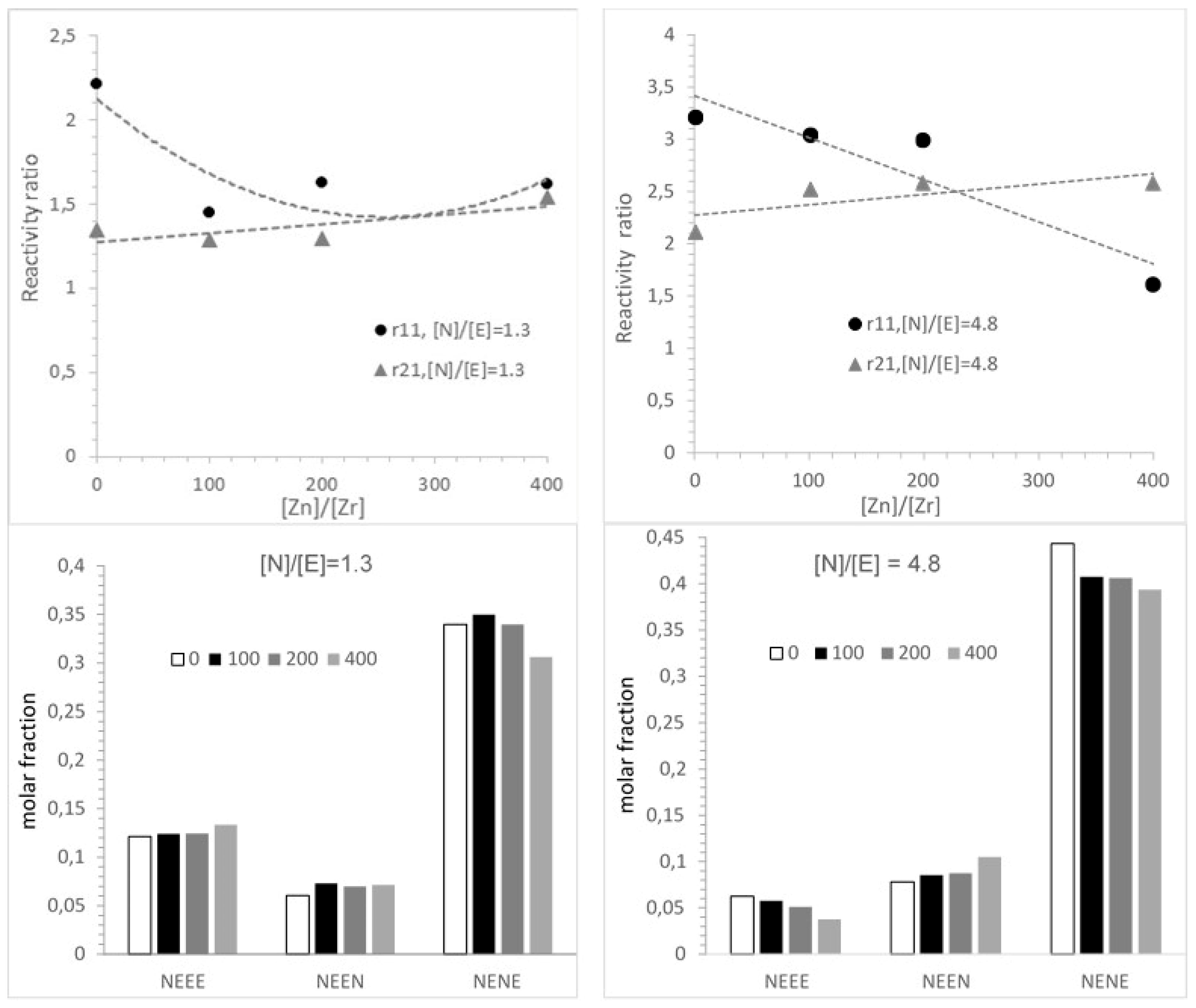
3.3. Microstructure
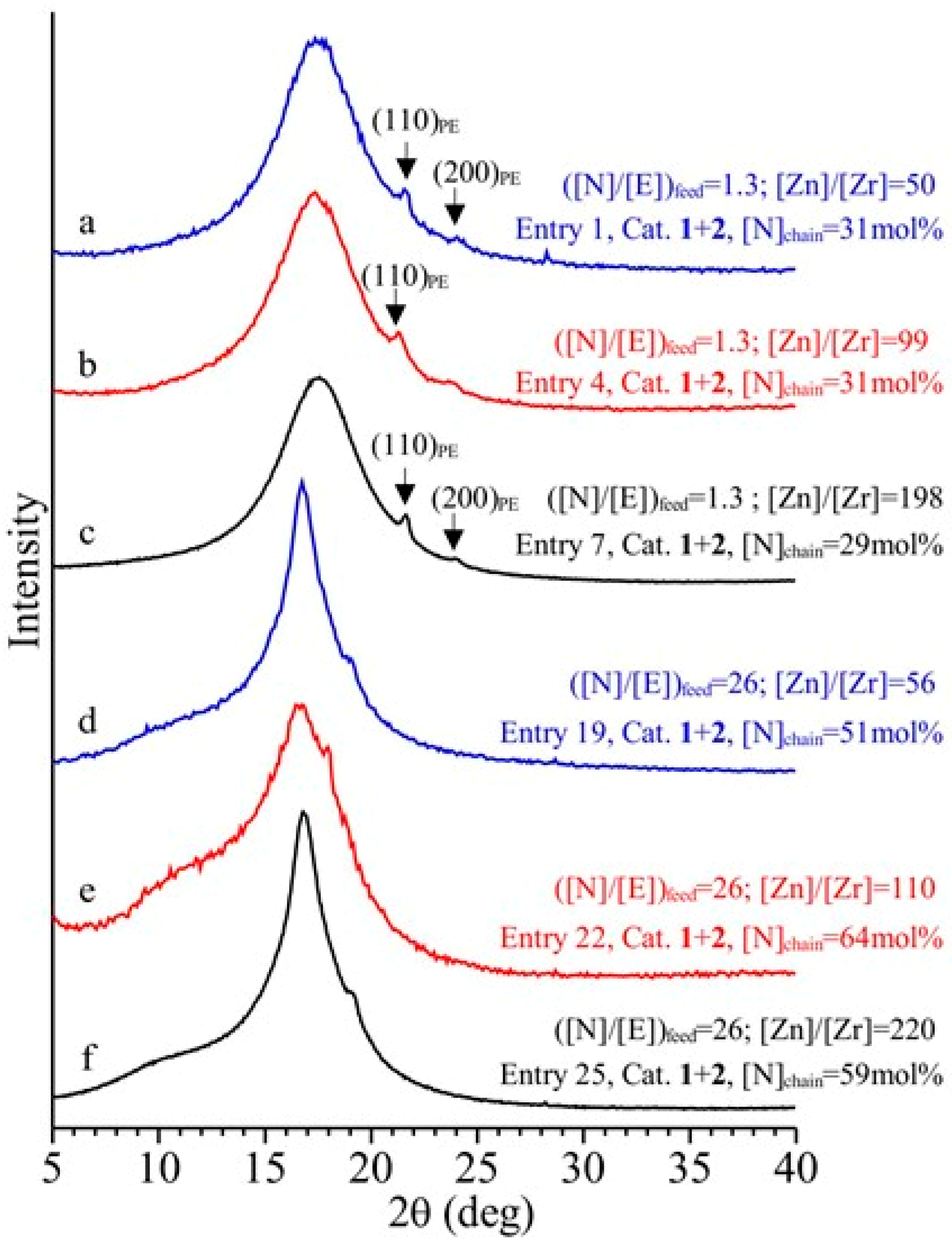
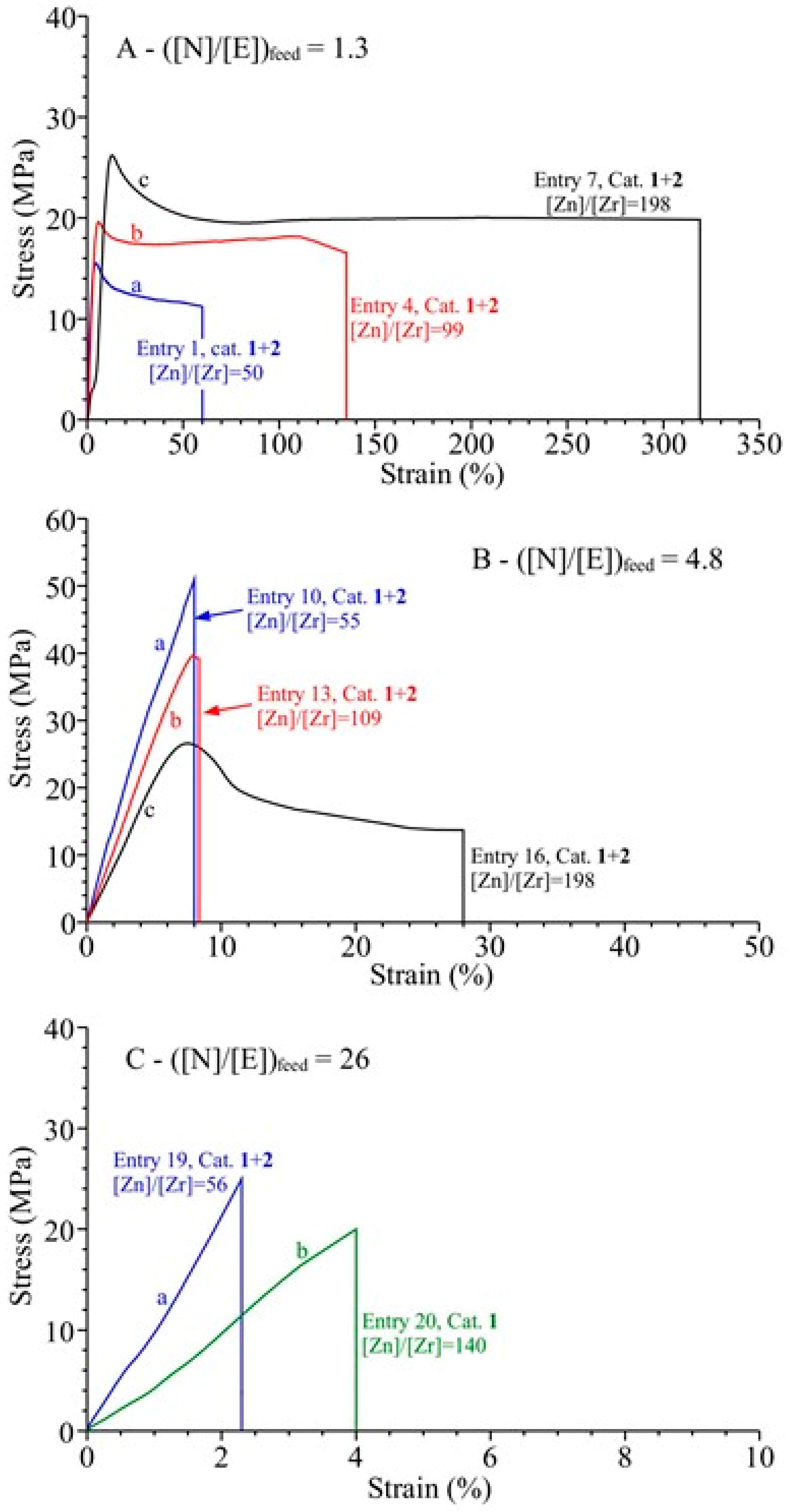
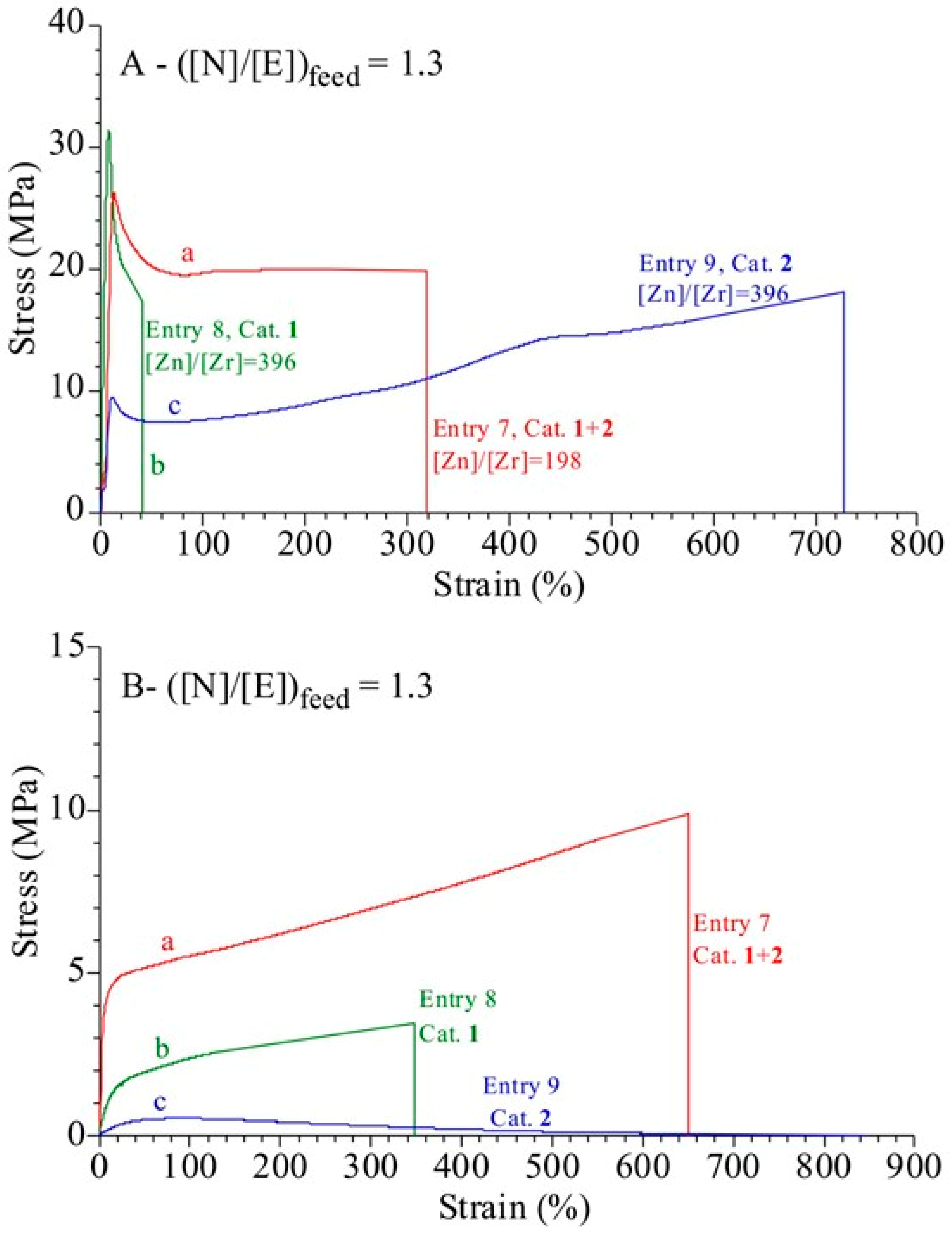
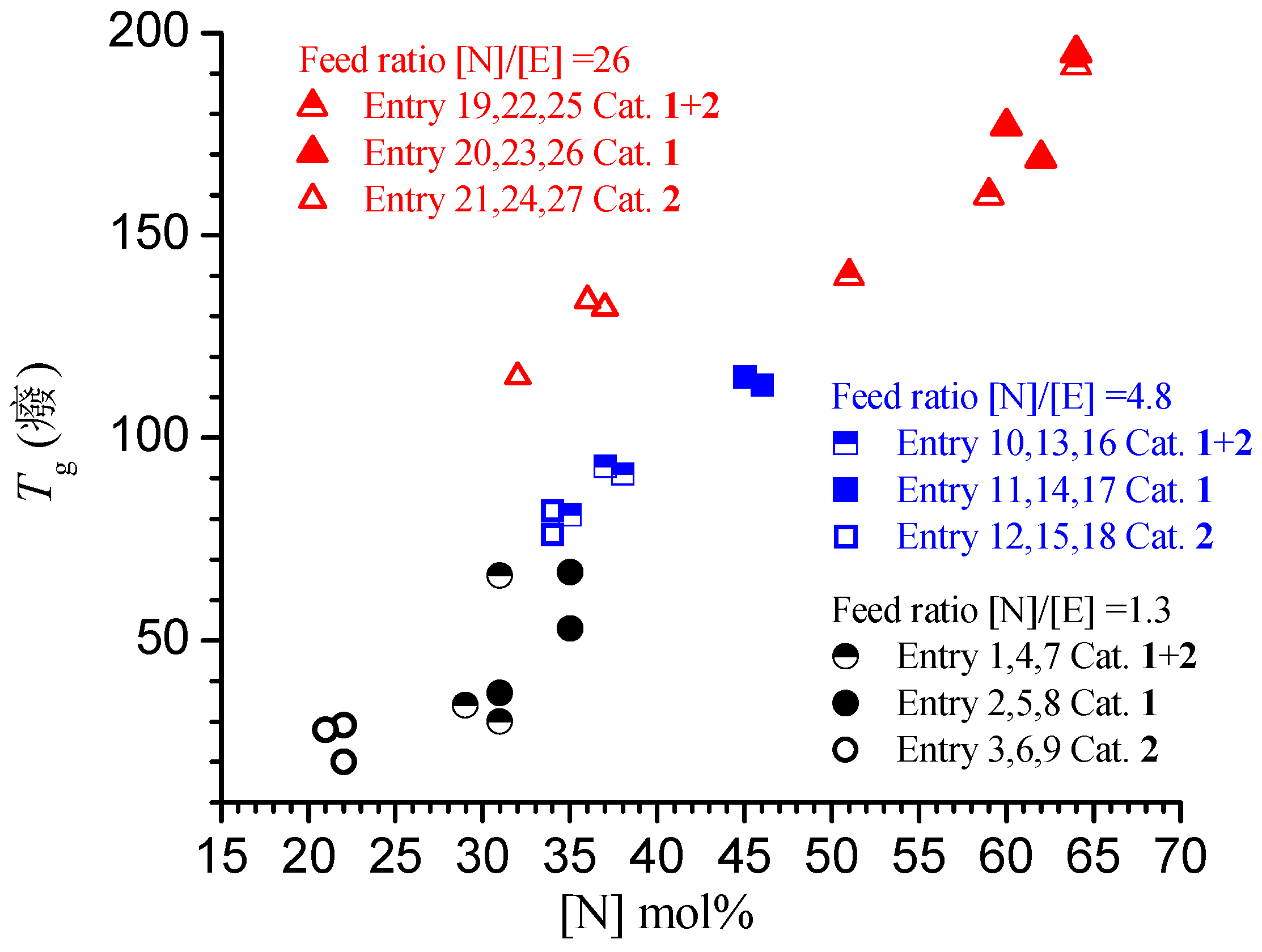
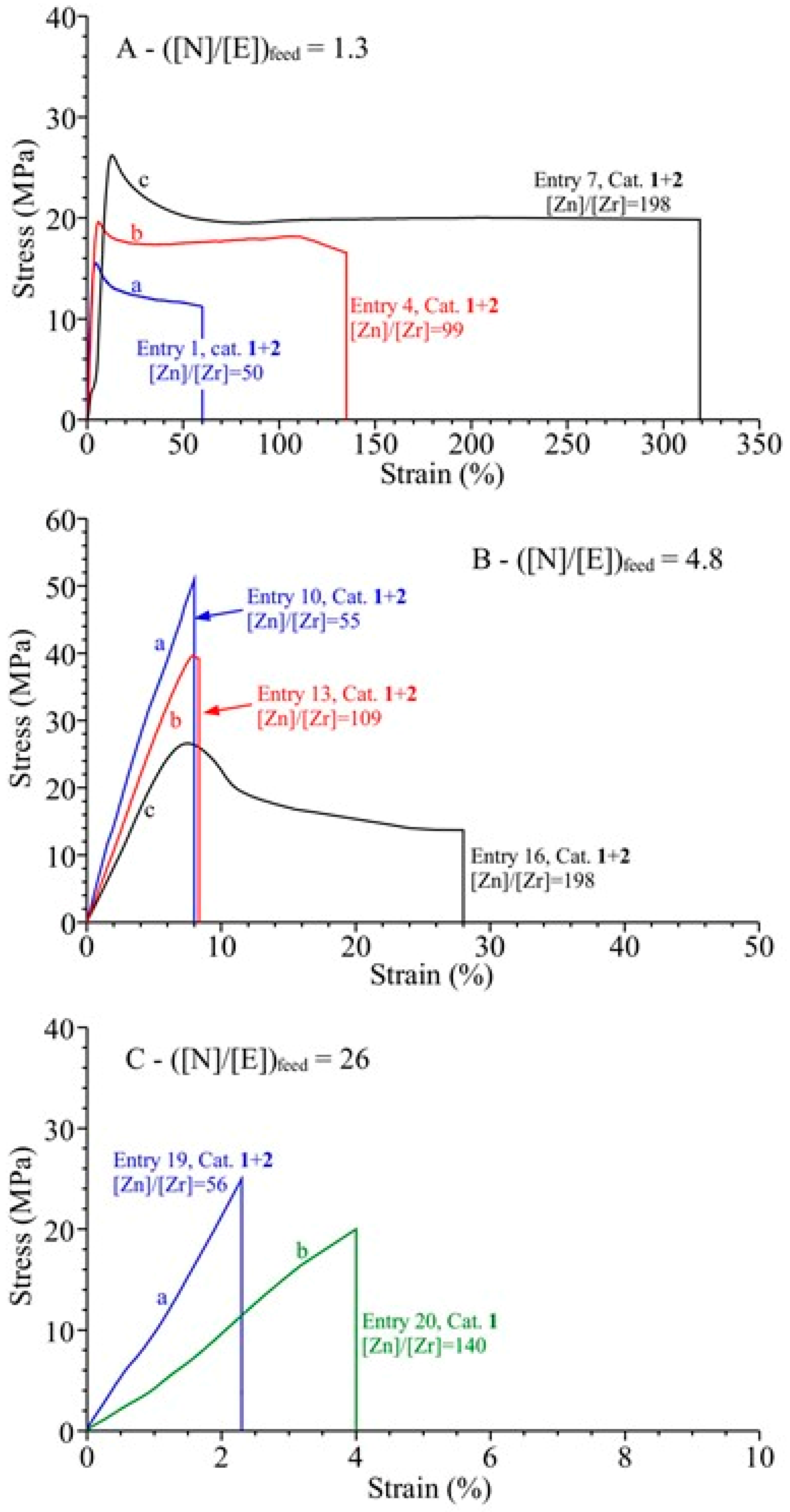
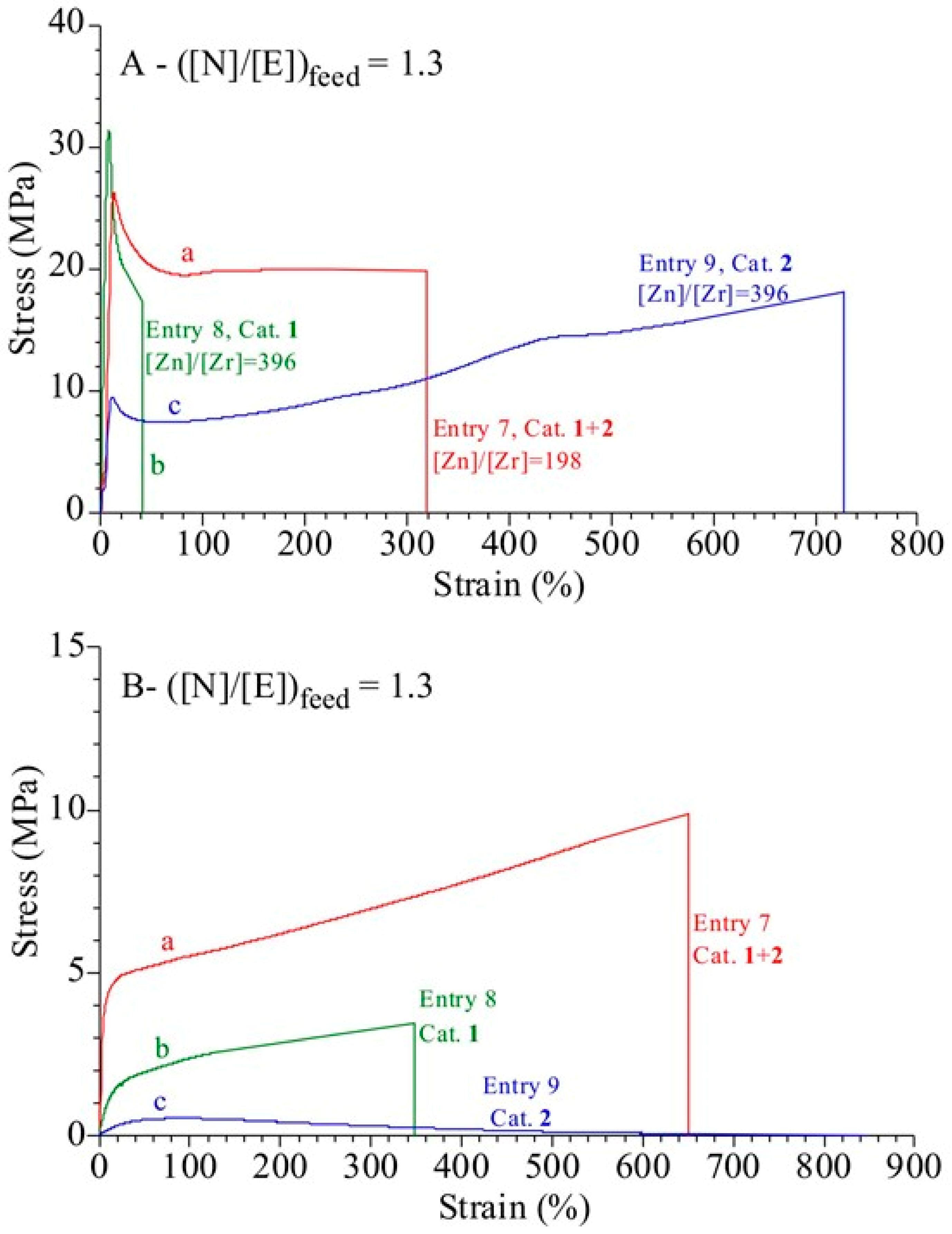
3.4. Analysis of the Structural, Thermal and Mechanical Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References and Notes
- Domski, G.J.; Rose, J.M.; Coates, G.W.; Bolig, A.D.; Brookhart, M. Living alkene polymerization: New methods for the precision synthesis of polyolefins. Prog. Polym. Sci. 2007, 32, 30–92. [Google Scholar] [CrossRef]
- Stürzel, M.; Mihan, S.; Mülhaupt, R. From multisite polymerization catalysis to sustainable materials and all-polyolefin composites. Chem. Rev. 2016, 116, 1398–1433. [Google Scholar] [CrossRef]
- Yasuda, H. Organo transition metal initiated living polymerizations. Prog. Polym. Sci. 2000, 25, 573–626. [Google Scholar] [CrossRef]
- Bates, F.S. Polymer–polymer Phase-behavior. Science 1991, 251, 898–905. [Google Scholar] [CrossRef]
- Kaminsky, W. (Ed.) Polyolefins: 50 Years after Ziegler and Natta I: Polyethylene and Polypropylene; Springer: New York, NY, USA, 2013; Volume 257. [Google Scholar]
- Saito, J.; Mitani, M.; Mohri, J.; Yoshida, Y.; Matsui, S.; Ishii, S.; Kojoh, S.; Kashiwa, N.; Fujita, T. Living polymerization of ethylene with a titanium complex containing two phenoxy-imine chelate ligands. Angew. Chem. Int. Ed. 2001, 40, 2918–2920. [Google Scholar] [CrossRef]
- Valente, A.; Mortreux, A.; Visseaux, M.; Zinck, P. Coordinative chain transfer polymerization. Chem. Rev. 2013, 113, 3836–3857. [Google Scholar] [CrossRef]
- Pelletier, J.F.; Mortreux, A.; Olonde, X.; Bujadoux, K. Synthesis of new dialkylmagnesium compounds by living transfer ethylene oligo- and polymerization with lanthanocene catalysts. Angew. Chem. 1996, 35, 1854–1856. [Google Scholar] [CrossRef]
- Britovsek, G.J.P.; Cohen, S.A.; Gibson, V.C.; van Meurs, M.J. Iron Catalyzed Polyethylene Chain Growth on Zinc: A Study of the Factors Delineating Chain Transfer versus Catalyzed Chain Growth in Zinc and Related Metal Alkyl Systems. J. Am. Chem. Soc. 2004, 126, 10701–10712. [Google Scholar] [CrossRef]
- van Meurs, M.; Britovsek, G.J.P.; Gibson, V.C.; Cohen, S.A. Polyethylene Chain Growth on Zinc Catalyzed by Olefin Polymerization Catalysts: A Comparative Investigation of Highly Active Catalyst Systems across the Transition Series. J. Am. Chem. Soc. 2005, 127, 9913–9923. [Google Scholar] [CrossRef]
- Kempe, R. How to polymerize ethylene in a highly controlled fashion? Chem. Eur. J. 2007, 13, 2764–2773. [Google Scholar] [CrossRef]
- Sita, L.R. Ex Uno Plures (“Out of One, Many”): New Paradigms for Expanding the Range of Polyolefins through Reversible Group Transfers. Angew. Chem. 2009, 48, 2464–2472. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, T.T.; Arriola, D.J.; Carnahan, E.M.; Hustad, P.D.; Kuhlman, R.L. Chain Shuttling Catalysis and Olefin Block Copolymers (OBCs). In Metal Catalysts in Olefin Polymerization; Springer: Berlin/Heidelberg, Germany, 2009; Volume 26, pp. 65–104. [Google Scholar]
- Arriola, D.J.; Carnahan, E.M.; Hustad, P.D.; Kuhlman, R.L.; Wenzel, T.T. Catalytic Production of Olefin Block Copolymers Via Chain Shuttling Polymerization. Science 2006, 312, 714–719. [Google Scholar] [CrossRef]
- Zinck, P. Unexpected Reactivities in Chain Shuttling Copolymerization. Polym. Int. 2016, 65, 11–15. [Google Scholar] [CrossRef]
- Zhang, W.; Sita, L.R. Highly Efficient, Living Coordinative Chain-Transfer Polymerization of Propene with ZnEt2: Practical Production of Ultrahigh to Very Low Molecular Weight Amorphous Atactic Polypropenes of Extremely Narrow Polydispersity. J. Am. Chem. Soc. 2008, 130, 442–443. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wei, J.; Sita, L.R. Living Coordinative Chain-Transfer Polymerization and Copolymerization of Ethene, α-Olefins, and α,ω-Nonconjugated Dienes using Dialkylzinc as “Surrogate” Chain-Growth Sites. Macromolecules 2008, 41, 7829–7833. [Google Scholar] [CrossRef]
- Valente, A.; Stoclet, G.; Bonnet, F.; Mortreux, A.; Visseaux, M.; Zinck, P. Isoprene- Styrene Chain Shuttling Copolymerization Mediated by a Lanthanide Half- Sandwich Complex and a Lanthanidocene: Straightforward Access to a New Type of Thermoplastic Elastomers. Angew. Chem. Int. Ed. 2014, 53, 4638–4641. [Google Scholar] [CrossRef] [PubMed]
- Johnson, H.C.; Cueny, E.S.; Landis, C.R. Chain Transfer with Dialkyl Zinc During Hafnium–Pyridyl Amido-Catalyzed Polymerization of 1-Octene: Relative Rates, Reversibility, and Kinetic Models. ACS Catal. 2018, 8, 4178–4188. [Google Scholar] [CrossRef]
- Cueny, E.S.; Johnson, H.C.; Landis, C.R. Selective Quench-Labeling of the Hafnium-Pyridyl Amido-Catalyzed Polymerization of 1-Octene in the Presence of Trialkyl-Aluminum Chain-Transfer Reagents. ACS Catal. 2018, 8, 11605–11614. [Google Scholar] [CrossRef]
- Vittoria, A.; Busico, V.; Cannavacciuolo, F.D.; Cipullo, R. Molecular Kinetic Study of “Chain Shuttling” Olefin Copolymerization. ACS Catal. 2018, 8, 5051–5061. [Google Scholar] [CrossRef]
- Tong, Z.Z.; Zhou, B.; Huang, J.; Xu, J.T.; Fan, Z.Q. Regulation of Crystallization Kinetics, Morphology, and Mechanical Properties of Olefinic Blocky Copolymers. Macromolecules 2014, 47, 333–346. [Google Scholar] [CrossRef]
- Auriemma, F.; De Rosa, C.; Scoti, M.; Di Girolamo, R.; Malafronte, A.; Talarico, G.; Carnahan, E. Unveiling the Molecular Structure of Ethylene/1-Octene Multi-block Copolymers from Chain Shuttling Technology. Polymer 2018, 154, 298–304. [Google Scholar] [CrossRef]
- Auriemma, F.; De Rosa, C.; Scoti, M.; Di Girolamo, R.; Malafronte, A.; Galotto, N. Structural Investigation at Nanometric Length Scale of Ethylene/1-Octene Multiblock Copolymers from Chain-Shuttling Technology. Macromolecules 2018, 51, 9613–9625. [Google Scholar] [CrossRef]
- Kaminsky, W.; Bark, A.; Arndt, M. New polymers by homogenous zirconocene/aluminoxane catalysts. Macromol. Symp. 1991, 47, 83–93. [Google Scholar] [CrossRef]
- Kaminsky, W.; Nolle, A. Copolymerization of Norbornene and Ethene with Homogenous Zirconocenes/Methylaluminoxane Catalysts. Polym. Bull. 1993, 31, 175–182. [Google Scholar] [CrossRef]
- Ruchatz, D.; Fink, G. Ethene-norbornene copolymerization using homogenous metallocene and half-sandwich catalysts: Kinetics and relationships between catalyst structure and polymer structure. 2. Comparative study of different metallocene- and half-sandwich methylaluminoxane catalysts and analysis of the copolymers by C-13 nuclear magnetic resonance spectroscopy. Macromolecules 1998, 31, 4674–4680. [Google Scholar] [CrossRef]
- Boggioni, L.; Tritto, I. The State of the Art of Cyclic Olefin Polymers. MRS Bull. 2013, 38, 245–251. [Google Scholar] [CrossRef]
- Tritto, I.; Boggioni, L.; Ferro, D.R. Metallocene Catalyzed Ethene- and Propene Co-Norbornene Polymerization: Mechanisms from a Detailed Microstructural Analysis. Coord. Chem. Rev. 2006, 250, 212–241. [Google Scholar] [CrossRef]
- Tritto, I.; Marestin, C.; Boggioni, L.; Sacchi, M.C.; Britzinger, H.H.; Ferro, D.R. Stereoregular and Stereoirregular Alternating Ethylene−Norbornene Copolymers. Macromolecules 2001, 34, 5770–5777. [Google Scholar] [CrossRef]
- Ravasio, A.; Zampa, C.; Boggioni, L.; Tritto, I.; Hitzbleck, J.; Okuda, J. Copolymerization of Ethylene with Norbornene Catalyzed by Cationic Rare-Earth Metal Half-Sandwich Complexes. Macromolecules 2008, 41, 956–9569. [Google Scholar] [CrossRef]
- Ní Bhriain, N.; Brintzinger, H.H.; Ruchatz, D.; Fink, G. Polymeryl Exchange between ansa-Zirconocene Catalysts for Norbornene−Ethene Copolymerization and Aluminum or Zinc Alkyls. Macromolecules 2005, 38, 2056–2063. [Google Scholar] [CrossRef]
- Boggioni, L.; Sidari, D.; Losio, S.; Stehling, U.M.; Auriemma, F.; Di Girolamo, R.; De Rosa, C.; Tritto, I. Ethylene–co–norbornene copolymerization in the presence of a Chain Transfer Agent. Eur. Pol. J. 2018, 107, 54–66. [Google Scholar] [CrossRef]
- Available online: http://www.topas.com/products/topas-coc-polymers (accessed on 20 March 2019).
- Available online: http://www.ptonline.com/articles/add-a-layer-of-coc-to-boost-polyolefin-film-properties (accessed on 20 March 2019).
- Available online: http://www.mitsuichemicals.com/apel.htm (accessed on 20 March 2019).
- Ruchatz, D.; Fink, G. Ethene−Norbornene Copolymerization Using Homogenous Metallocene and Half-Sandwich Catalysts: Kinetics and Relationships between Catalyst Structure and Polymer Structure. 1. Kinetics of the Ethene−Norbornene Copolymerization Using the [(Isopropylidene)(η5-inden-1-ylidene-η5-cyclopentadienyl)]zirconium Dichloride/Methylaluminoxane Catalyst. Macromolecules 1998, 31, 4669–4673. [Google Scholar] [CrossRef] [PubMed]
- Arndt, M.; Beulich, I. C1-Symmetric Metallocenes for Olefin Polymerisation, 1. Catalytic Performance of [Me2C(3-tertBuCp)(Flu)]ZrCl2 in Ethene/Norbornene Copolymerisation. Macromol. Chem. Phys. 1998, 199, 1221–1232. [Google Scholar] [CrossRef]
- Herfert, N.; Montag, P.; Fink, G. Elementary Processes of the Ziegler Catalysis, 7. Ethylene, Alpha-Olefin and Norbornene Copolymerization with the Stereorigid Catalyst Systems Ipr[FluCp]ZrCl2/MAO and Me2Si[Ind]2ZrCl2/MAO. Makromol. Chem. 1993, 94, 3167–3182. [Google Scholar] [CrossRef]
- Tritto, I.; Boggioni, L.; Ferro, D.R. Alternating Isotactic Ethylene-Norbornene Copolymers by C1-symmetric Metallocenes: Determination of the Copolymerization Parameters and Mechanistic Considerations on the Basis of Pentad Analysis. Macromolecules 2004, 37, 9681–9693. [Google Scholar] [CrossRef]
- Götz, C.; Rau, A.; Luft, G. Ternary metallocene catalyst systems based on metallocene dichlorides and AliBu3/[PhNMe2H][B(C6F5)4]: NMR investigations of the influence of Al/Zr ratios on alkylation and on formation of the precursor of the active metallocene species. J. Mol. Catal. A Chem. 2002, 184, 95–110. [Google Scholar] [CrossRef]
- Chen, E.Y.X.; Marks, T.J. Cocatalysts for Metal-Catalyzed Olefin Polymerization: Activators, Activation Processes, and Structure−Activity Relationships. Chem. Rev. 2000, 100, 1391–1434. [Google Scholar] [CrossRef] [PubMed]
- Bochmann, M. Kinetic and mechanistic aspects of metallocene polymerisation catalysts. J. Organomet. Chem. 2004, 689, 3982–3998. [Google Scholar] [CrossRef]
- Bochmann, M. The Chemistry of Catalyst Activation: The Case of Group 4 Polymerization Catalysts. Organometallics 2010, 29, 4711–4740. [Google Scholar] [CrossRef]
- In a study of the effect of chain transfer by R3Al in ethylene-norbornene copolymerization with Ph2C(Flu)(3-RCp)ZrCl2/Mehylaluminoxane catalysts, it was concluded that the addition of TIBA increases the molar masses of copolymers since TIBA addition has an influence on the active ion pair and increases the propagation rate more than the chain transfer rate.
- Tada, T.; Cai, Z.; Nakayama, T.; Shiono, T. Efficient Molecular Weight Control with Trialkylaluminum in Ethylene/Norbornene Copolymerization by [Ph2C(Flu)(3-MeCp)]ZrCl2/Methylaluminoxane Catalyst. Macromol. Chem. Phys. 2010, 211, 2132–2137. [Google Scholar] [CrossRef]
- Norbornene and ethylene have opposite refractive index, thus there is a copolymer composition, around 20 mol % of N in our conditions, where copolymers are isorefractive and it is not possible to observe a SEC signal. In our lab we have our own calibration curve. See Bergstrom et al. [48].
- Bergstrom, C.; Ruotoistenmaki, J.; SeppaIa, J. Test Method: A GPC Method to Determine the Amount of Comonomer in an Ethylene-Norbornene-Copolymer. Polym. Test. 1997, 16, 43–48. [Google Scholar] [CrossRef]
- “A reviewer observed that catalytic activities of 1+2 in Table 2 are higher than those in Table 1. Activities depend from a number of factors. Here it is worth observing that at higher [N]/[E] feed ratios activities tend to decrease, while at higher catalyst concentrations can increase the activities. We have used higher catalyst concentration going from Table 1, to Table 2 and Table 3. It is possible that at [N]/[E] ratio of 4.8, the balance among these factors results in greater activities.”.
- Tritto, I.; Boggioni, L.; Jansen, J.C.; Thorshaug, K.; Sacchi, M.C.; Ferro, D.R. Ethylene-Norbornene Copolymers from Metallocene-Based Catalysts:Microstructure at Tetrad Level and Reactivity Ratios. Macromolecules 2002, 35, 616–623. [Google Scholar] [CrossRef]
- De Rosa, C.; Buono, A.; Auriemma, F.; Grassi, A. Crystal structure of alternating ethylene−norbornene copolymer. Macromolecules 2004, 37, 9489–9502. [Google Scholar] [CrossRef]
- De Rosa, C.; Corradini, P.; Buono, A.; Auriemma, F.; Grassi, A.; Altamura, P. Crystalline ethylene−norbornene copolymers: Plastic crystals from macromolecules. Macromolecules 2003, 36, 3789–3792. [Google Scholar] [CrossRef]
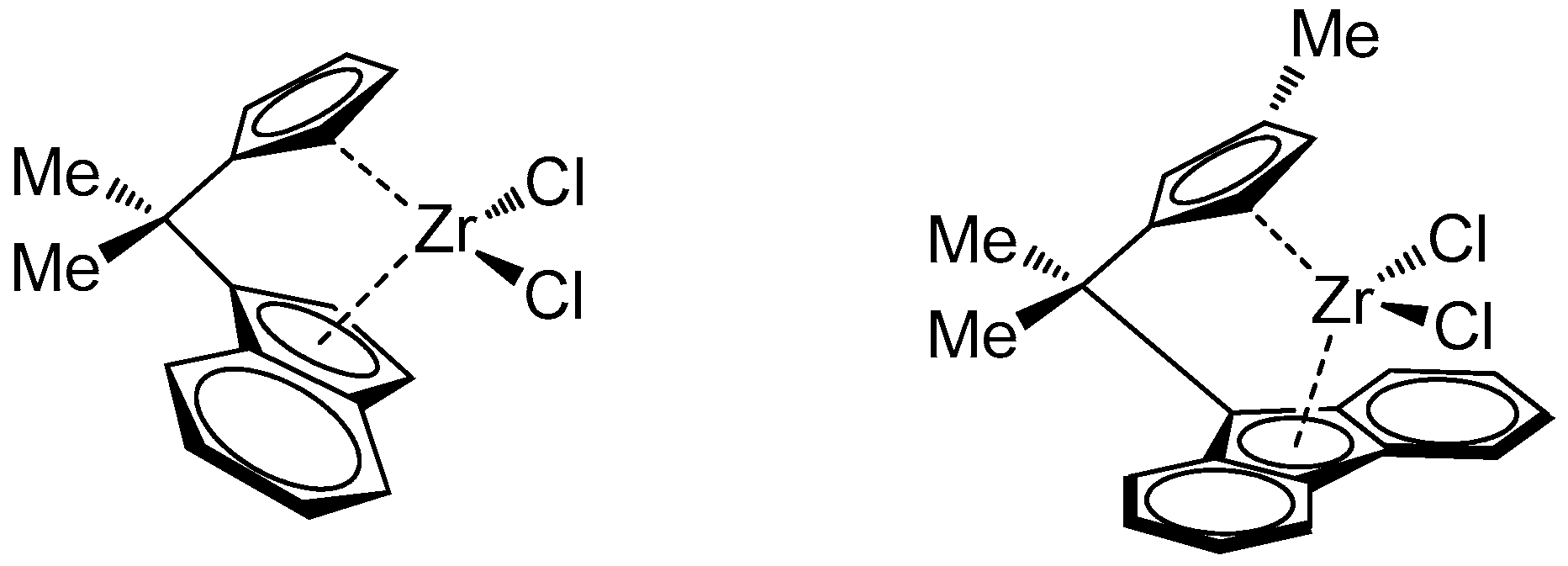


{kind=link}
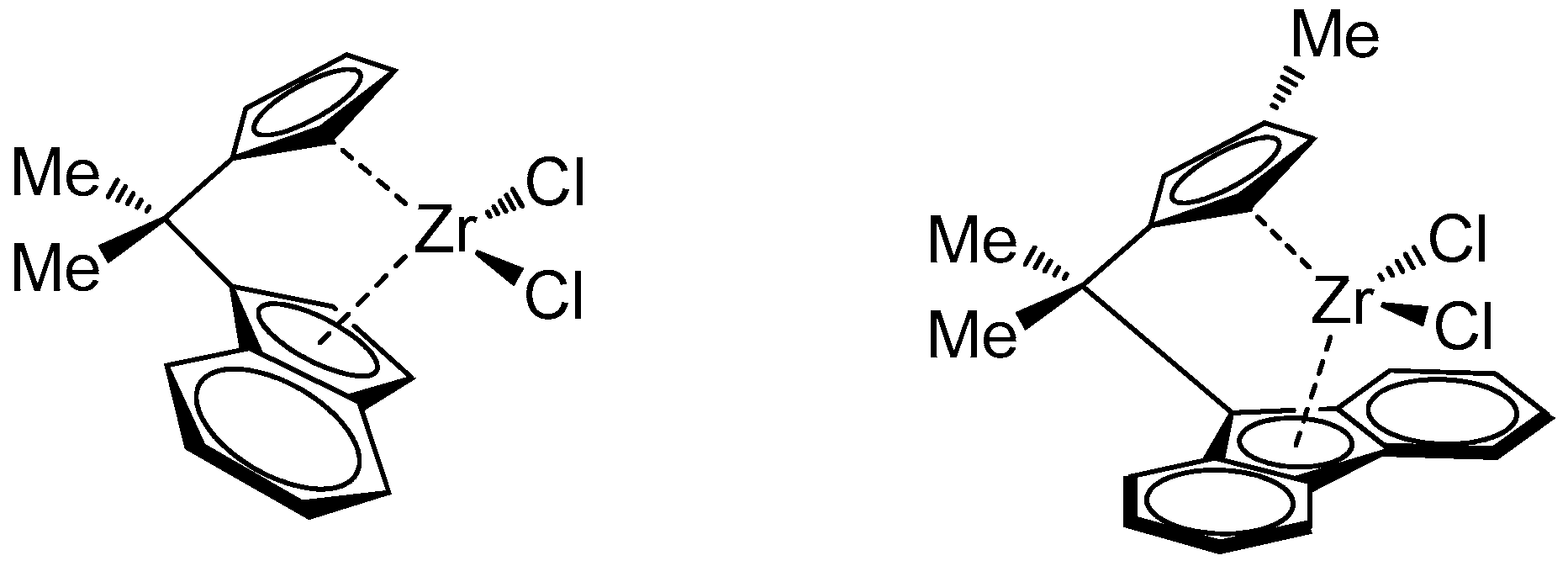{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry a | Catalyst | Zn (mmol) | [Zn]/[Zr] | Yield (g) | Activity b | N (mol %) c | Conv (%) | Tg (°C) d | Mw × 10−3 (Kg/mol) e | De | Chain Number for Catalytic Center f | Chain End per 10,000 units g |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1+2 | 0.8 | 50 | 18.4 | 242 | 31 | 42 | 66 | 27 | 1.8 | 77 | 8.6 |
| 2 | 1 | 0.8 | 99 | 9.3 | 244 | 35 | 23 | 67 | 30 | 1.9 | 72 | 14.1 |
| 3 | 2 | 0.8 | 99 | 9.7 | 255 | 22 | 18 | 29 | --- | --- | -- | 4 |
| 4 | 1+2 | 1.6 | 99 | 16.8 | 221 | 31 | 36 | 30 | 25 | 1.8 | 77 | 6.2 |
| 5 | 1 | 1.6 | 198 | 7.2 | 189 | 35 | 17.5 | 53 | 29 | 1.9 | 59 | 13 |
| 6 | 2 | 1.6 | 198 | 9.5 | 250 | 21 | 17.1 | 28 | --- | --- | --- | 3.8 |
| 7 | 1+2 | 3.2 | 198 | 8.4 | 110 | 29 | 18 | 34 | 23 | 1.7 | 39 | 8.7 |
| 8 | 1 | 3.2 | 396 | 5.3 | 139 | 31 | 12 | 37 | 29 | 1.8 | 41 | 11.8 |
| 9 | 2 | 3.2 | 396 | 9.8 | 258 | 22 | 18 | 20 | --- | --- | --- | 3.6 |
| Entry a | Catalyst | Zn (mmol) | [Zn]/[Zr] | Yield (g) | Activity b | N (mol %) c | Conv (%) | Tg (°C) d | Mw × 10−3 (Kg/mol) e | De | Chain Number for Catalytic Center f |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 10 | 1+2 | 1.2 | 55 | 16 | 290 | 37 | 20.6 | 93 | 239 | 2.9 | 9 |
| 11 | 1 | 1 | 100 | 1.7 | 68 | 46 | 2.4 | 113 | 61 | 1.9 | 5 |
| 12 | 2 | 1.2 | 100 | 5.9 | 197 | 34 | 7.2 | 82 | 309 | 1.7 | 3 |
| 13 | 1+2 | 2.4 | 109 | 15.9 | 289 | 38 | 21 | 91 | 155 | 2.7 | 13 |
| 14 | 1 | 2 | 200 | 11 | 440 | 45 | 16 | 115 | 64 | 2 | 34 |
| 15 | 2 | 2.4 | 200 | 1.5 | 50 | 34 | 1.8 | 76 | 170 | 2.9 | 2 |
| 16 | 1+2 | 4.8 | 198 | 9.7 | 176 | 35 | 12 | 81 | 113 | 2.9 | 11 |
| 17 | 1 | 4 | 400 | 0.7 | 28 | 46 | 1 | 113 | 48 | 2 | 3 |
| 18 | 2 | 4.8 | 400 | 3.9 | 130 | 34 | 4.8 | 76 | 97 | 2.6 | 9 |
| Entry a | Catalyst | Zn (mmol) | [Zn]/[Zr] | Yield (g) | Activity b | N (mol %) c | Conv (%) | Tg (°C) d | Mw × 10−3 (Kg/mol) e | De | Chain Number for Catalytic Center f |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 19 | 1+2 | 2.8 | 56 | 7.9 | 158 | 51 | 5.5 | 140 | 99 | 2.2 | 4 |
| 20 | 1 | 2.8 | 140 | 15.8 | 790 | 64 | 12.1 | 195 | 146 | 5.4 | 13 |
| 21 | 2 | 2.8 | 93 | 1 | 33 | 37 | 0.6 | 132 | --- | --- | --- |
| 22 | 1+2 | 5.5 | 110 | 21.6 | 432 | 64 | 16.6 | 192 | 53 | 1.9 | 16 |
| 23 | 1 | 5.5 | 275 | 1.7 | 85 | 60 | 1.3 | 177 | 54 | 2 | 3 |
| 24 | 2 | 5.5 | 183 | 2 | 67 | 36 | 1.2 | 134 | 122 | 2 | 1 |
| 25 | 1+2 | 11 | 220 | 8.8 | 176 | 59 | 6.5 | 160 | 27 | 2 | 13 |
| 26 | 1 | 11 | 550 | 0.2 | 10 | 62 | 0.2 | 169 | 40 | 3.1 | 1 |
| 27 | 2 | 11 | 367 | 0.4 | 13 | 32 | 0.2 | 115 | 77 | 4.8 | 1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boggioni, L.; Sidari, D.; Losio, S.; Stehling, U.M.; Auriemma, F.; Malafronte, A.; Di Girolamo, R.; De Rosa, C.; Tritto, I. Ethylene-co-norbornene Copolymerization Using a Dual Catalyst System in the Presence of a Chain Transfer Agent. Polymers 2019, 11, 554. https://doi.org/10.3390/polym11030554
Boggioni L, Sidari D, Losio S, Stehling UM, Auriemma F, Malafronte A, Di Girolamo R, De Rosa C, Tritto I. Ethylene-co-norbornene Copolymerization Using a Dual Catalyst System in the Presence of a Chain Transfer Agent. Polymers. 2019; 11(3):554. https://doi.org/10.3390/polym11030554
Chicago/Turabian StyleBoggioni, Laura, Diego Sidari, Simona Losio, Udo M. Stehling, Finizia Auriemma, Anna Malafronte, Rocco Di Girolamo, Claudio De Rosa, and Incoronata Tritto. 2019. "Ethylene-co-norbornene Copolymerization Using a Dual Catalyst System in the Presence of a Chain Transfer Agent" Polymers 11, no. 3: 554. https://doi.org/10.3390/polym11030554
APA StyleBoggioni, L., Sidari, D., Losio, S., Stehling, U. M., Auriemma, F., Malafronte, A., Di Girolamo, R., De Rosa, C., & Tritto, I. (2019). Ethylene-co-norbornene Copolymerization Using a Dual Catalyst System in the Presence of a Chain Transfer Agent. Polymers, 11(3), 554. https://doi.org/10.3390/polym11030554