Assessment of the Tumbling-Snake Model against Linear and Nonlinear Rheological Data of Bidisperse Polymer Blends



Abstract

{kind=link}
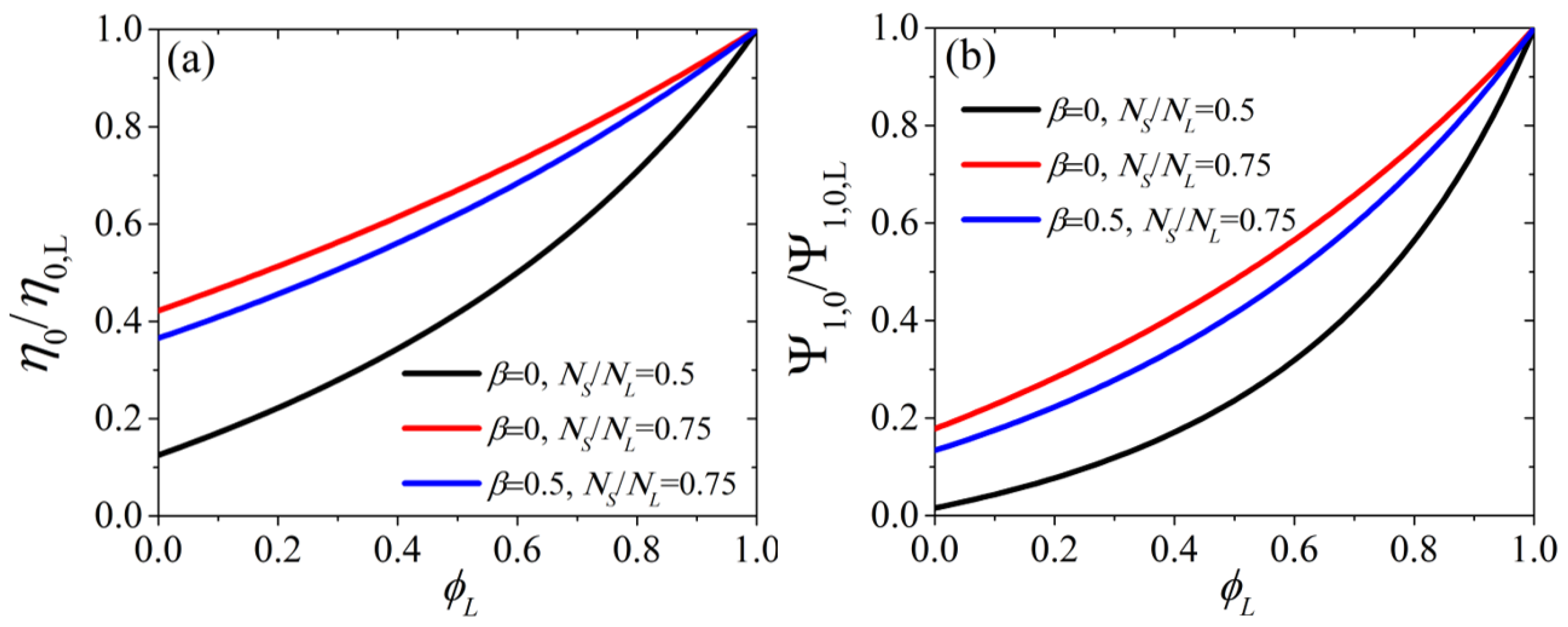{kind=link}
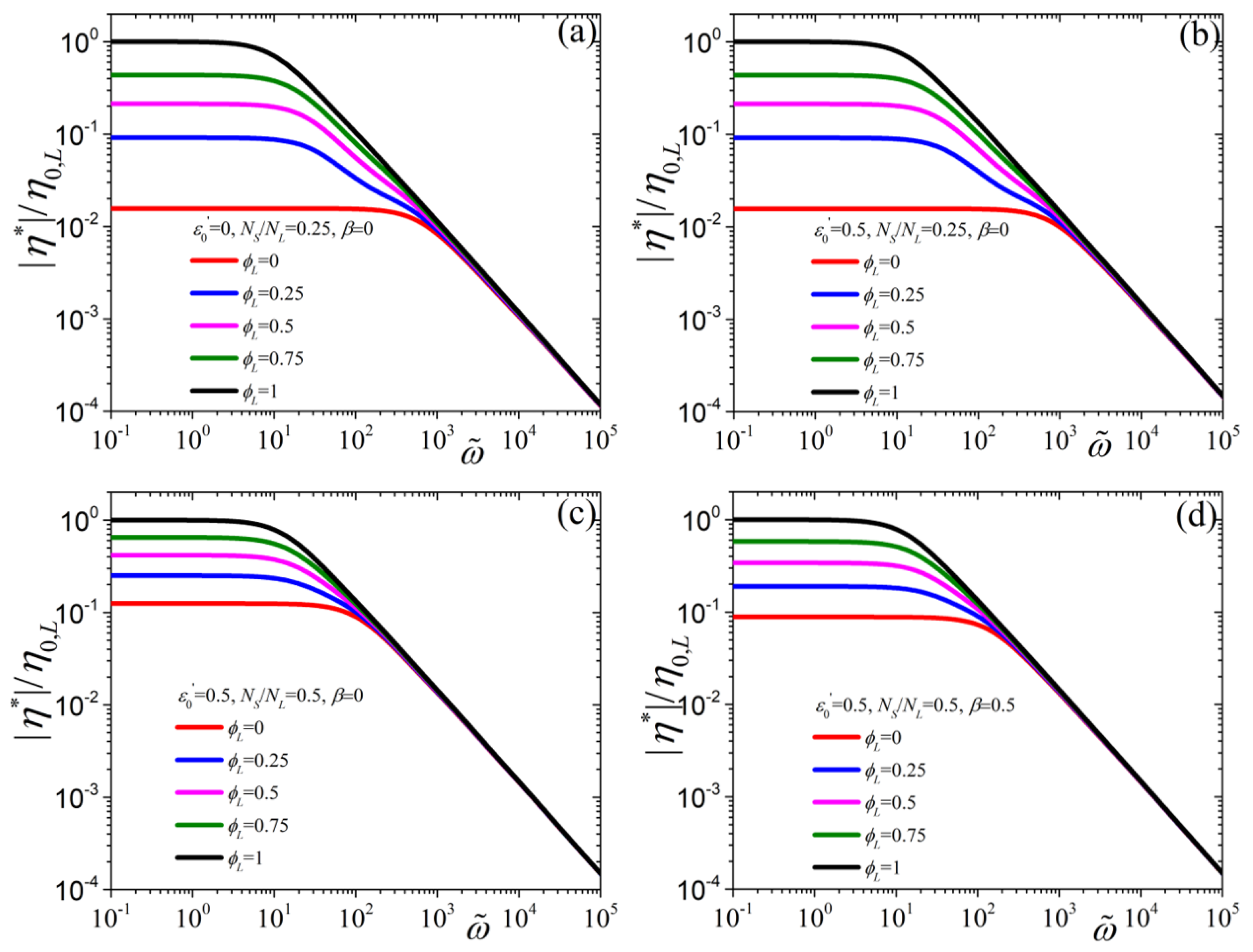{kind=link}
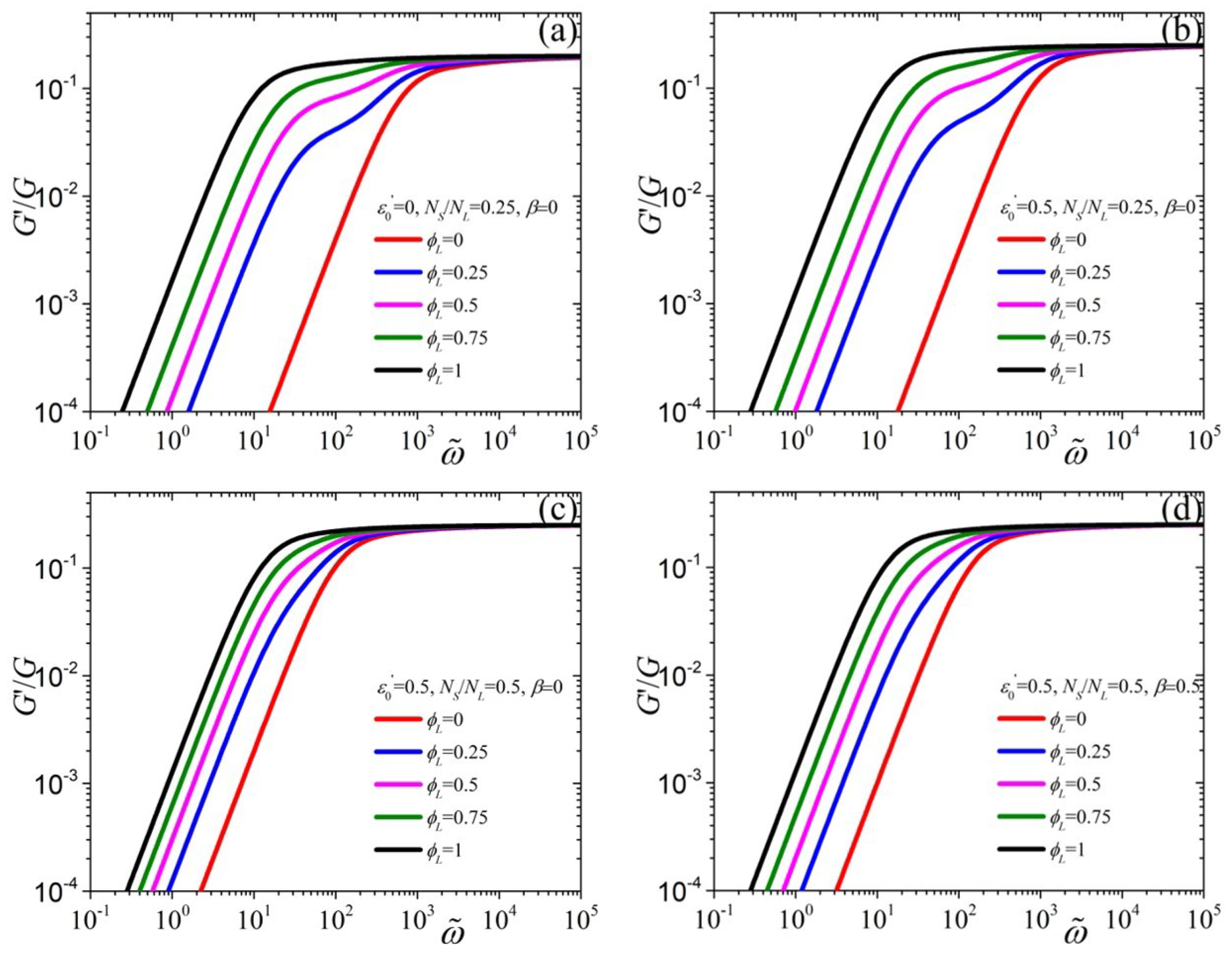{kind=link}
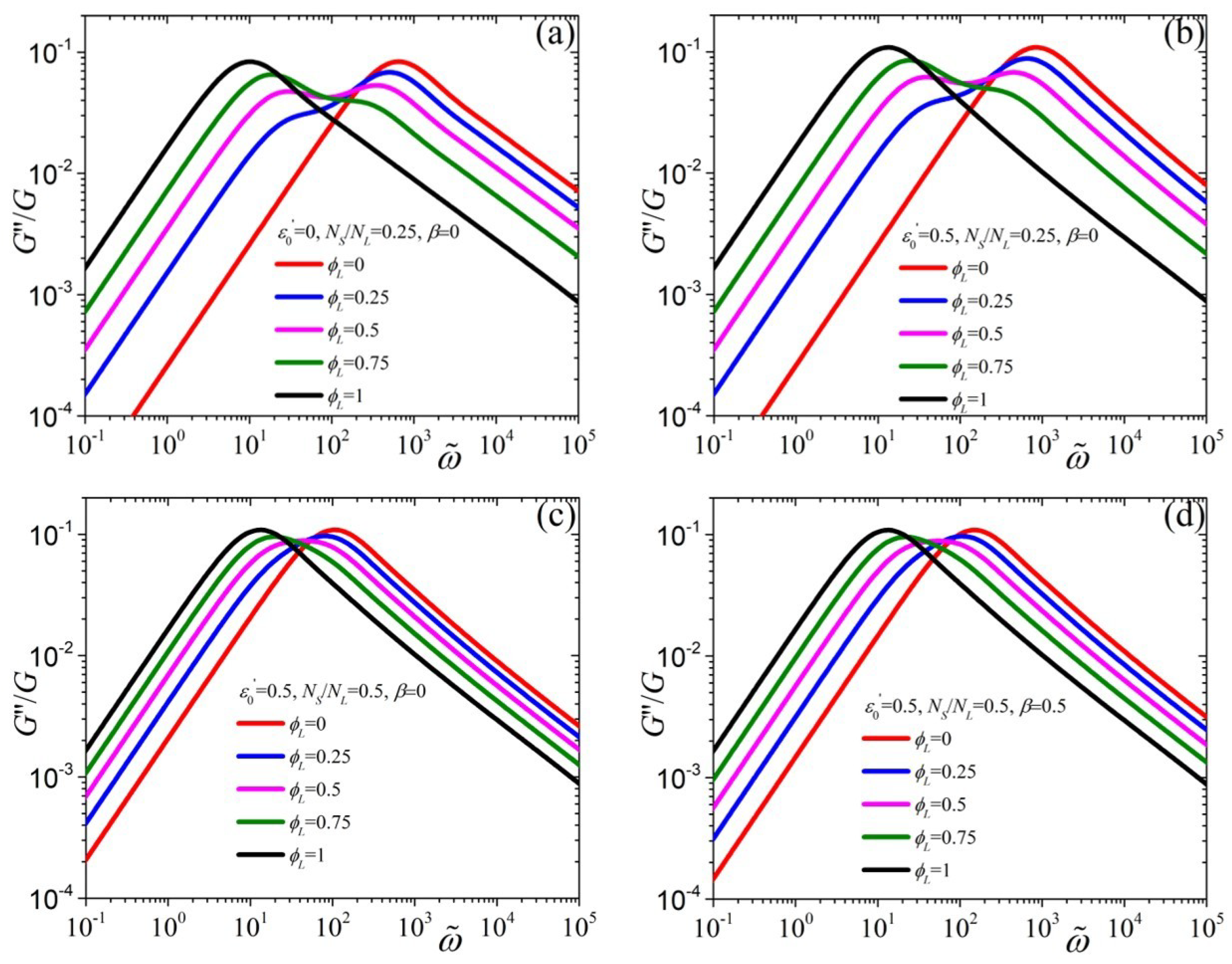{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Stress Tensor
3. Small Shear Rate Expansion in the Stationary State
3.1. Stationary Regime, Constant
3.2. Stationary Regime, Variable
4. Linear Viscoelastic Regime
5. Brownian Dynamics Results
5.1. Linear Viscoelastic Behavior
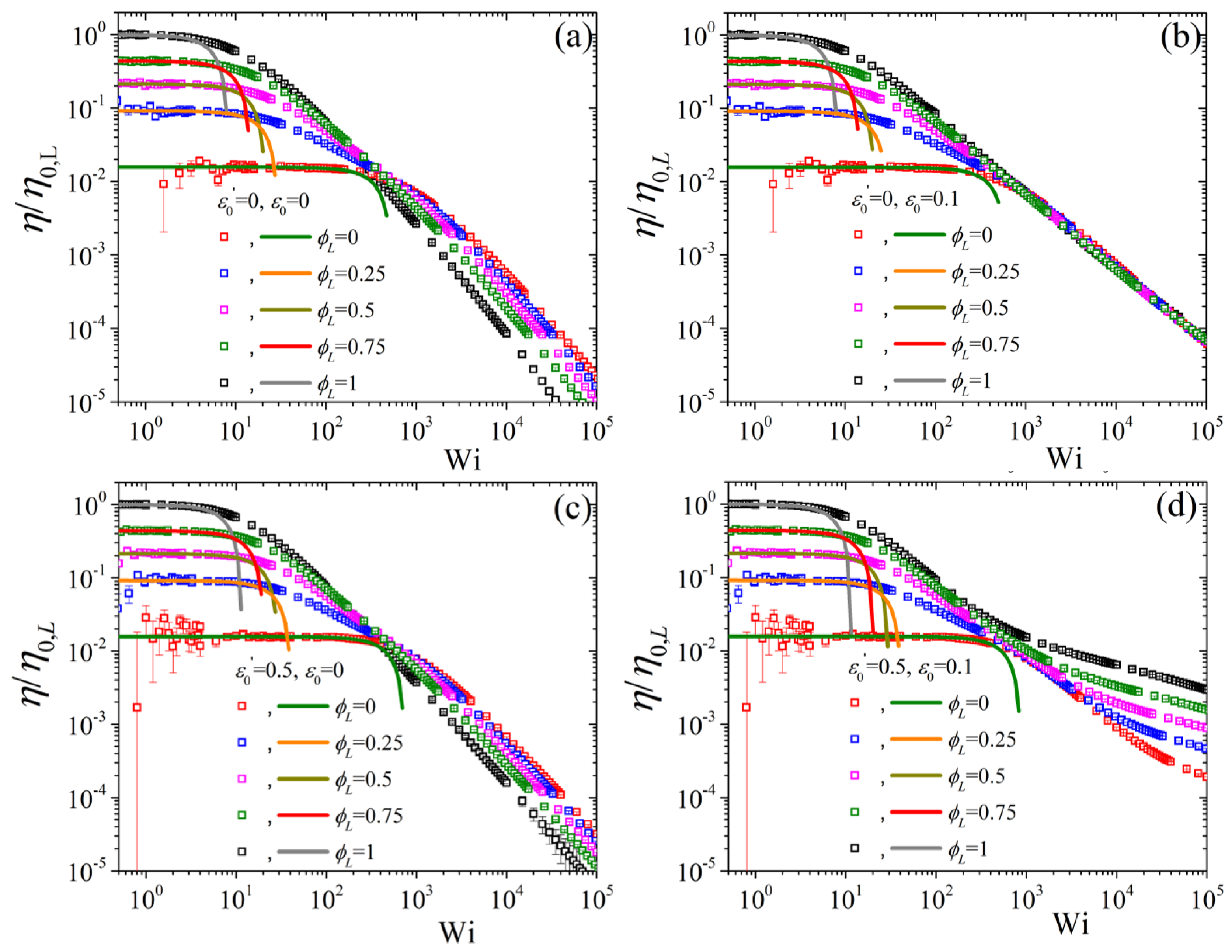
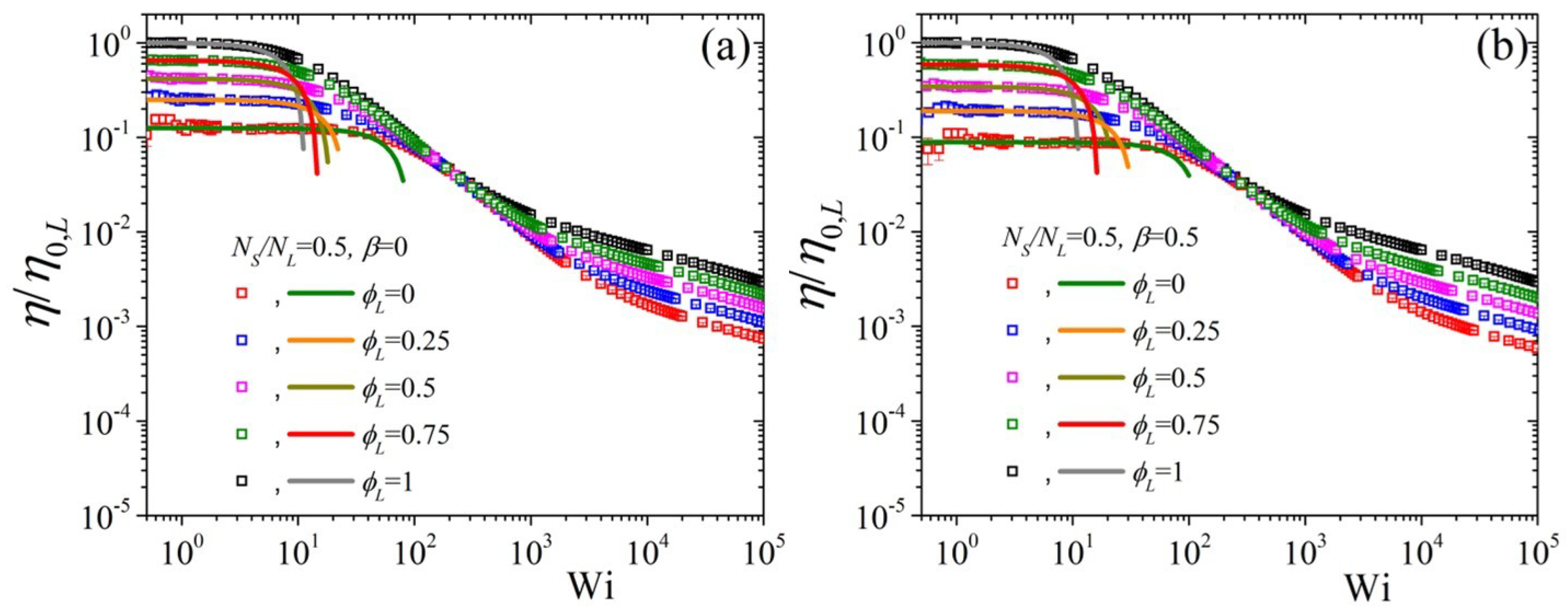
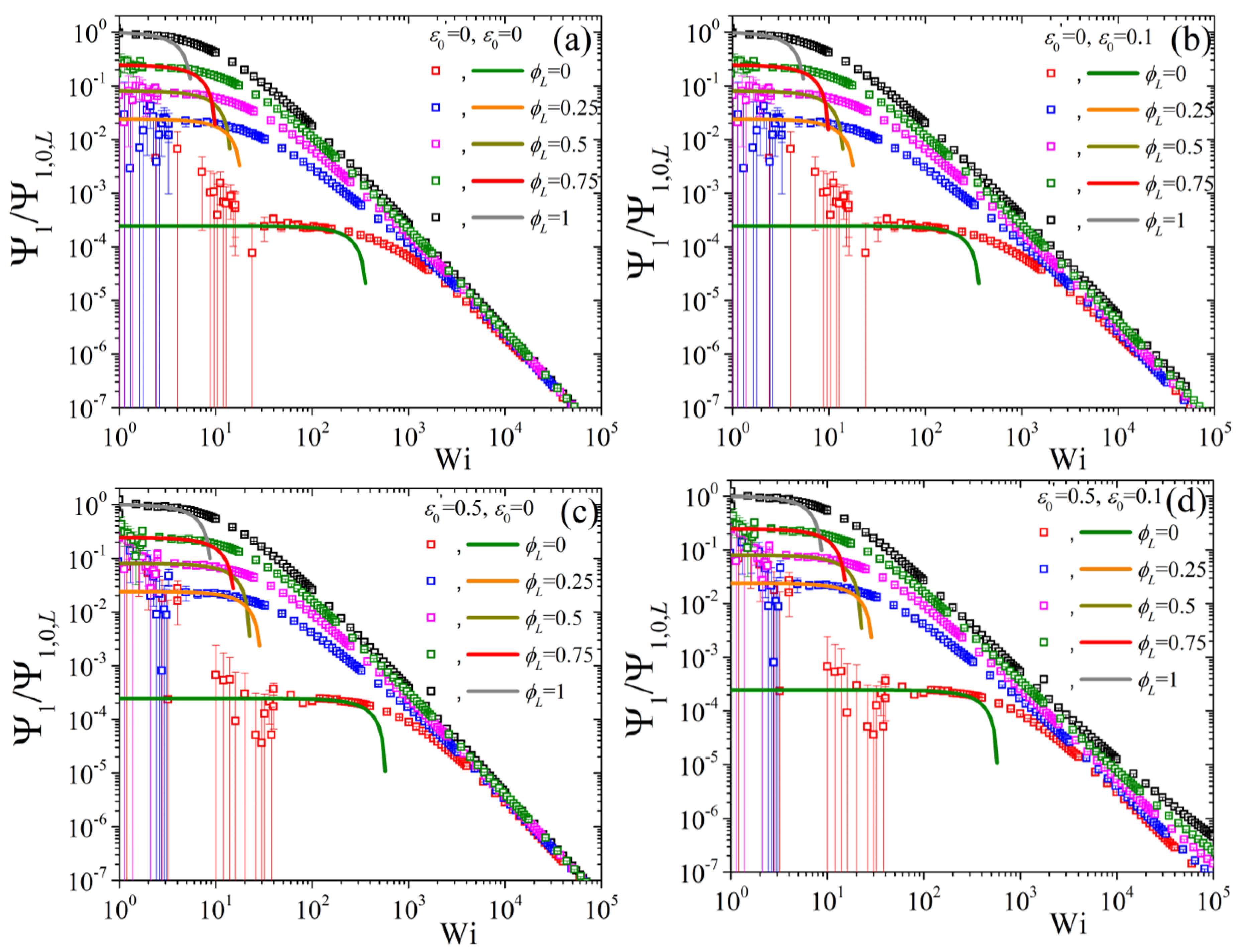
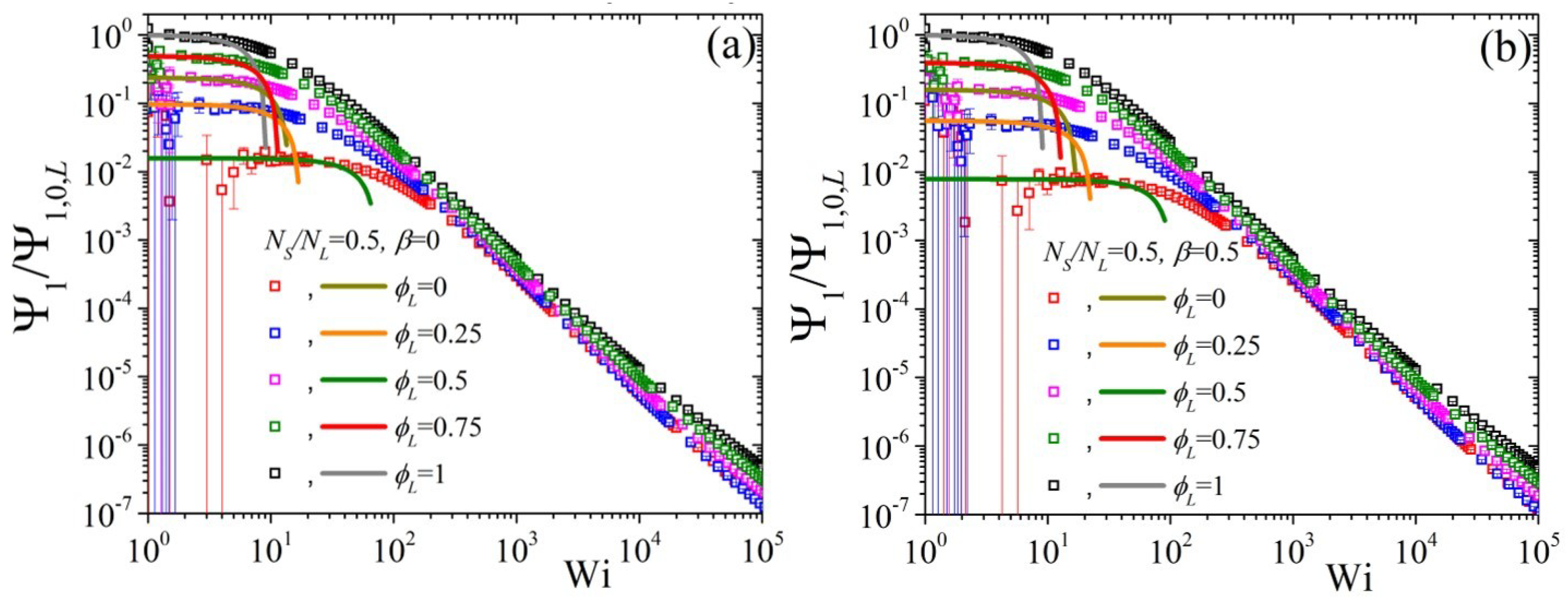
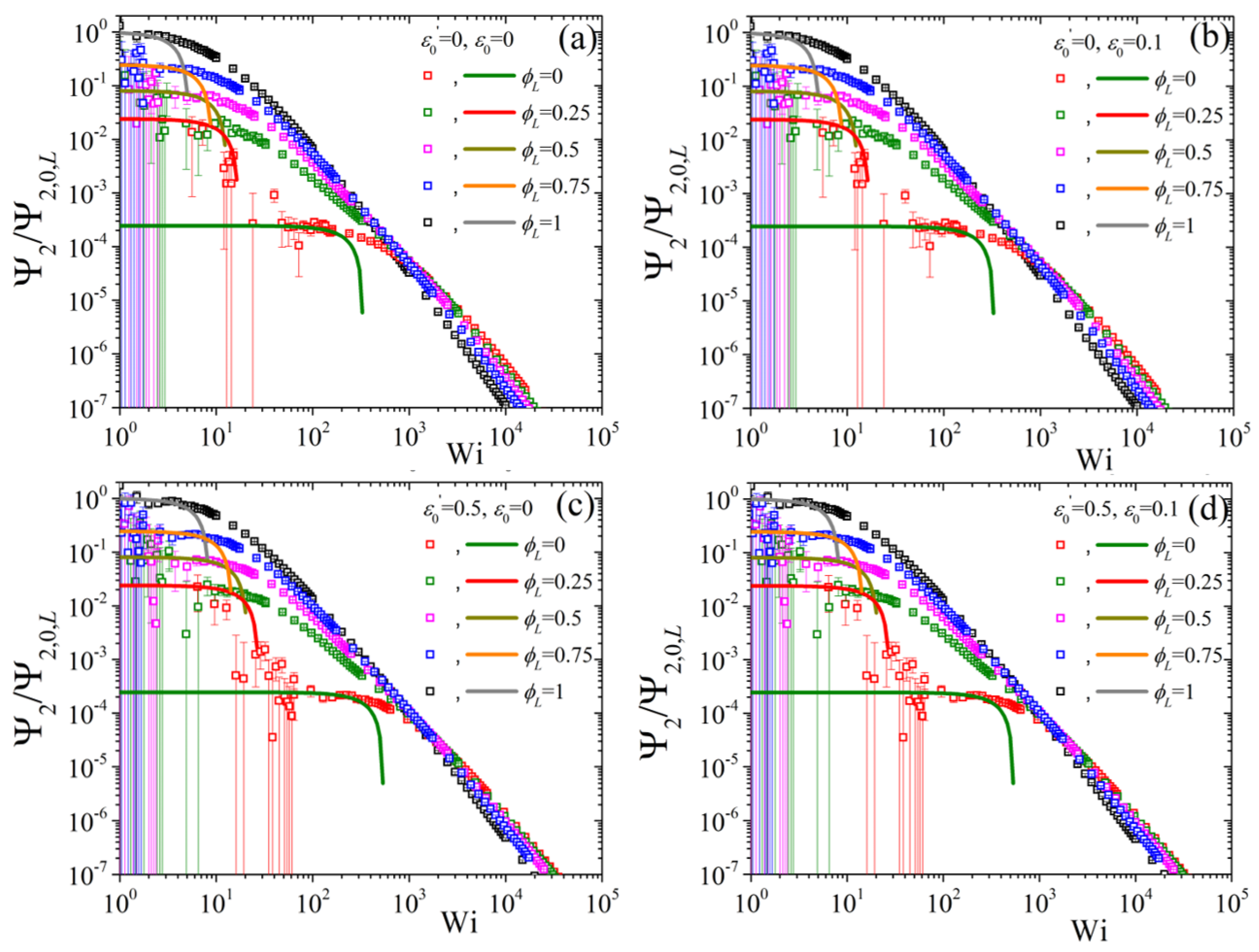
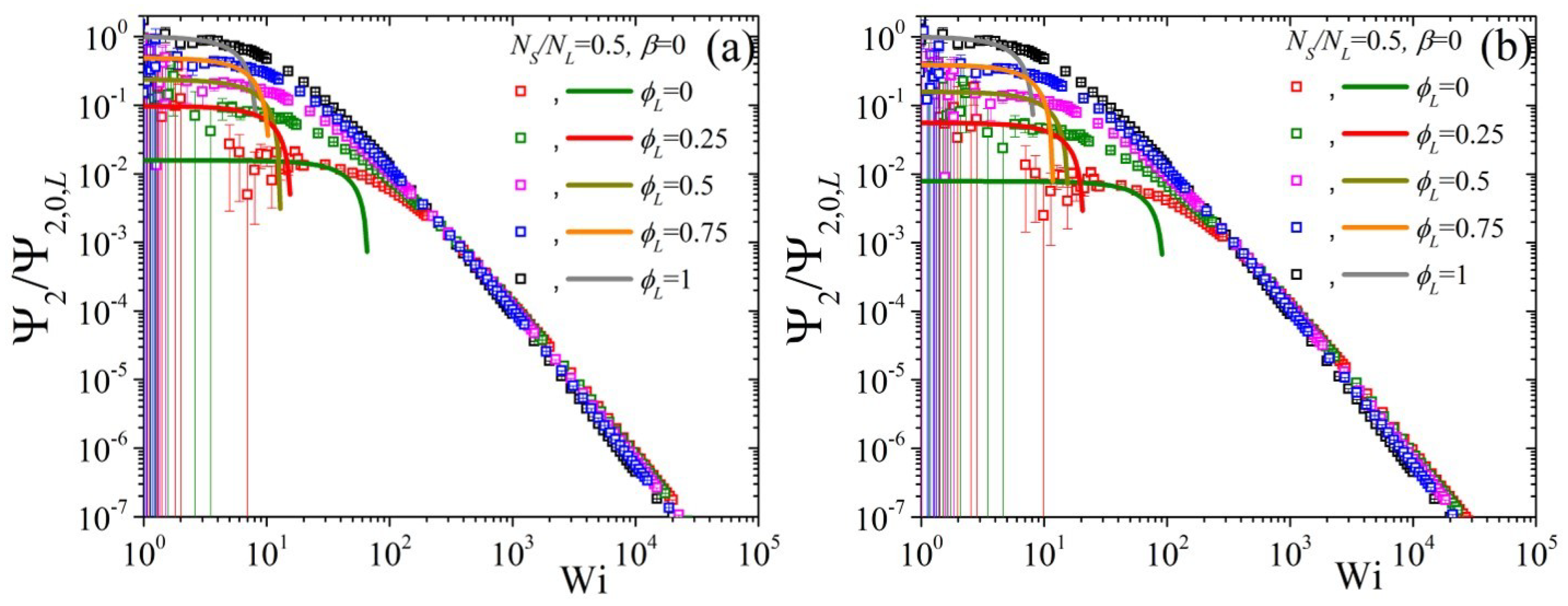
5.2. Non-Linear Regime
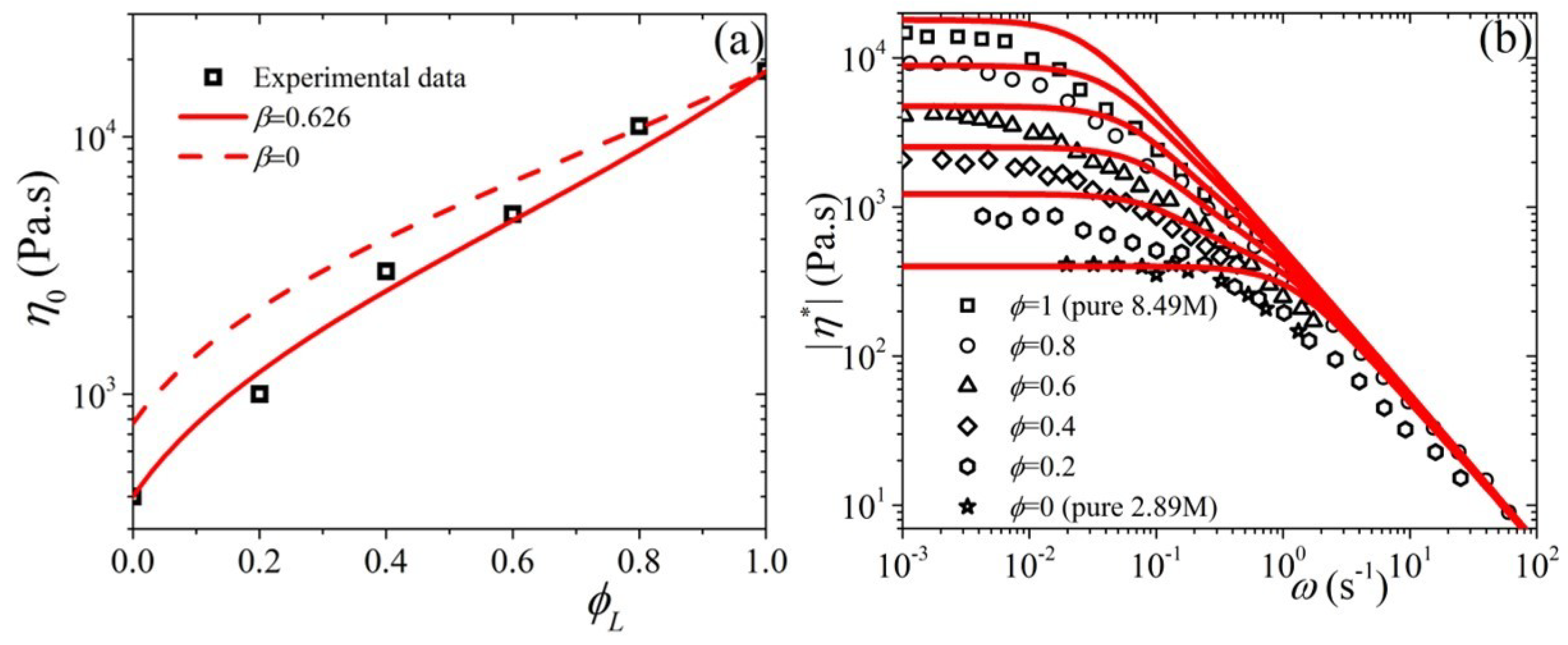
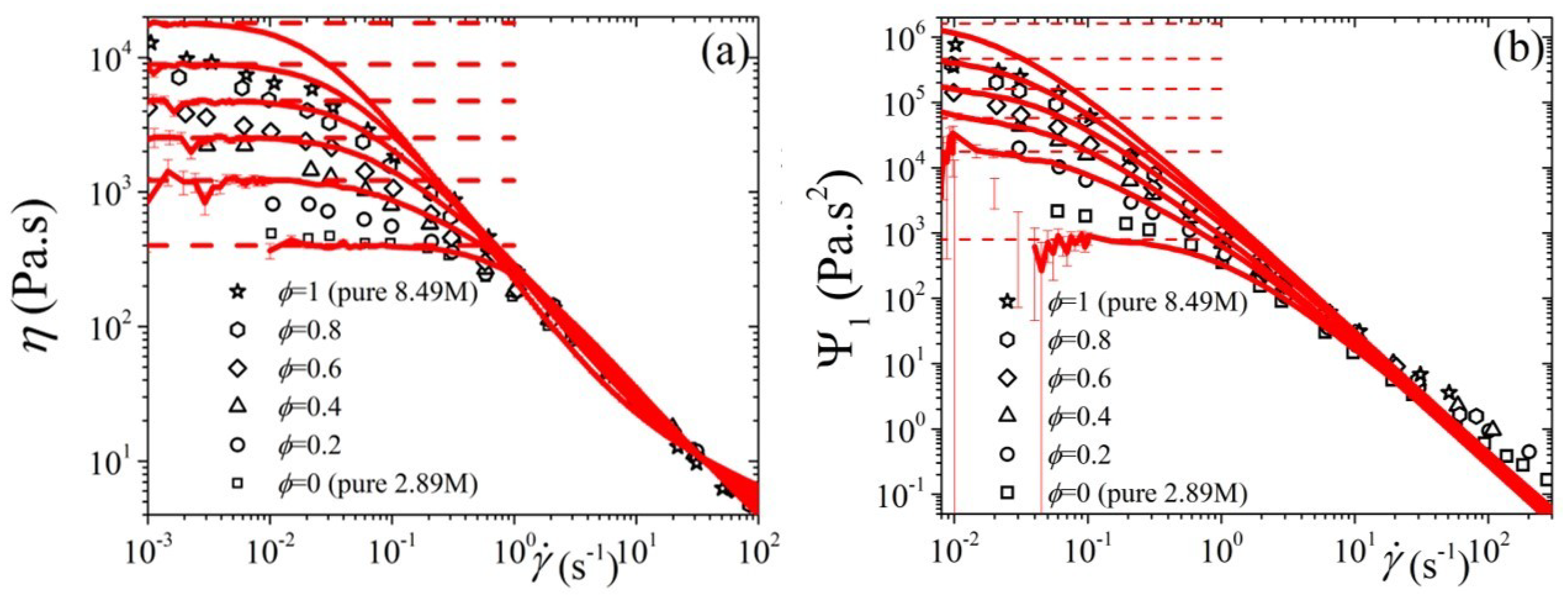
5.3. Comparison with Experimental Data
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Doi, M.; Edwards, S.F. Dynamics of concentrated polymer systems. 1. Brownian-motion in equilibrium state. J. Chem. Soc. Faraday Trans. 2 1978, 74, 1789–1801. [Google Scholar] [CrossRef]
- Doi, M.; Edwards, S.F. The Theory of Polymer Dynamics; Clarendon: Oxford, UK, 1986. [Google Scholar]
- De Gennes, P.G. Reptation of a polymer chain in presence of fixed obstacles. J. Chem. Phys. 1971, 55, 572–579. [Google Scholar] [CrossRef]
- Watanabe, H. Viscoelasticity and dynamics of entangled polymers. Prog. Polym. Sci. 1999, 24, 1253. [Google Scholar] [CrossRef]
- McLeish, T.C.B. Tube theory of entangled polymer dynamics. Adv. Phys. 2002, 51, 1379–1527. [Google Scholar] [CrossRef]
- Stephanou, P.S.; Mavrantzas, V.G. Quantitative predictions of the linear viscoelastic properties of entangled polyethylene and polybutadiene melts via modified versions of modern tube models on the basis of atomistic simulation data. J. Non-Newton. Fluid Mech. 2013, 200, 111–130. [Google Scholar] [CrossRef]
- Stephanou, P.S.; Mavrantzas, V.G. Accurate prediction of the linear viscoelastic properties of highly entangled mono and bidisperse polymer melts. J. Chem. Phys. 2014, 140, 214903. [Google Scholar] [CrossRef] [PubMed]
- Marrucci, G.; Grizzuti, N. Fast flows of concentrated polymers—Predictions of the tube model on chain stretching. Gazz. Chim. Ital. 1988, 118, 179–185. [Google Scholar]
- Ianniruberto, G.; Marrucci, G. A simple constitutive equation for entangled polymers with chain stretch. J. Rheol. 2001, 45, 1305–1318. [Google Scholar] [CrossRef]
- Stephanou, P.S.; Tsimouri, I.C.; Mavrantzas, V.G. Flow-induced orientation and stretching of entangled polymers in the framework of nonequilibrium thermodynamics. Macromolecules 2016, 49, 3161–3173. [Google Scholar] [CrossRef]
- Marrucci, G. Dynamics of entanglements: A nonlinear model consistent with the Cox-Merz rule. J. Non-Newton. Fluid Mech. 1996, 62, 279–289. [Google Scholar] [CrossRef]
- Ianniruberto, G.; Marrucci, G. On compatibility of the Cox-Merz rule with the model of Doi and Edwards. J. Non-Newton. Fluid Mech. 1996, 65, 241–246. [Google Scholar] [CrossRef]
- Ianniruberto, G.; Marrucci, G. Flow-induced orientation and stretching of entangled polymers. Philos. Trans. R. Soc. A 2003, 361, 677–687. [Google Scholar]
- Pattamaprom, C.; Larson, R.G. Constraint Release Effects in Monodisperse and Bidisperse Polystyrenes in Fast Transient Shearing Flows. Macromolecules 2001, 34, 5229–5237. [Google Scholar] [CrossRef]
- Pearson, D.S.; Kiss, A.D.; Fetters, L.J.; Doi, M. Flow-induced birefringence of concentrated polyisoprene solutions. J. Rheol. 1989, 33, 517–535. [Google Scholar] [CrossRef]
- Mead, D.W.; Larson, R.G.; Doi, M. A Molecular Theory for Fast Flows of Entangled Polymers. Macromolecules 1998, 31, 7895–7914. [Google Scholar] [CrossRef]
- Ye, X.; Larson, R.G.; Pattamaprom, C.; Sridhar, T. Extensional properties of monodisperse and bidisperse polystyrene solutions. J. Rheol. 2003, 47, 443–448. [Google Scholar] [CrossRef]
- Leygue, A.; Bailly, C.; Keunings, R. A tube-based constitutive equation for polydisperse entangled linear polymers. J. Non-Newton. Fluid Mech. 2006, 136, 1–16. [Google Scholar] [CrossRef]
- Read, D.J.; Jagannathan, K.; Sukumaran, S.K.; Auhl, D. A full-chain constitutive model for bidisperse blends of linear polymers. J. Rheol. 2012, 56, 823–873. [Google Scholar] [CrossRef]
- Graham, R.S.; Likhtman, A.E.; McLeish, T.C.B.; Milner, S.T. Microscopic theory of linear, entangled polymer chains under rapid deformation including chain stretch and convective constraint release. J. Rheol. 2003, 47, 1171–1200. [Google Scholar] [CrossRef]
- Bird, R.B.; Armstrong, R.C.; Hassager, O. Dynamics of Polymeric Liquids: Vol. 2, Kinetic Theory; John Wiley & Sons: New York, NY, USA, 1987. [Google Scholar]
- Curtiss, C.F.; Bird, R.B. A kinetic-theory for polymer melts. 1. The equation for the single-link orientational distribution function. J. Chem. Phys. 1981, 74, 2016–2025. [Google Scholar] [CrossRef]
- Curtiss, C.F.; Bird, R.B. A kinetic-theory for polymer melts. 2. The stress tensor and the rheological equation of state. J. Chem. Phys. 1981, 74, 2026–2033. [Google Scholar] [CrossRef]
- Stephanou, P.S.; Schweizer, T.; Kröger, M. Communication: Appearance of undershoots in start-up shear: Experimental findings captured by tumbling-snake dynamics. J. Chem. Phys. 2017, 146, 161101. [Google Scholar] [CrossRef] [PubMed]
- Bird, R.B.; Saab, H.H.; Curtiss, C.F. A kinetic-theory for polymer melts. 4. Rheological properties for shear flows. J. Chem. Phys 1982, 77, 4747–4757. [Google Scholar] [CrossRef]
- Bird, R.B.; Saab, H.H.; Curtiss, C.F. A kinetic-theory for polymer melts. 3. Elongational flows. J. Phys. Chem. 1982, 86, 1102–1106. [Google Scholar] [CrossRef]
- Stephanou, P.S.; Kröger, M. Solution of the complete Curtiss-Bird model for polymeric liquids subjected to simple shear flow. J. Chem. Phys. 2016, 144, 124905. [Google Scholar] [CrossRef]
- Stephanou, P.S.; Kröger, M. Non-constant link tension coefficient in the tumbling-snake model subjected to simple shear. J. Chem. Phys. 2017, 147, 174903. [Google Scholar] [CrossRef] [PubMed]
- Stephanou, P.S.; Kröger, M. From intermediate anisotropic to isotropic friction at large strain rates to account for viscosity thickening in polymer solutions. J. Chem. Phys. 2018, 148, 184903. [Google Scholar] [CrossRef] [PubMed]
- Stephanou, P.S.; Kröger, M. Tumbling-Snake Model for Polymeric Liquids Subjected to Biaxial Elongational Flows with a Focus on Planar Elongation. Polymers 2018, 10, 329. [Google Scholar] [CrossRef]
- Kröger, M. Models for Polymeric and Anisotropic Liquids; Springer: New York, NY, USA, 2005; Volume 675. [Google Scholar]
- Luap, C.; Müller, C.; Schweizer, T.; Venerus, D.C. Simultaneous stress and birefringence measurements during uniaxial elongation of polystyrene melts with narrow molecular weight distribution. Rheol. Acta 2005, 45, 83–91. [Google Scholar] [CrossRef]
- Schweizer, T.; Hostettler, J.; Mettler, F. A shear rheometer for measuring shear stress and both normal stress differences in polymer melts simultaneously: The MTR 25. Rheol. Acta 2008, 47, 943–957. [Google Scholar] [CrossRef]
- Auhl, D.; Ramirez, J.; Likhtman, A.E.; Chambon, P.; Fernyhough, C. Linear and nonlinear shear flow behavior of monodisperse polyisoprene melts with a large range of molecular weights. J. Rheol. 2008, 52, 801–835. [Google Scholar] [CrossRef]
- Costanzo, S.; Huang, Q.; Ianniruberto, G.; Marrucci, G.; Hassager, O.; Vlassopoulos, D. Shear and extensional rheology of polystyrene melts and solutions with the same number of entanglements. Macromolecules 2016, 49, 3925–3935. [Google Scholar] [CrossRef]
- Sefiddashti, M.H.N.; Edwards, B.J.; Khomami, B. Individual chain dynamics of a polyethylene melt undergoing steady shear flow. J. Rheol. 2015, 59, 1–35. [Google Scholar] [CrossRef]
- Sefiddashti, M.H.N.; Edwards, B.J.; Khomami, B. Steady shearing flow of a moderately entangled polyethylene liquid. J. Rheol. 2016, 60, 1227–1244. [Google Scholar] [CrossRef]
- Kim, J.M.; Baig, C. Precise analyis of polymer rotational dynamics. Sci. Rep. 2016, 6, 19127. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Alvarez, N.J.; Matsumiya, Y.; Rasmussen, H.K.; Watanabe, H.; Hassager, O. Extensional rheology of entangled polystyrene solutions suggests importance of nematic interactions. ACS Macro Lett. 2013, 2, 741–744. [Google Scholar] [CrossRef]
- Huang, Q.; Mednova, O.; Rasmussen, H.K.; Alvarez, N.J.; Skov, A.L.; Almdal, K.; Hassager, O. Concentrated polymer solutions are different from melts: Role of entanglement molecular weight. Macromolecules 2013, 46, 5026–5035. [Google Scholar] [CrossRef]
- Huang, Q.; Hengeller, L.; Alvarez, N.J.; Hassager, O. Bridging the gap between polymer melts and solutions in extensional rheology. Macromolecules 2015, 48, 4158–4163. [Google Scholar] [CrossRef]
- Schieber, J.D.; Curtiss, C.F.; Bird, R.B. Kinetic Theory of Polymer melts. 7. Polydisprese Effects. Ind. Chem. Fundam. 1986, 25, 471–475. [Google Scholar] [CrossRef]
- Schieber, J.D. Kinetic theory of polymer melts. VIII. Rheological properties of polydisperse mixtures. J. Chem. Phys. 1987, 87, 4917–4927. [Google Scholar] [CrossRef]
- Schieber, J.D. Kinetic theory of polymer melts. IX. Comparisons with experimental data. J. Chem. Phys. 1987, 87, 4928–4936. [Google Scholar] [CrossRef]
- Rubinstein, M.; Colby, R.H. Polymer Physics; Oxford University Press: Oxford, UK, 2003. [Google Scholar]
- Stephanou, P.S.; Baig, C.; Mavrantzas, V.G. Toward an improved description of constraint release and contour length fluctuations in tube models for entangled polymer melts guided by atomistic simulations. Macromol. Theory Simul. 2011, 20, 752–768. [Google Scholar] [CrossRef]
- Stephanou, P.S.; Baig, C.; Mavrantzas, V.G. Projection of atomistic simulation data for the dynamics of entangled polymers onto the tube theory: Calculation of the segment survival probability function and comparison with modern tube models. Soft Matter 2011, 7, 380–395. [Google Scholar] [CrossRef]
- Kröger, M.; Hess, S. Viscoelasticity of polymeric melts and concentrated solutions. The effect of flow-induced alignment of chain ends. Physica A 1993, 195, 336–353. [Google Scholar] [CrossRef]
- Fang, J.; Kröger, M.; Öttinger, H.C. A thermodynamically admissible reptation model for fast flows of entangled polymers. II. Model predictions for shear and extensional flows. J. Rheol. 2000, 44, 1293–1317. [Google Scholar] [CrossRef]
- Öttinger, H.C. Thermodynamically admissible reptation models with anisotropic tube cross sections and convective constraint release. J. Non-Newton. Fluid Mech. 2000, 89, 165–185. [Google Scholar] [CrossRef]












© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stephanou, P.S.; Kröger, M. Assessment of the Tumbling-Snake Model against Linear and Nonlinear Rheological Data of Bidisperse Polymer Blends. Polymers 2019, 11, 376. https://doi.org/10.3390/polym11020376
Stephanou PS, Kröger M. Assessment of the Tumbling-Snake Model against Linear and Nonlinear Rheological Data of Bidisperse Polymer Blends. Polymers. 2019; 11(2):376. https://doi.org/10.3390/polym11020376
Chicago/Turabian StyleStephanou, Pavlos S., and Martin Kröger. 2019. "Assessment of the Tumbling-Snake Model against Linear and Nonlinear Rheological Data of Bidisperse Polymer Blends" Polymers 11, no. 2: 376. https://doi.org/10.3390/polym11020376
APA StyleStephanou, P. S., & Kröger, M. (2019). Assessment of the Tumbling-Snake Model against Linear and Nonlinear Rheological Data of Bidisperse Polymer Blends. Polymers, 11(2), 376. https://doi.org/10.3390/polym11020376