Chemical Modification and Foam Processing of Polylactide (PLA)



Abstract
:
1. Introduction
2. Chemical Modifications
2.1. Functional Group Reaction
2.1.1. Epoxide
2.1.2. Diisocyanate
2.1.3. Dianhydride
2.1.4. Oxazoline
2.1.5. Carbodiimide
2.1.6. Phosphite
2.2. Free-Radical Reaction
2.2.1. Peroxide
2.2.2. Grafting
3. Rheological Behavior
3.1. Shear Rheology
3.1.1. Increased Zero Shear Viscosity
3.1.2. Pronounced Shear Thinning Effect
3.1.3. Increased Shear Viscosity
3.1.4. Improved Melt Elasticity
3.1.5. Enhanced Melt Stability
3.2. Elongational Rheology
3.2.1. Improved Melt Strength
3.2.2. Strain Hardening
4. Crystallization Behavior
4.1. Influence of Chemical Modification
4.2. Influence of Nucleating Agents
4.3. Influence of Plasticization
4.4. Influence of Deformation
4.5. Influence of Thermal Treatment
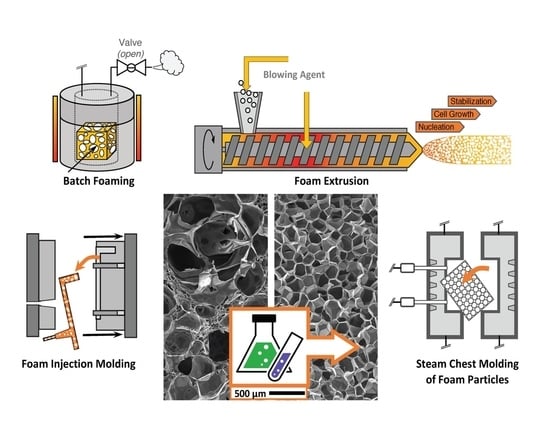
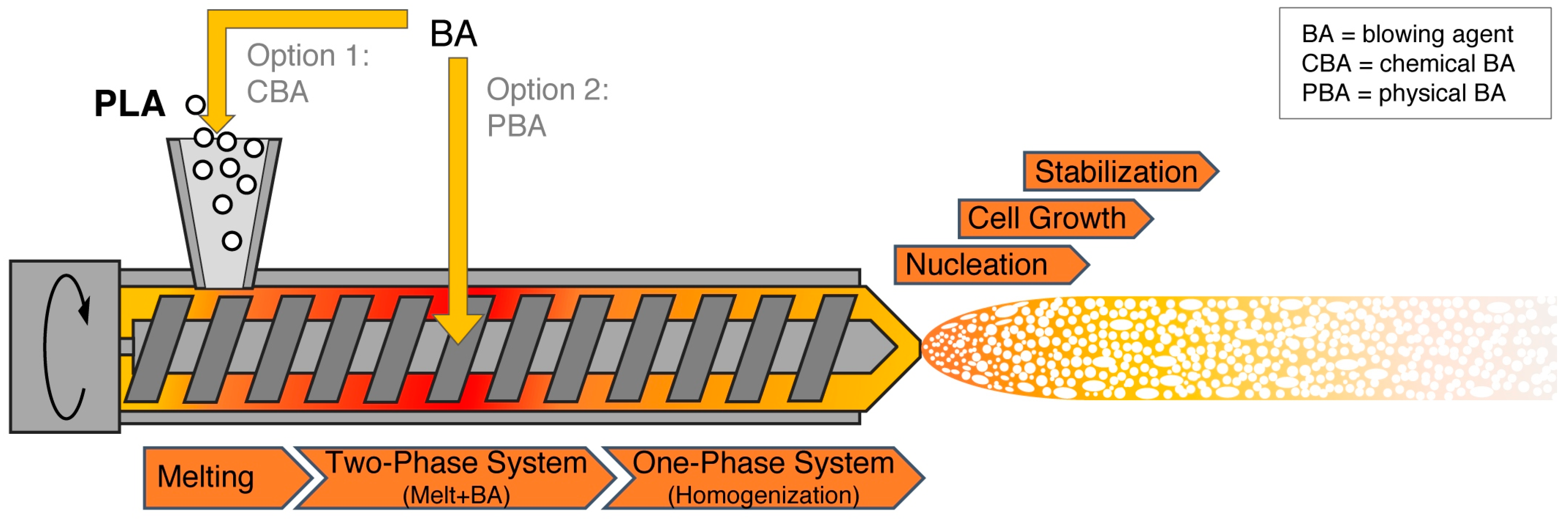
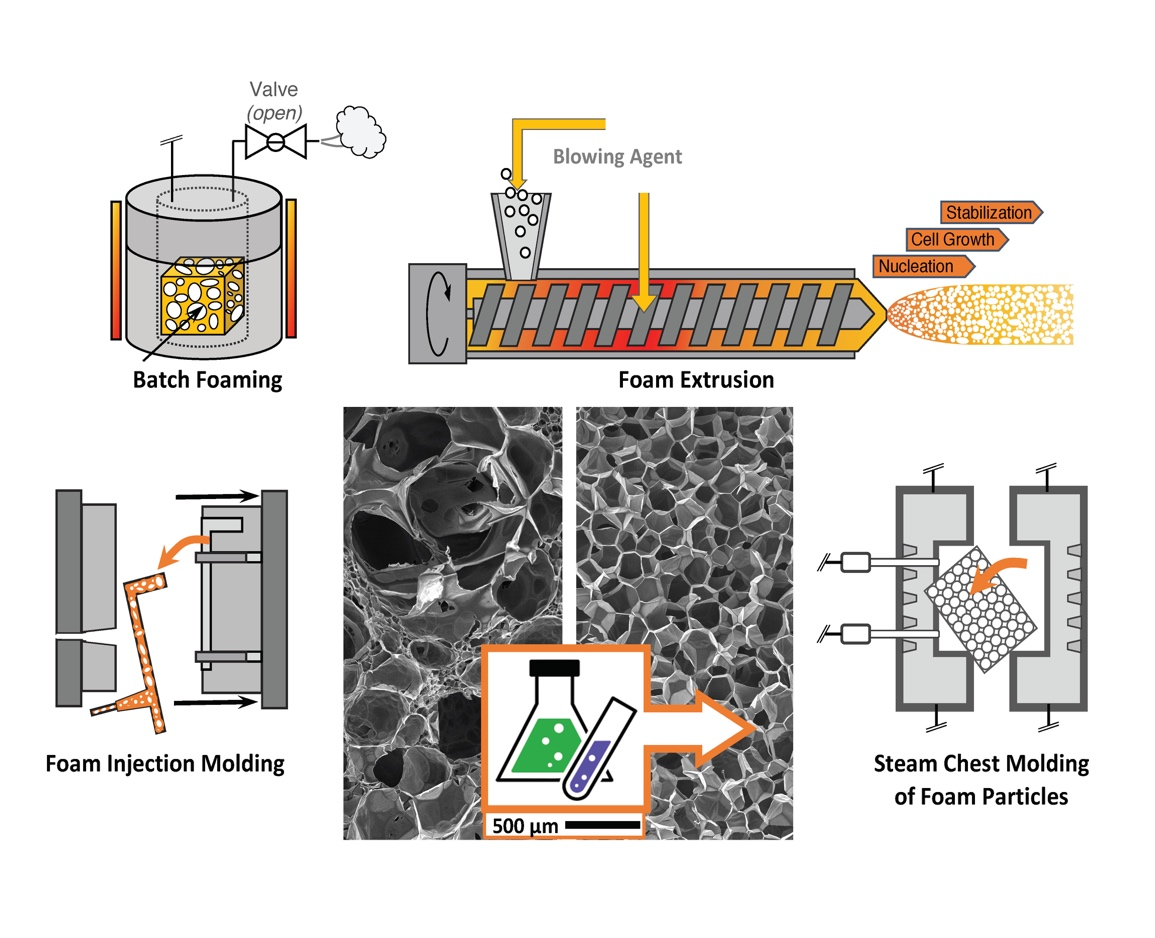
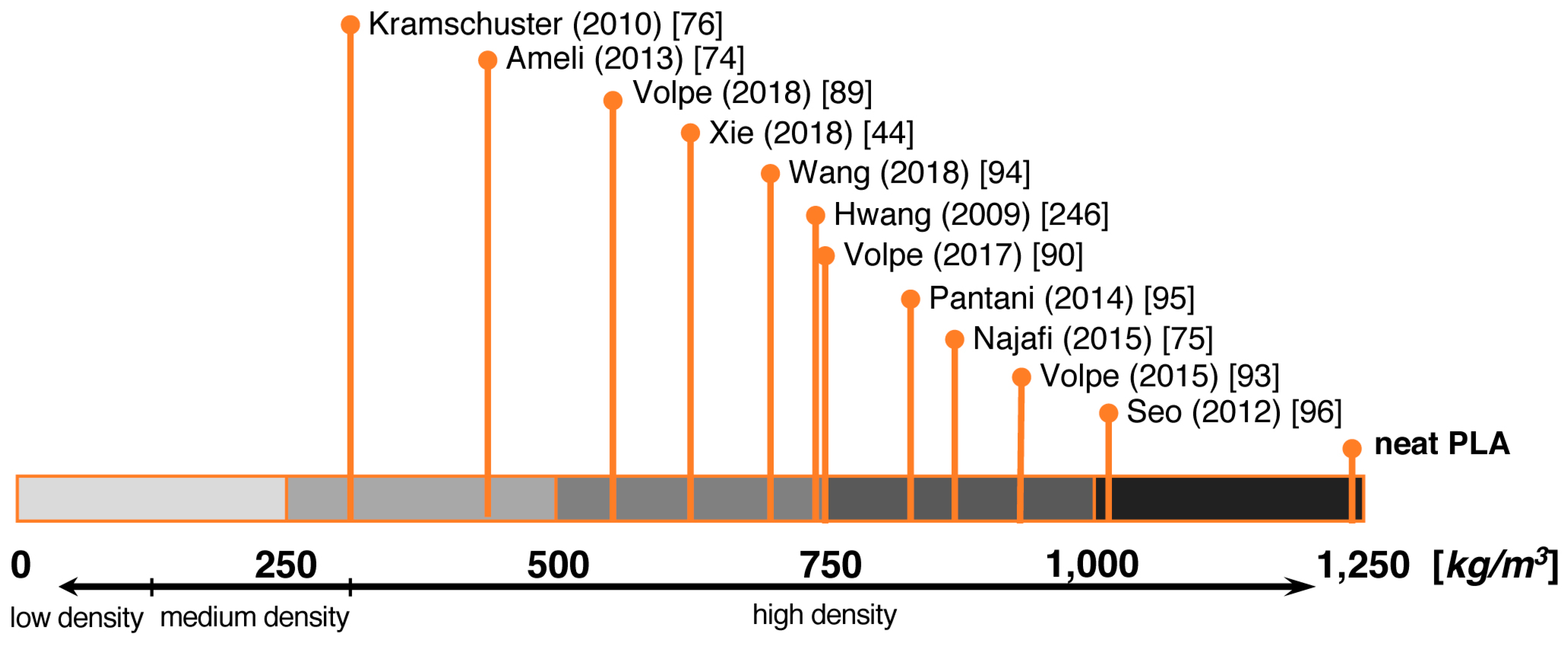
5. Processes
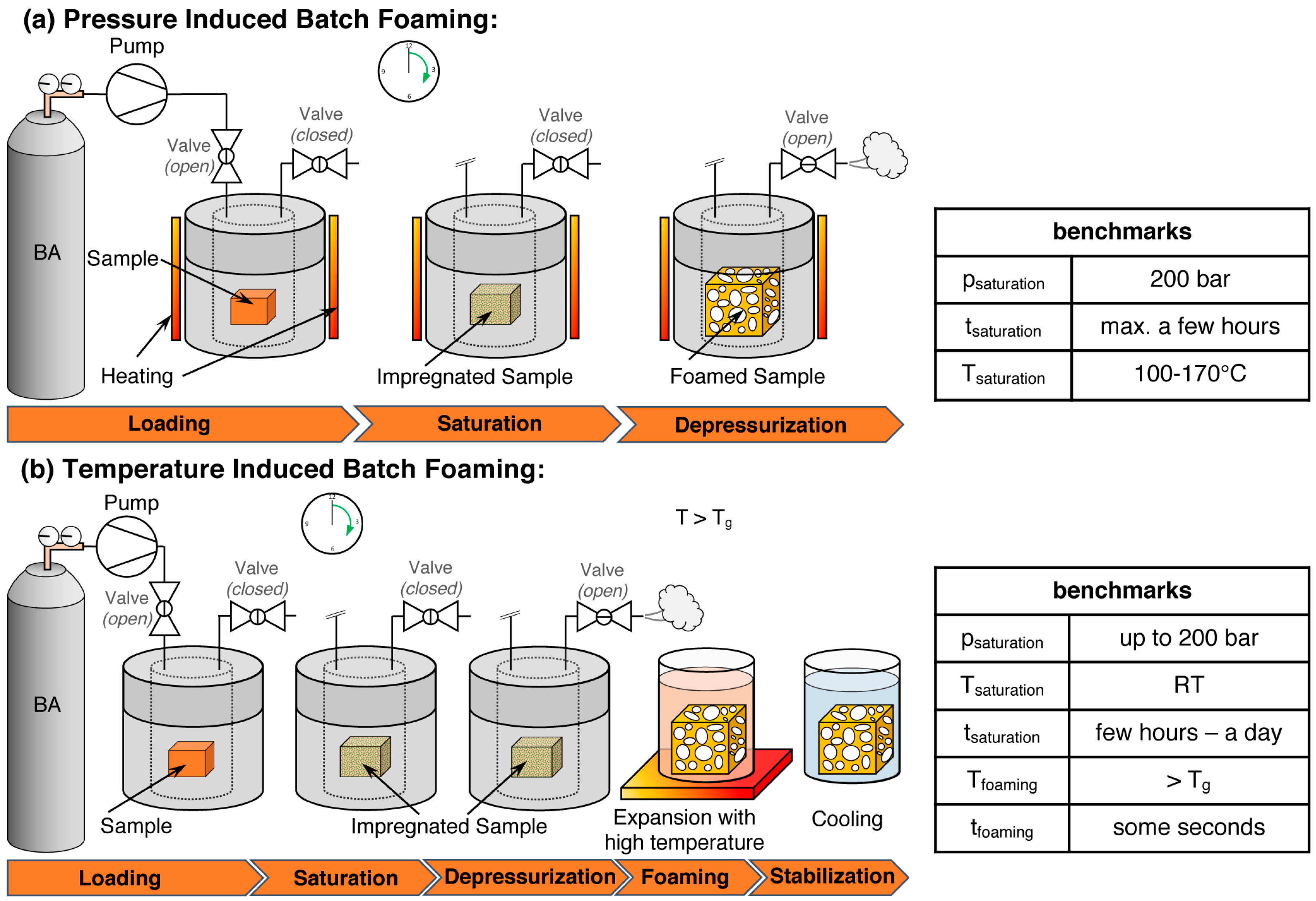
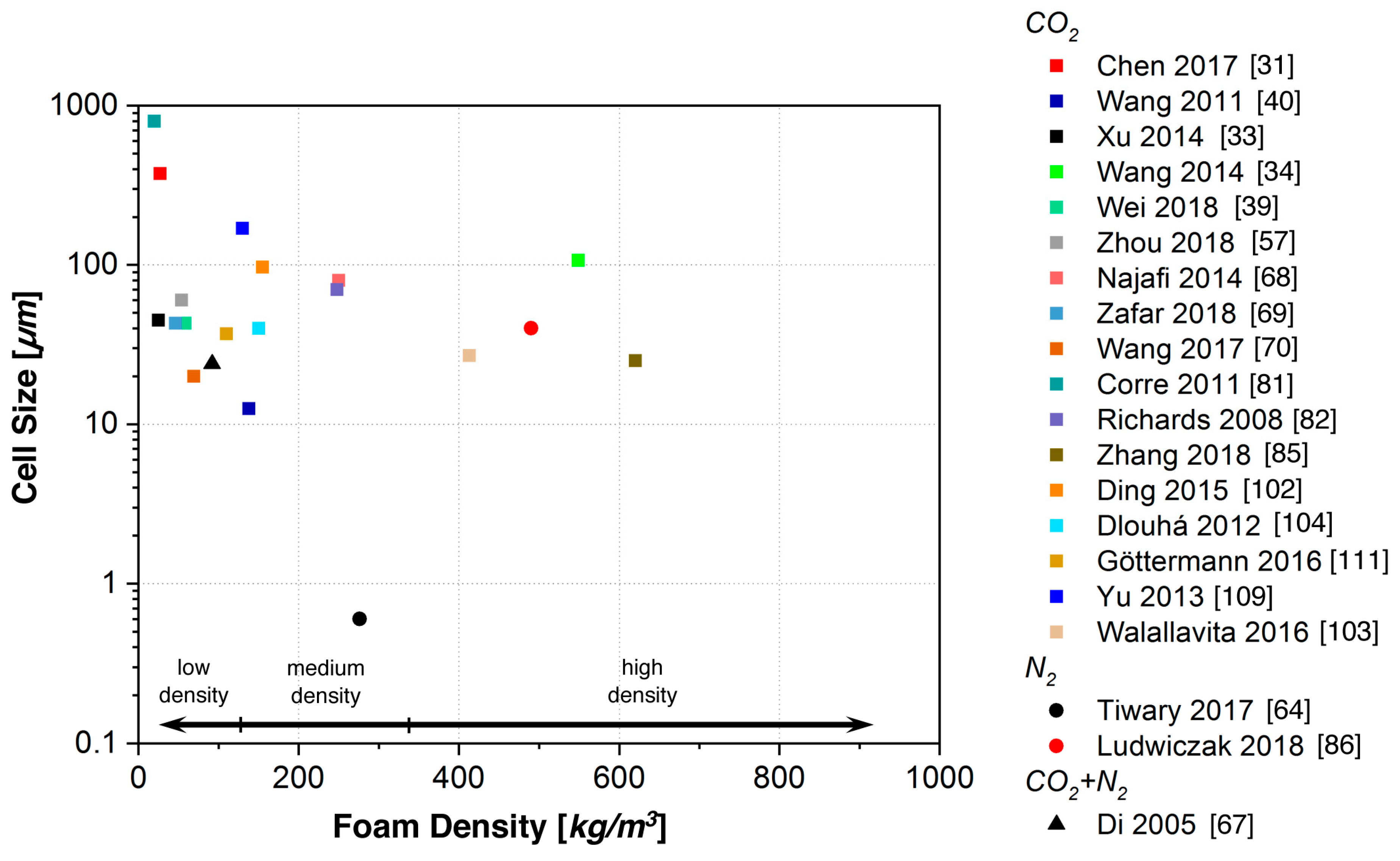
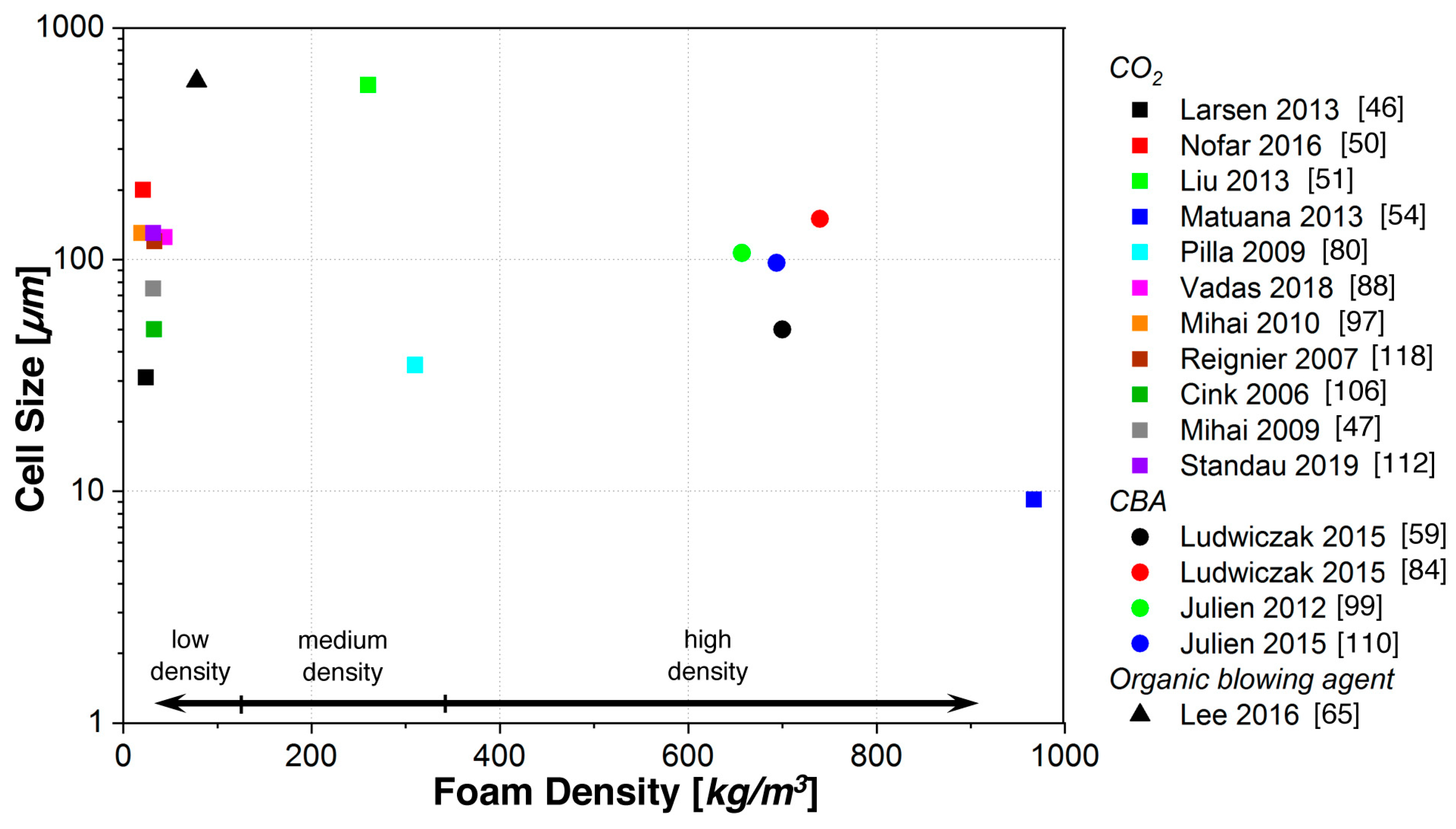
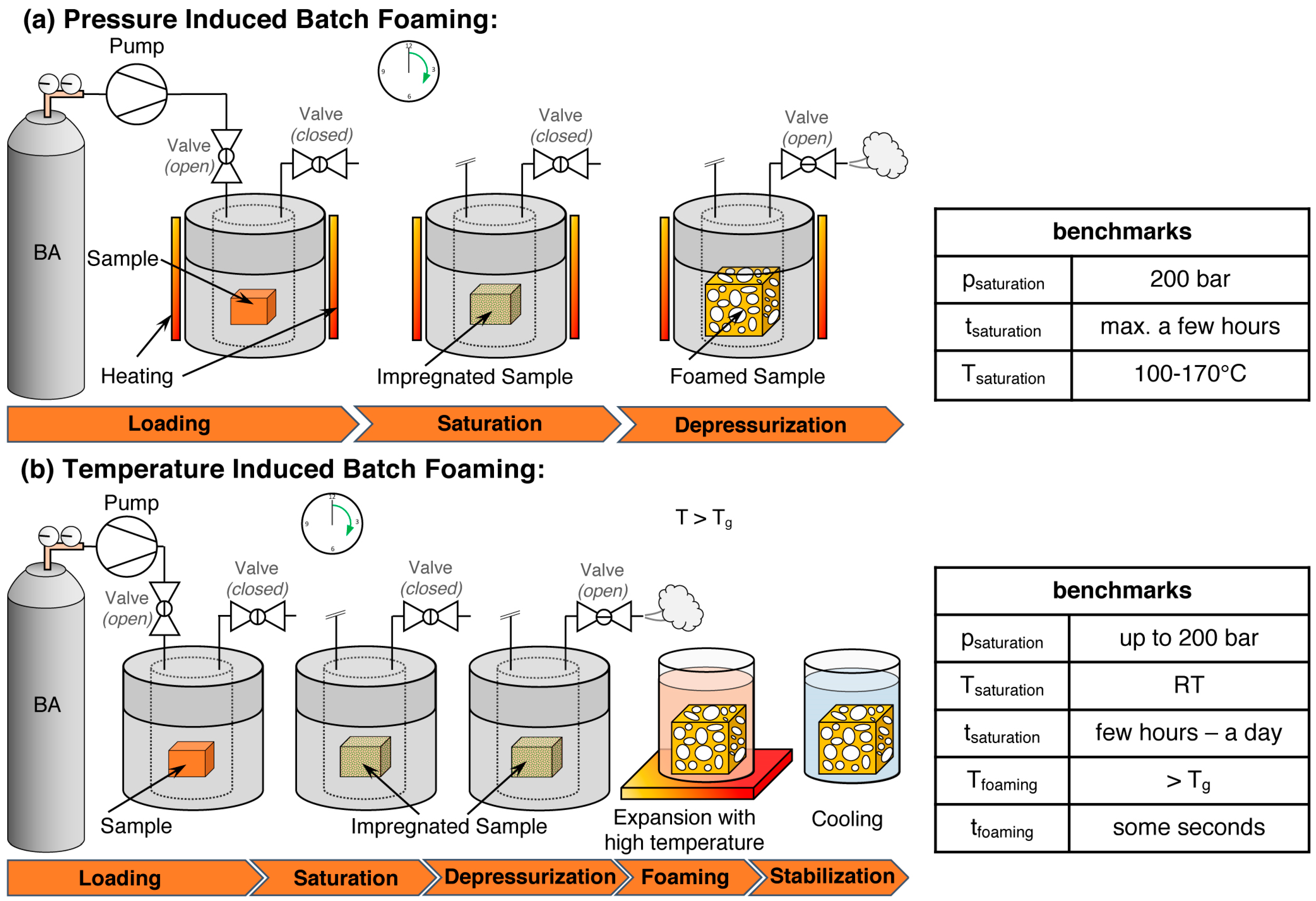
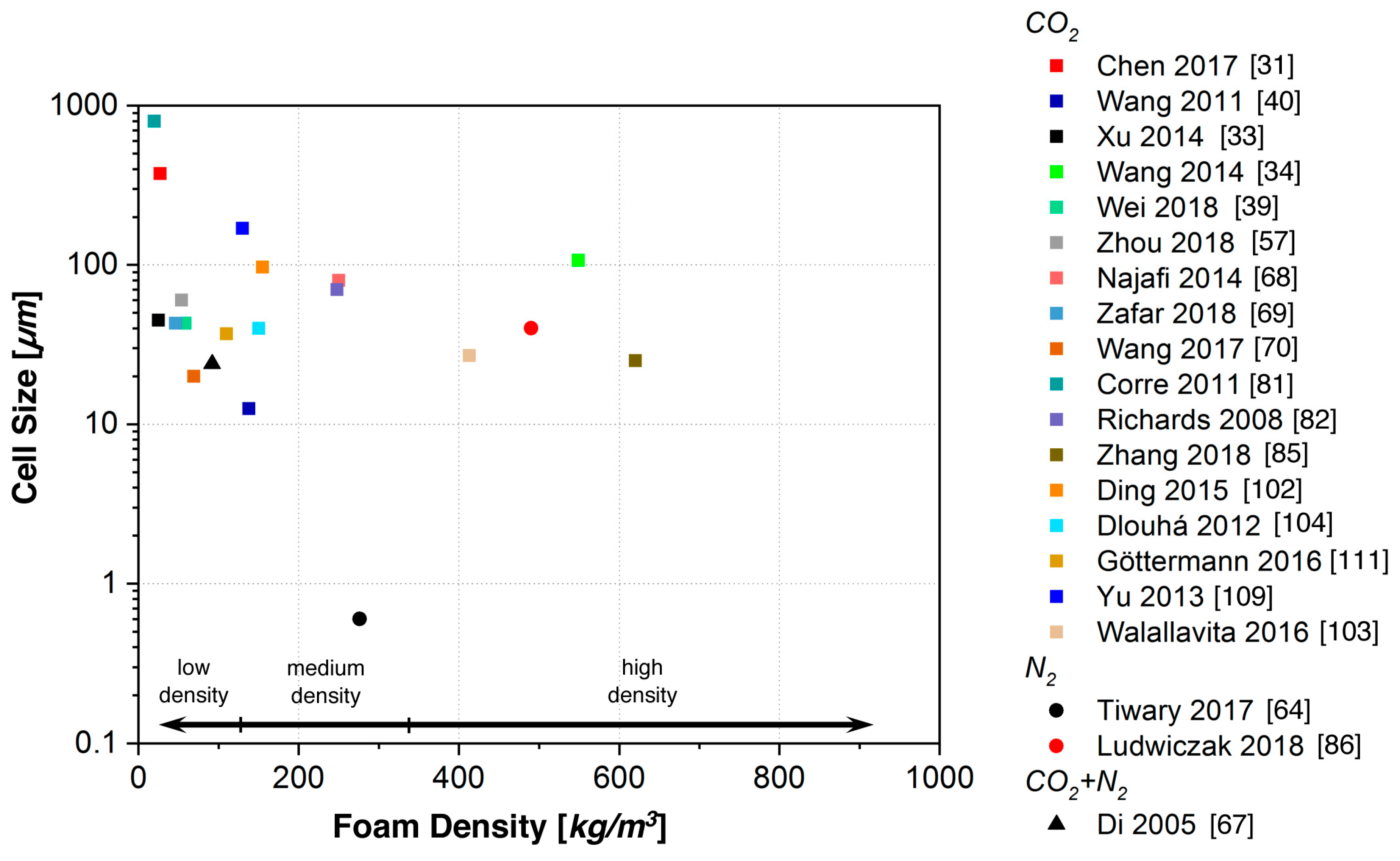
5.1. Batch Foaming
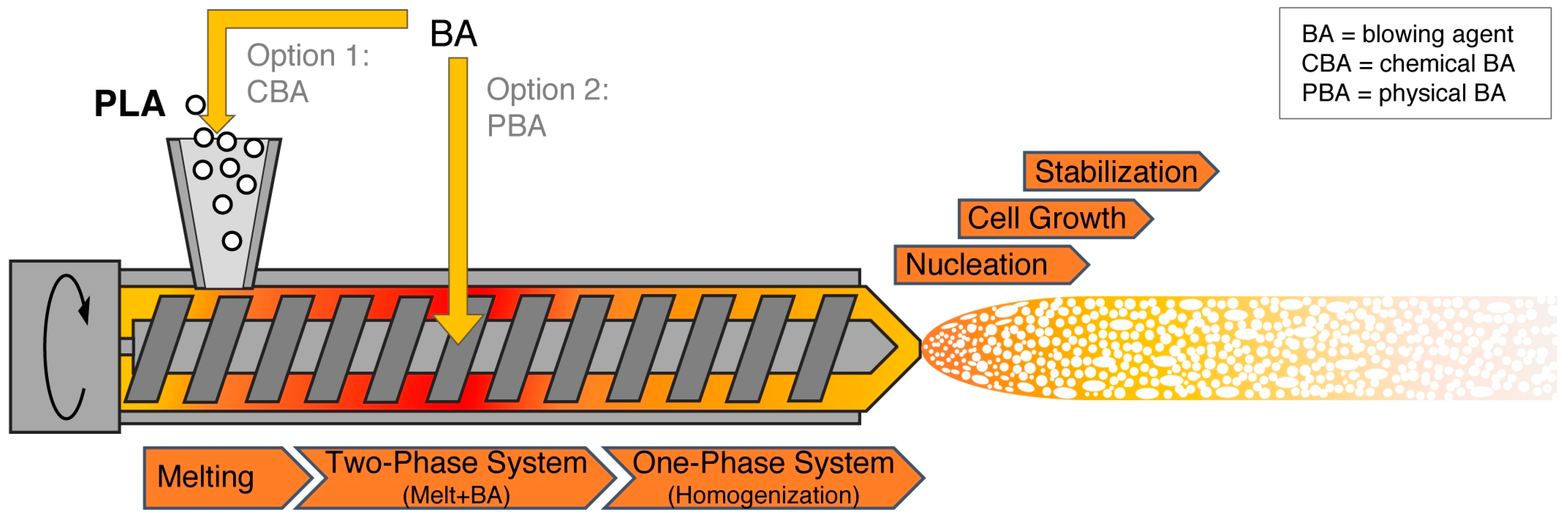
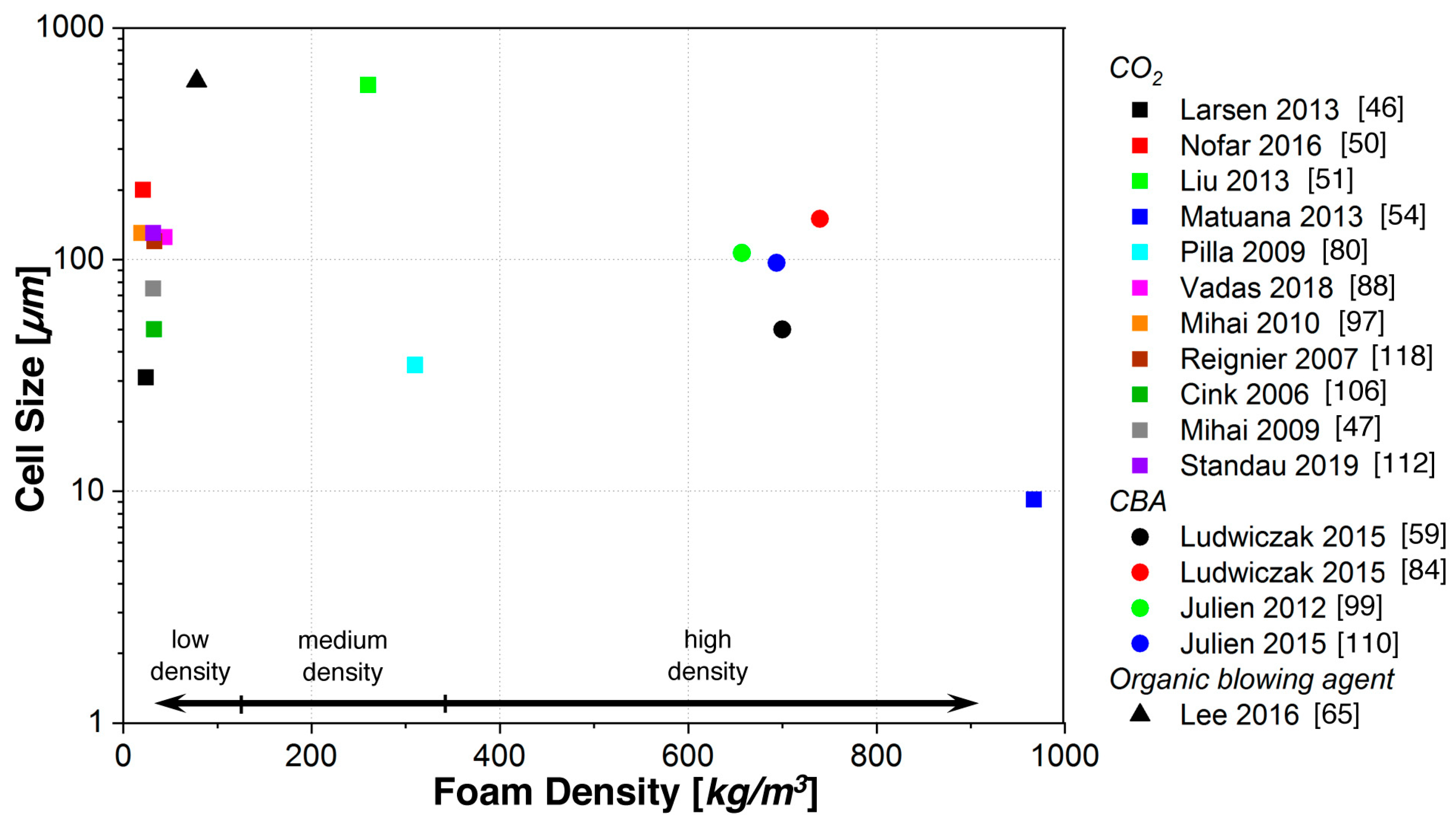
5.2. Foam Extrusion
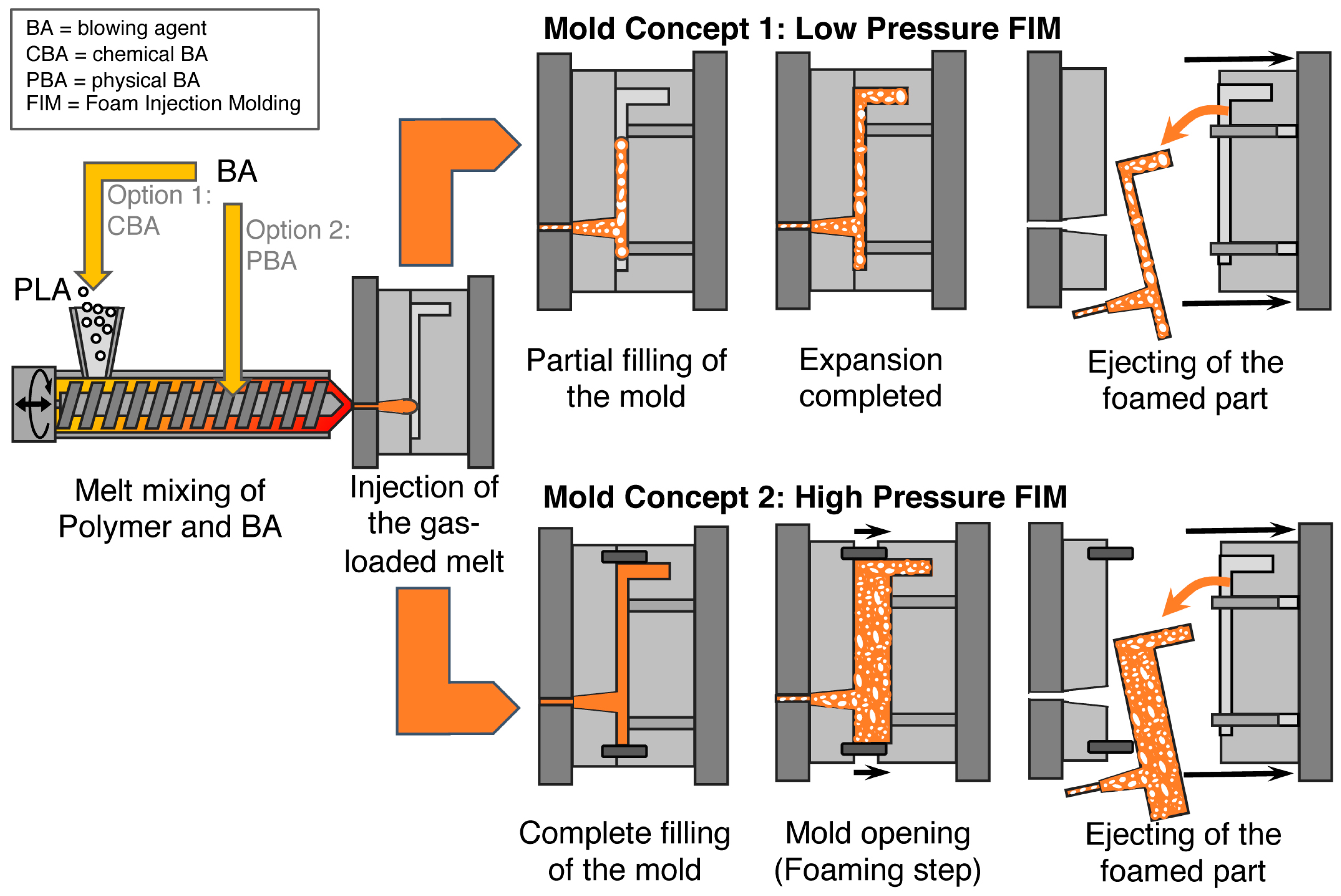
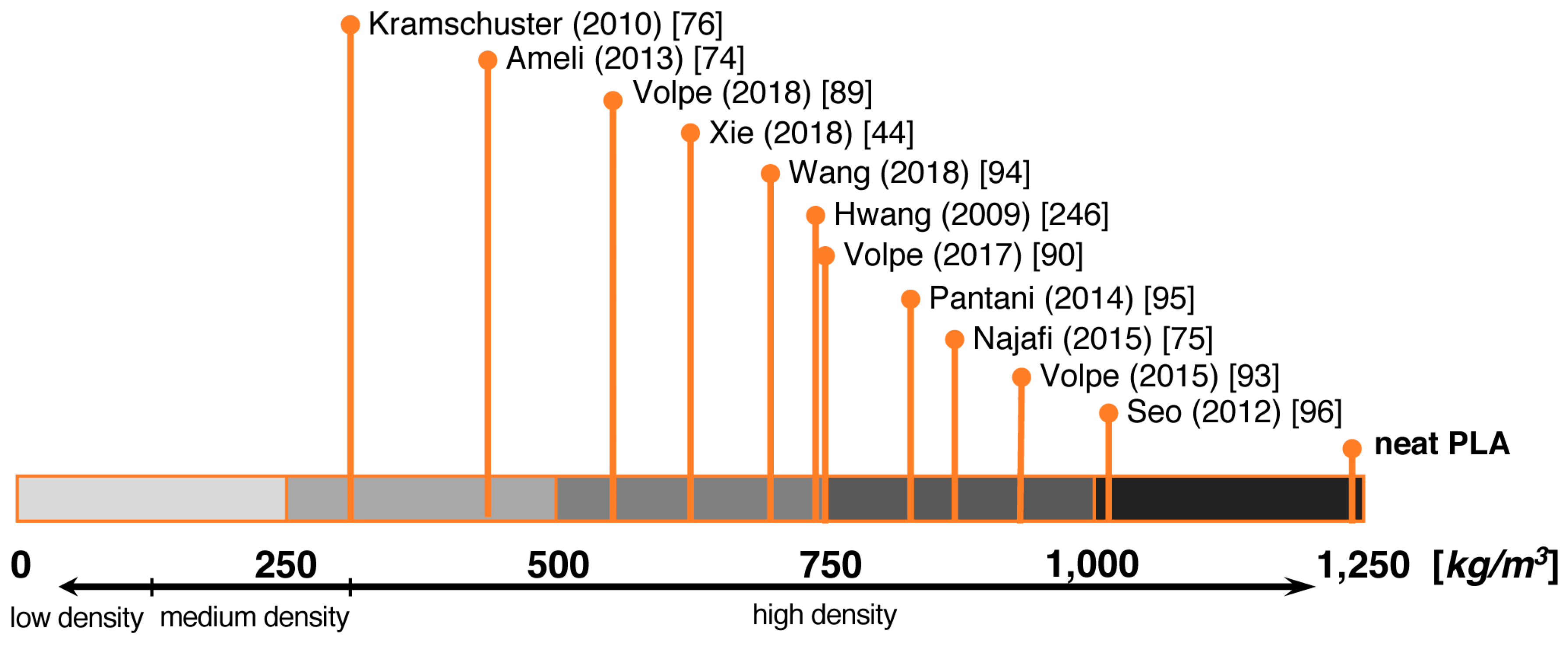
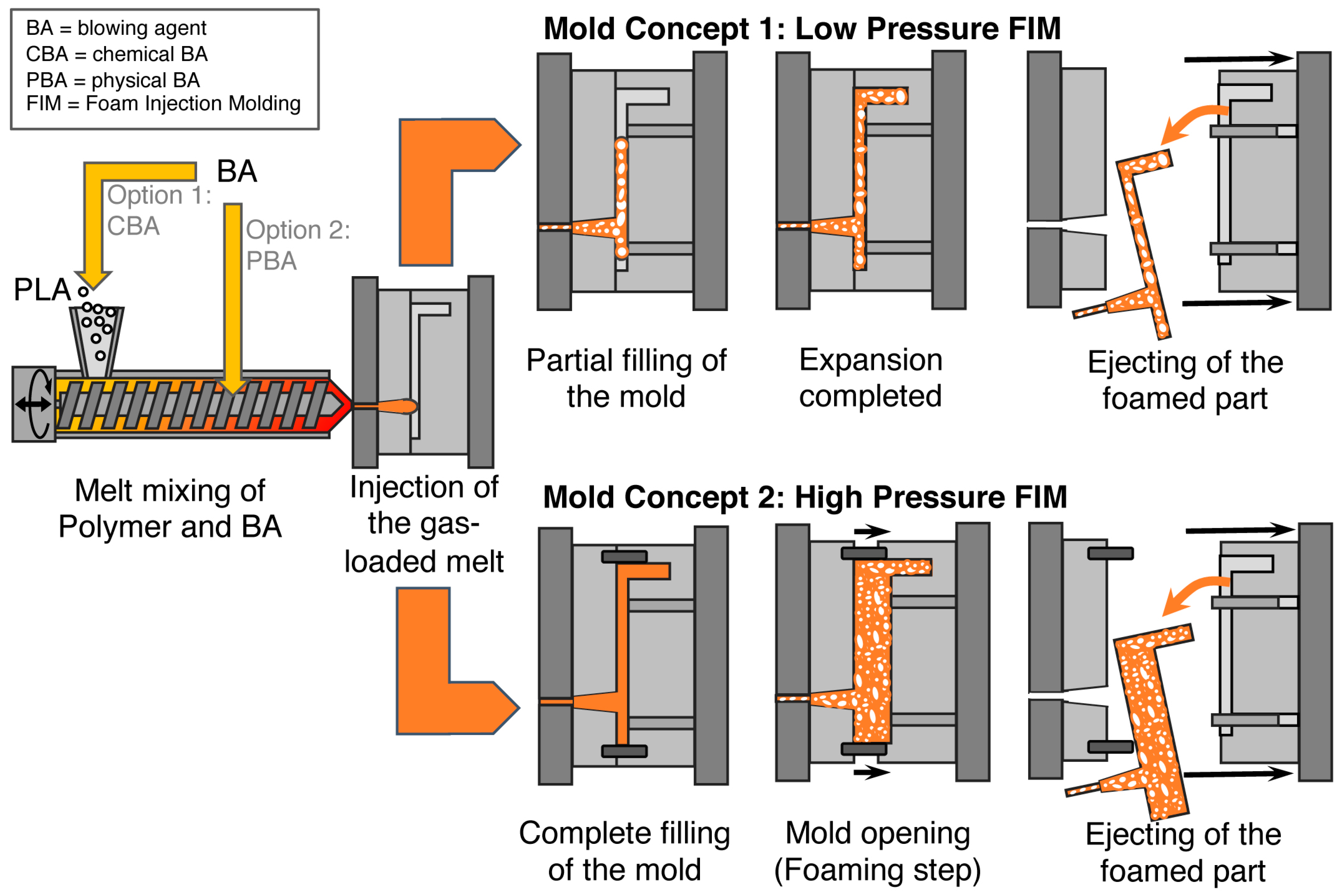
5.3. Foam Injection Molding (FIM)
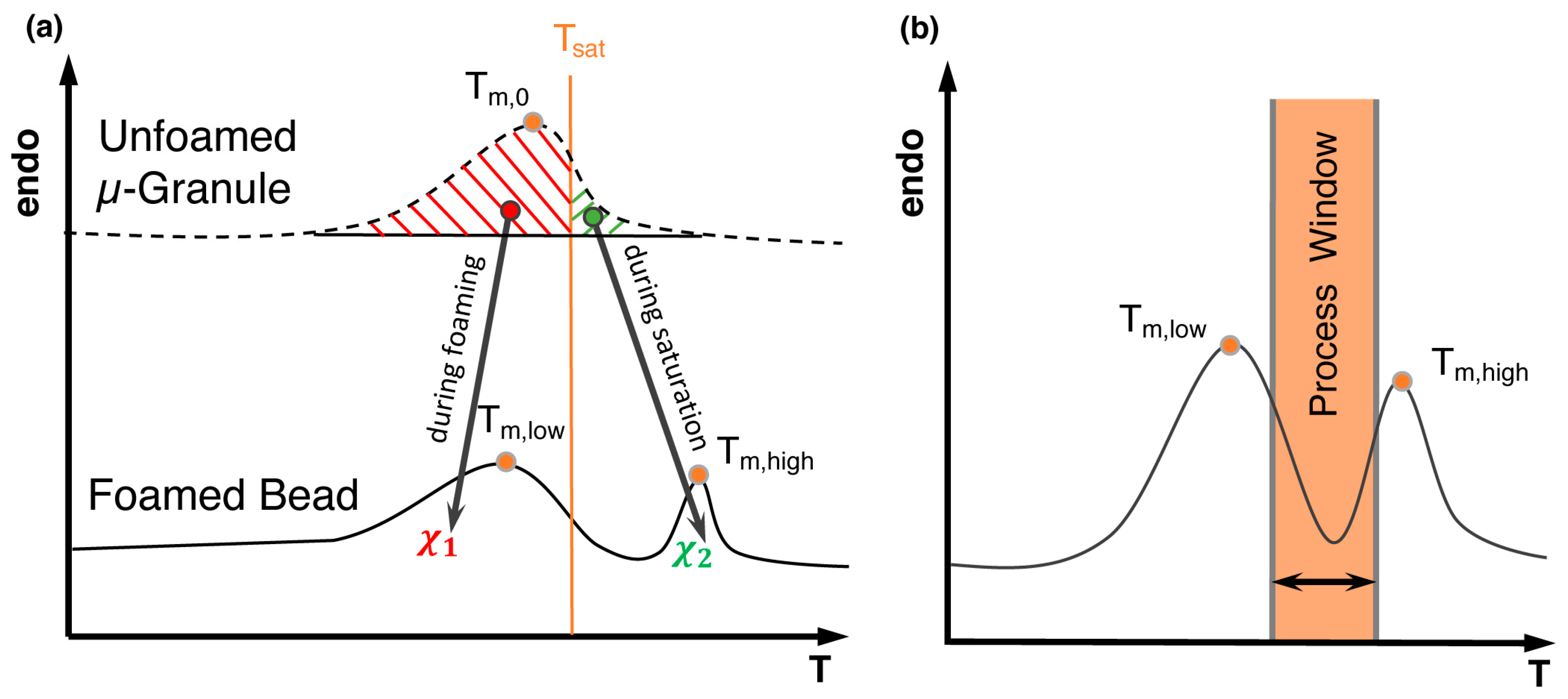
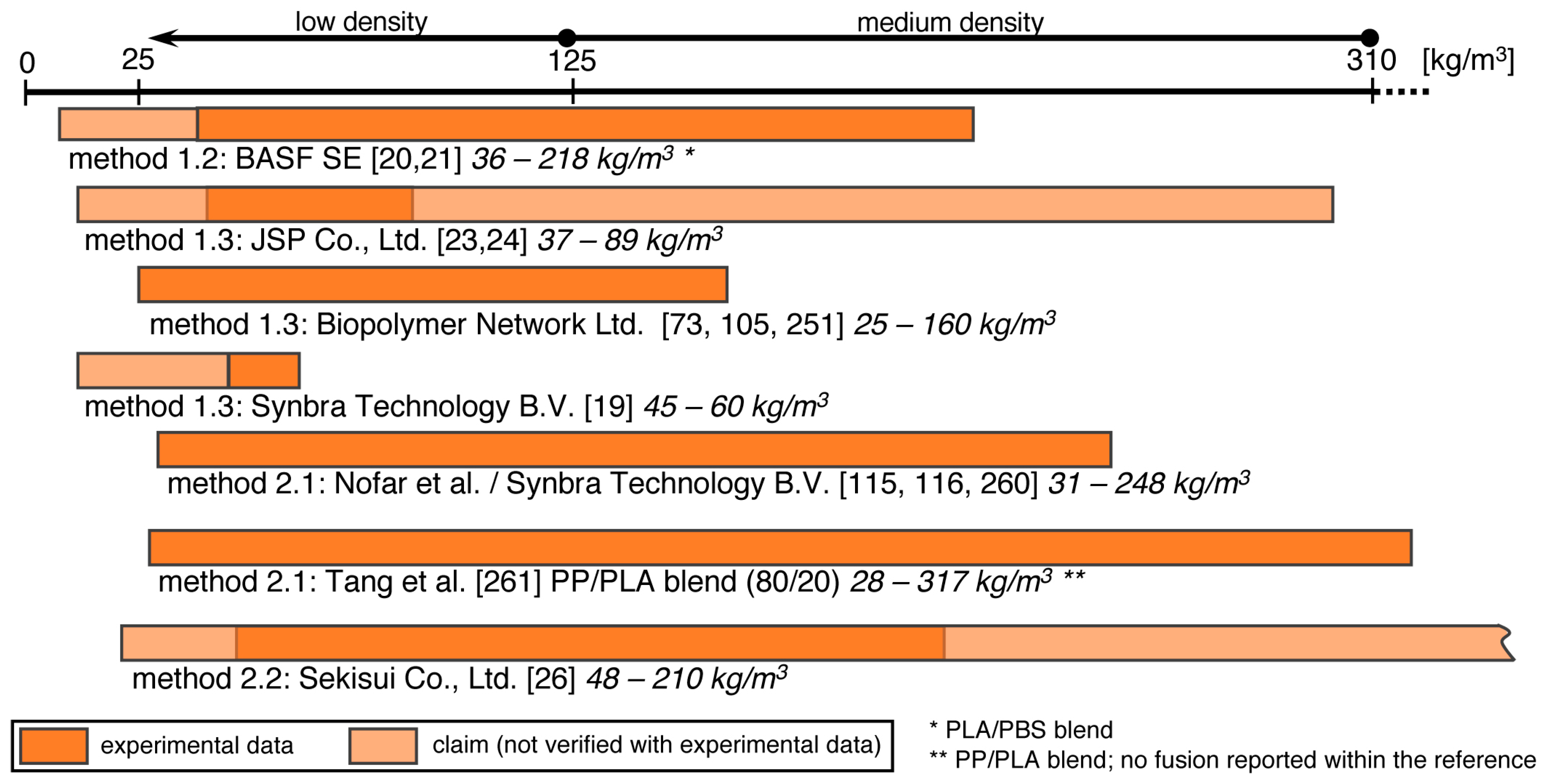
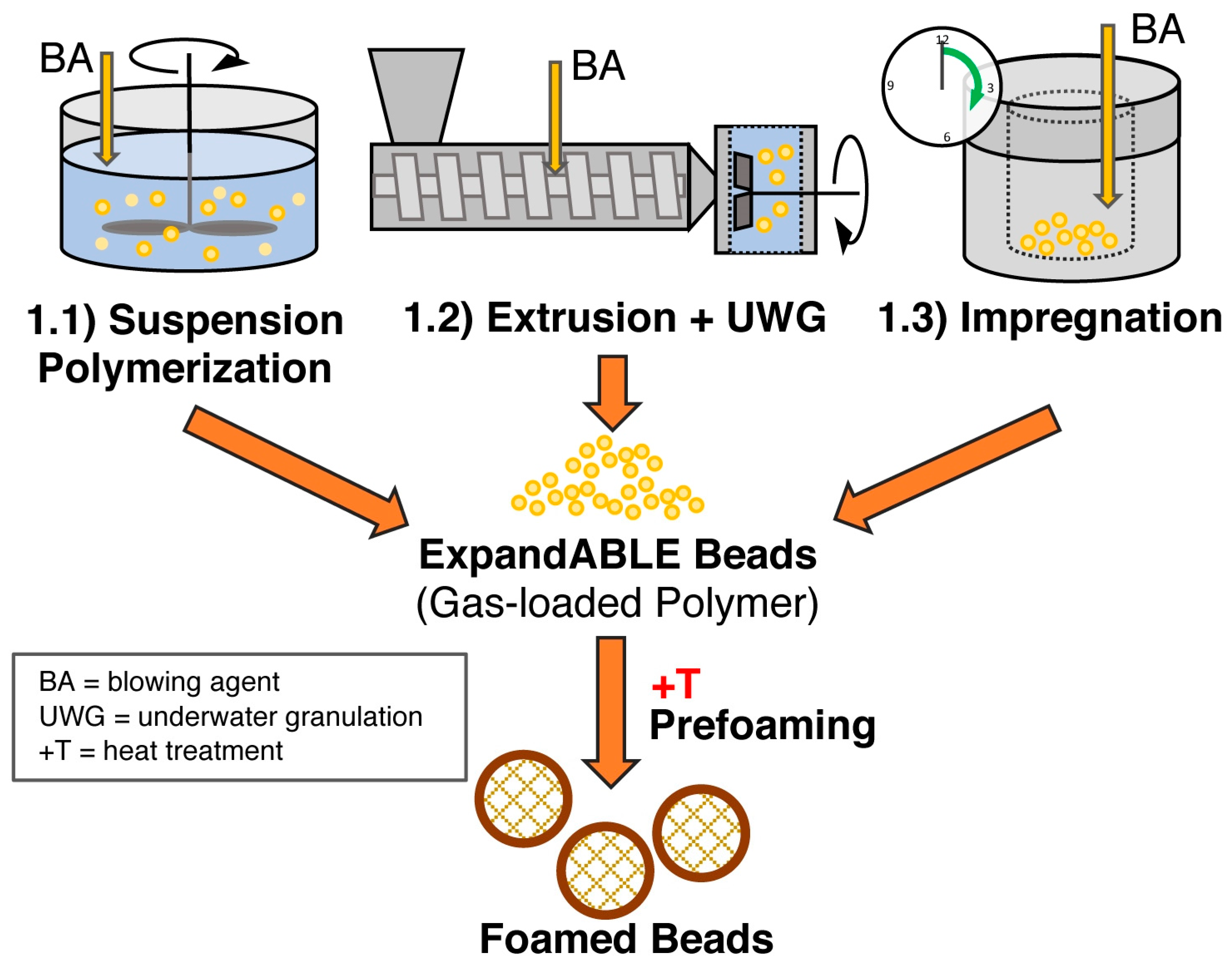
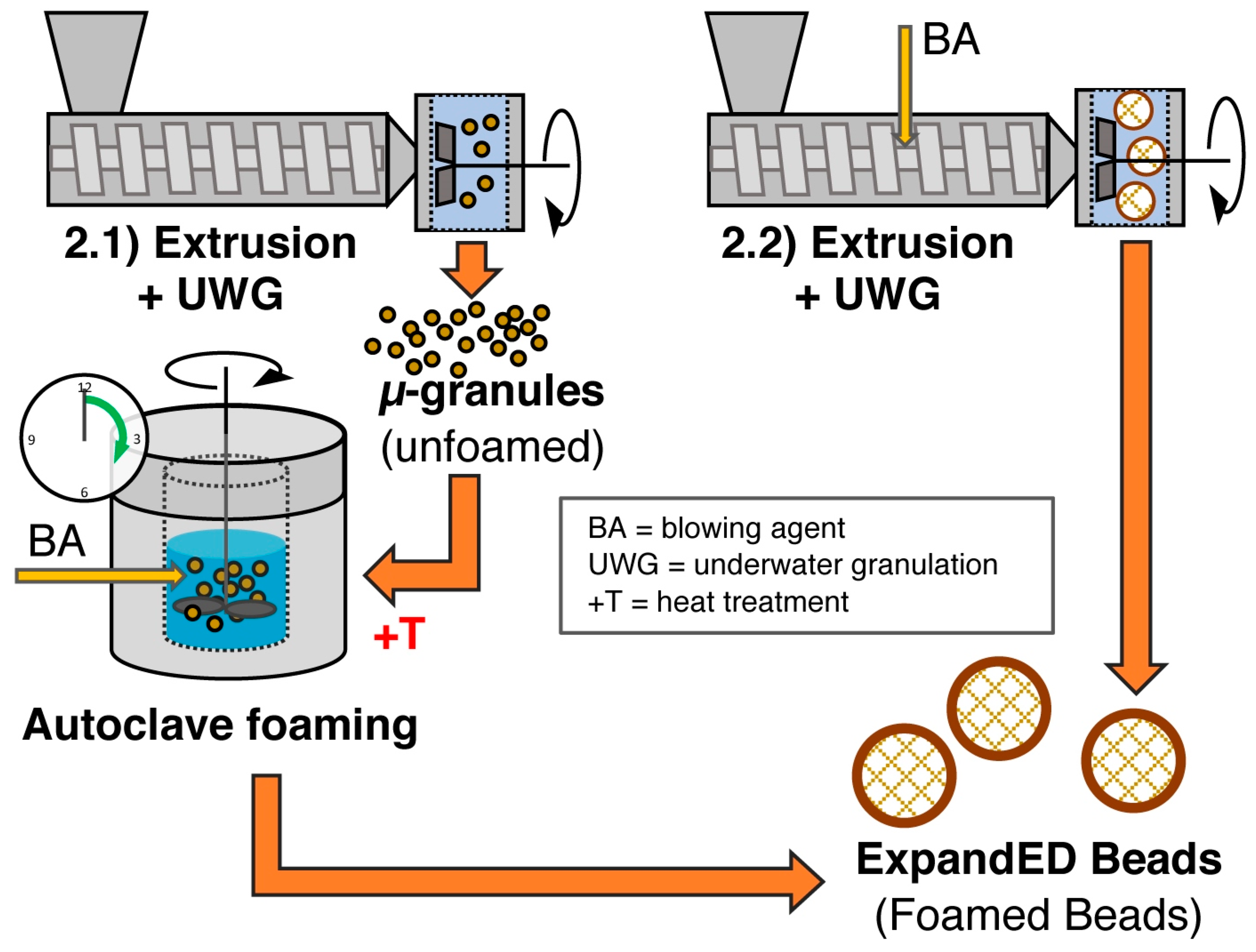
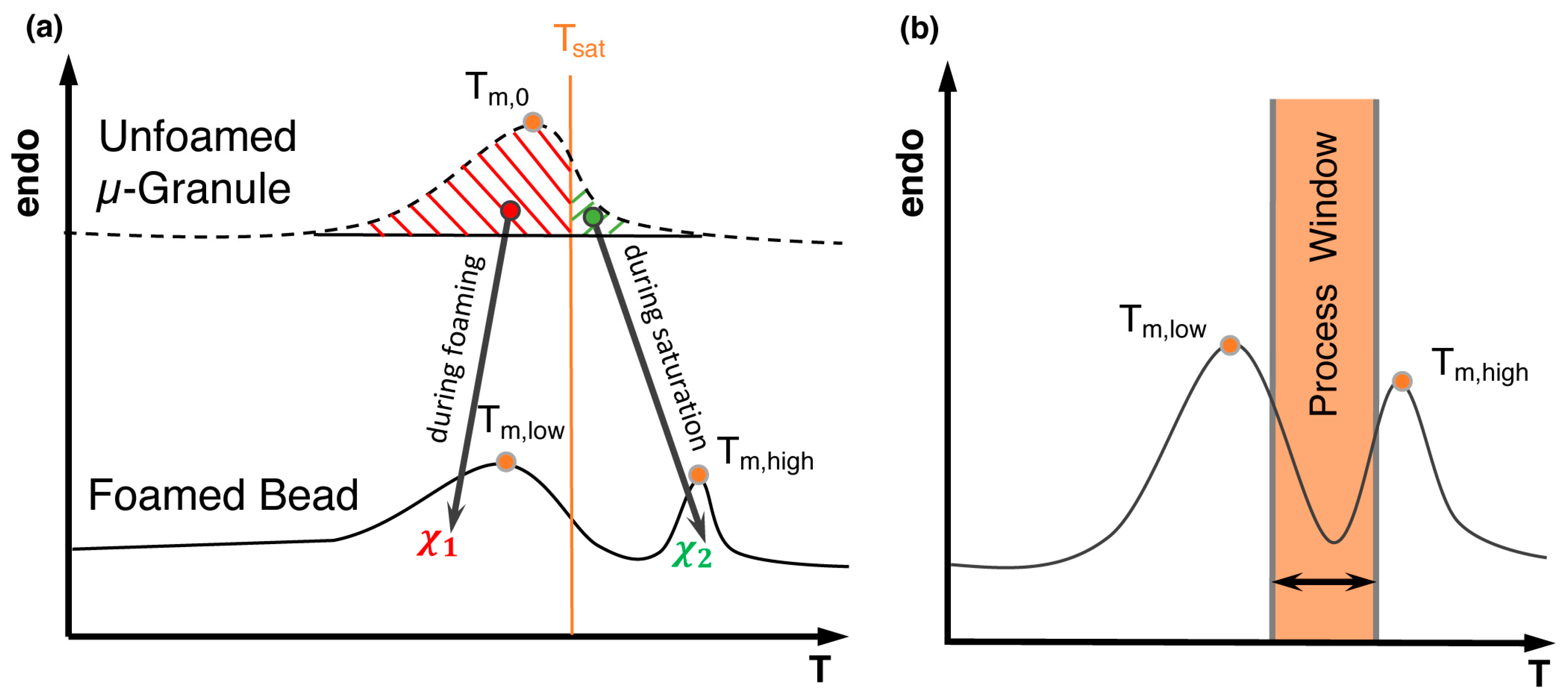
5.4. Bead Foaming
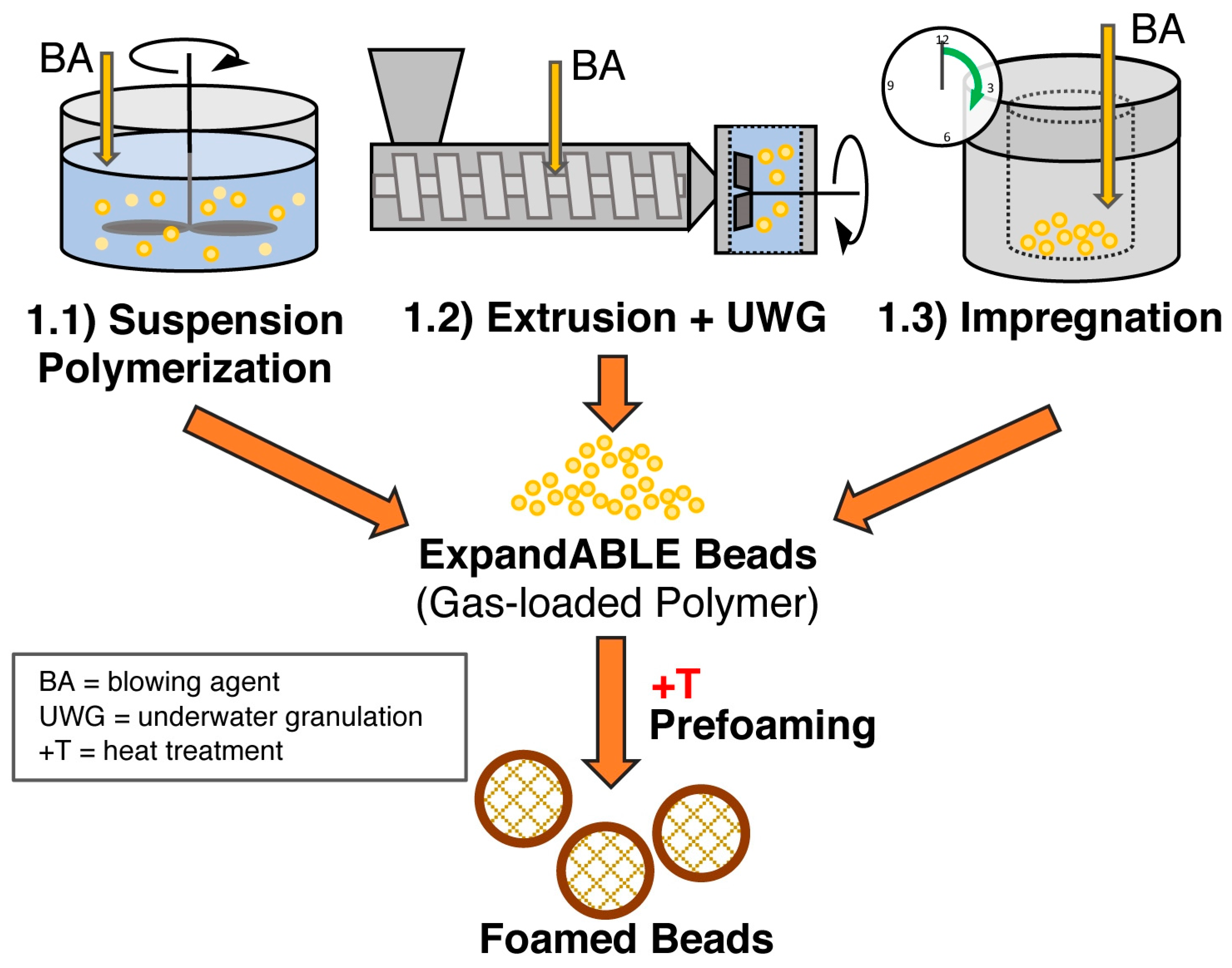
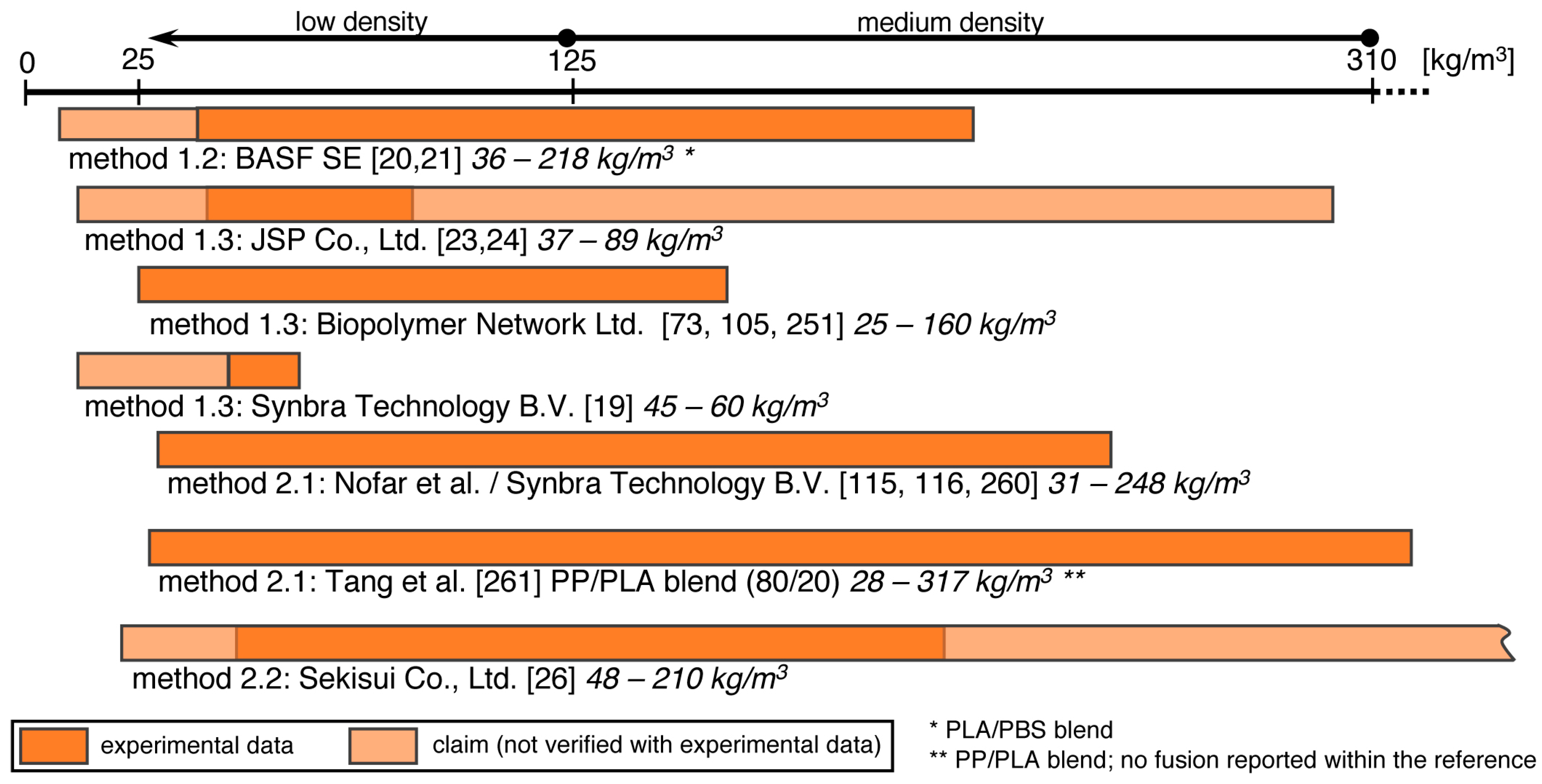
5.5. Method 1.1—Suspension Polymerization with Organic Blowing Agents
5.6. Method 1.2—Extrusion with Blowing Agent Combined with UWG and Suppressed Expansion
5.7. Method 1.3—Impregnation
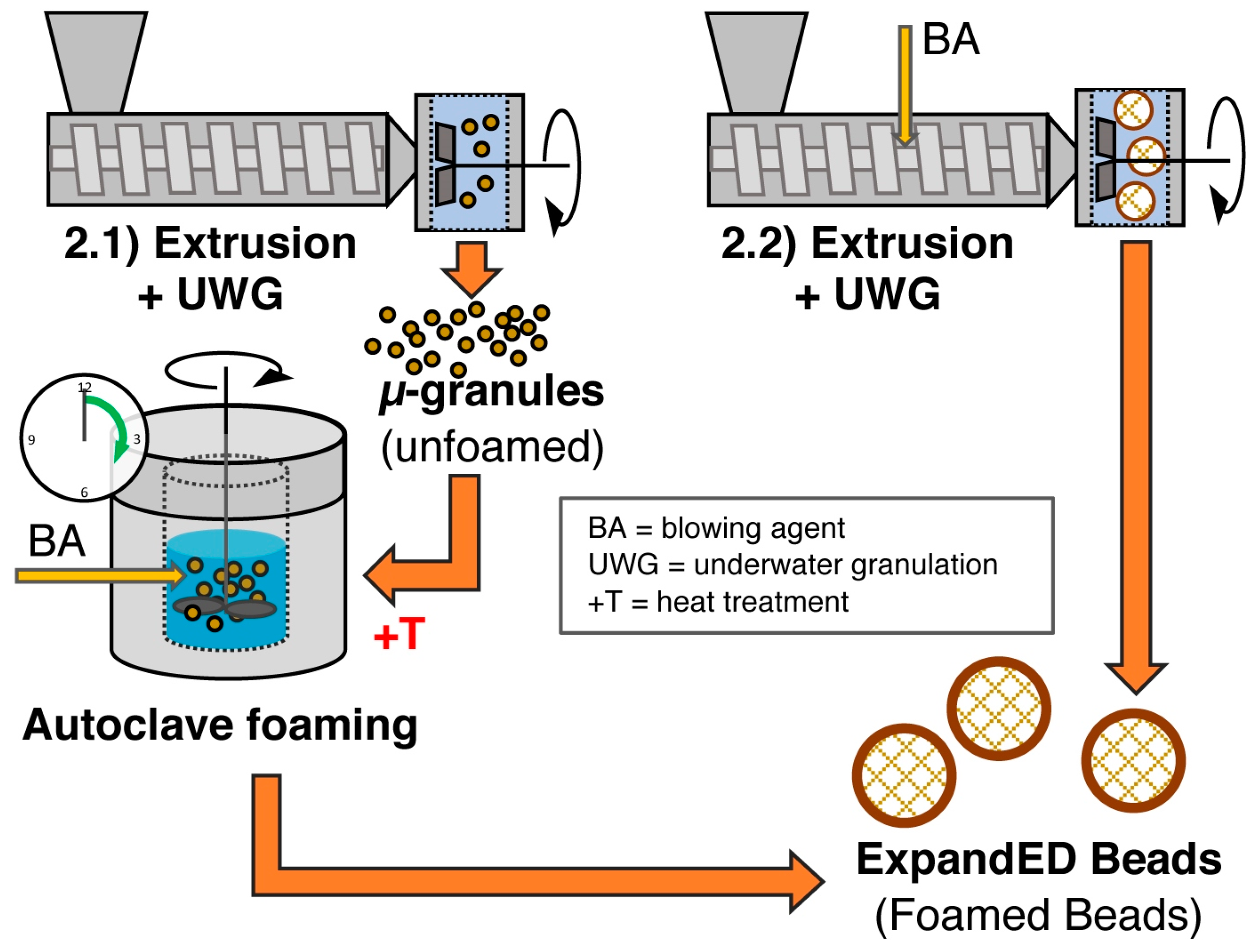
5.8. Method 2.1—Extrusion with UWG and Autoclave Foaming
5.9. Method 2.2—Extrusion with Blowing Agent Combined with UWG and Expansion
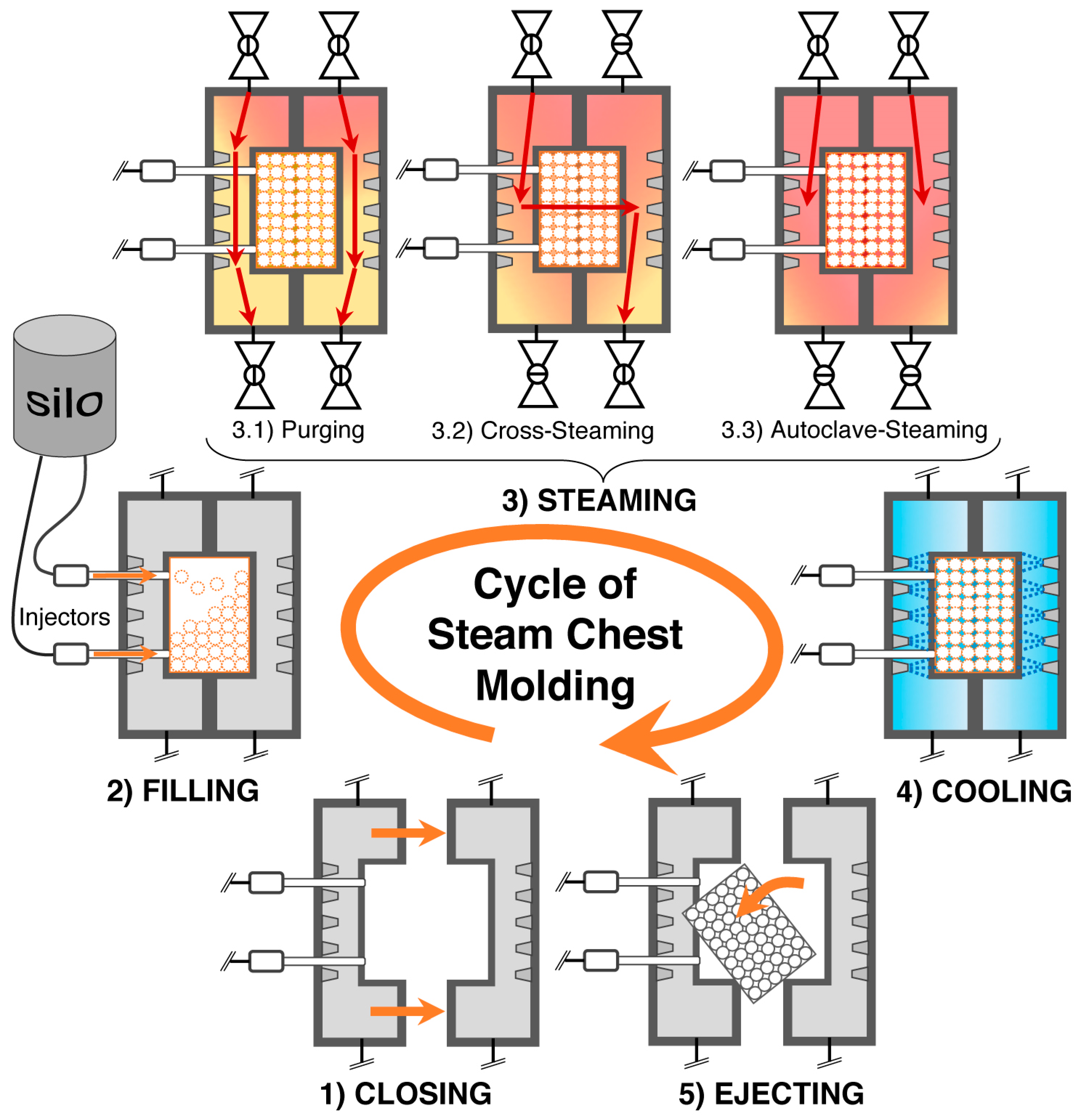
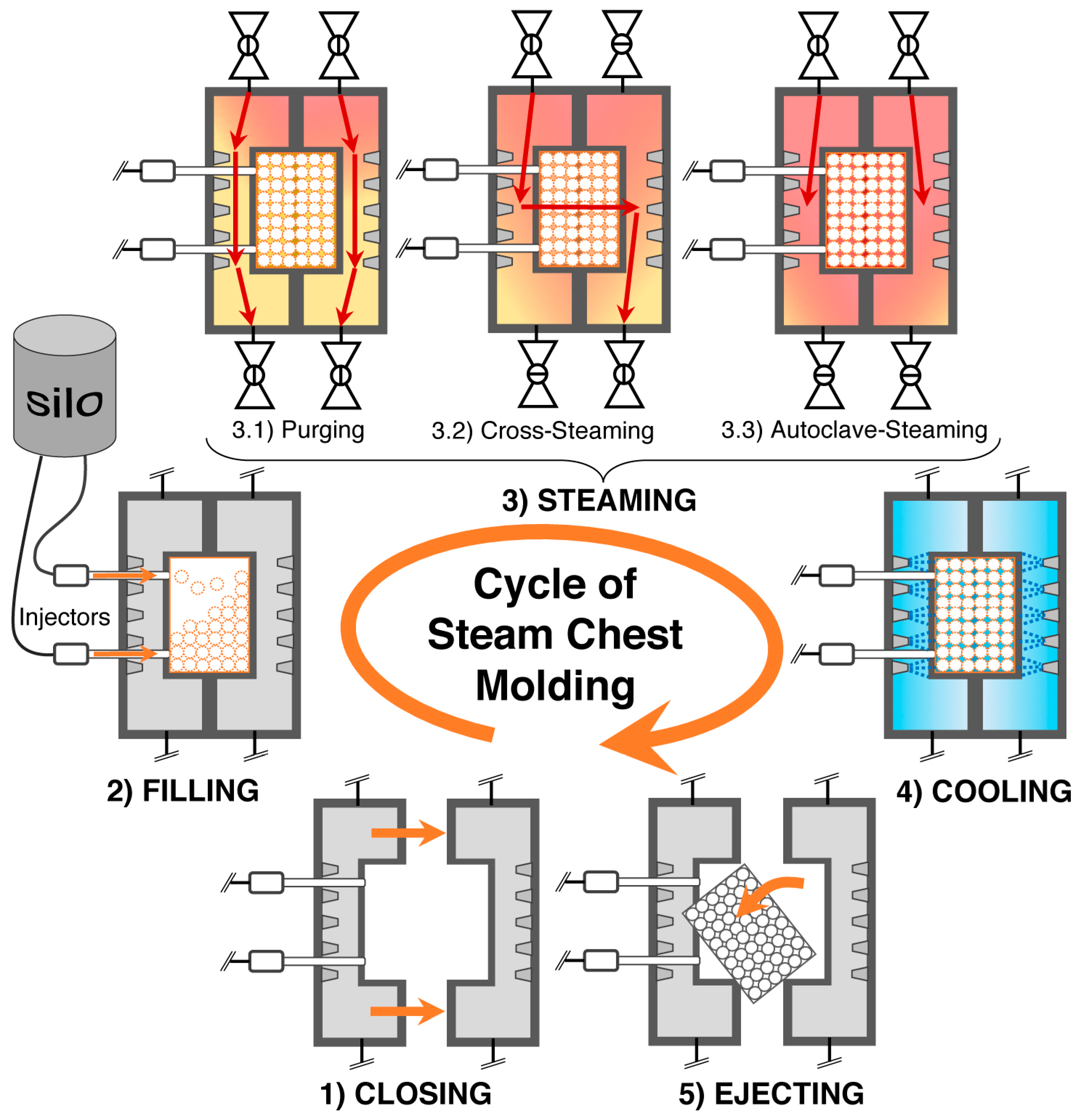
5.10. Steam Chest Molding
6. Trends and Perspectives
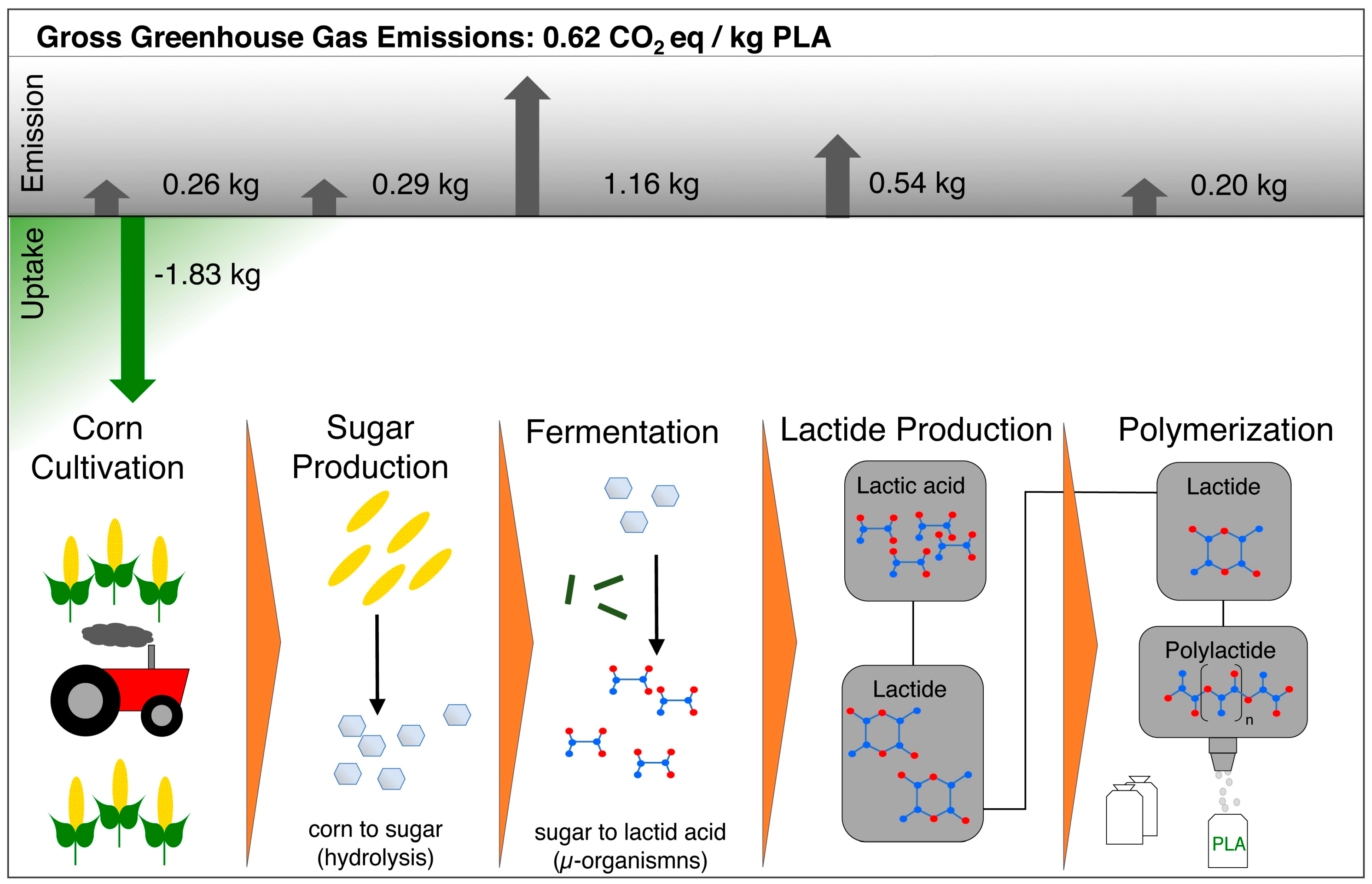
6.1. Biodegradability
6.2. Medicine
6.3. Thermal Properties
6.4. Flame Retardancy
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Carothers, W.H.; Borough, G.L.; Natta, F.J. Studies of polymerization and ring formation. X. The reversible polymerization of six-membered cyclic esters. J. Am. Chem. Soc. 1932, 54, 761–772. [Google Scholar] [CrossRef]
- Lowe, C.E. Preparation of High Molecular Weight Polyhydroxyacetic Ester. U.S. Patent US 2668162, 2 February 1954. [Google Scholar]
- Vink, E.T.H.; Rábago, K.R.; Glassner, D.A.; Springs, B.; O’Connor, R.P.; Kolstad, J.; Gruber, P.R. The sustainability of nature worksTM polylactide polymers and ingeoTM polylactide fibers: An update of the future. Initiated by the 1st International Conference on Bio-based Polymers (ICBP 2003), November 12–14 2003, Saitama, Japan. Macromol. Biosci. 2004, 4, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Vink, E.T.H.; Davies, S. Life Cycle Inventory and Impact Assessment Data for 2014 IngeoTM Polylactide Production. Ind. Biotechnol. 2015, 11, 167–180. [Google Scholar] [CrossRef]
- Lunt, J. Large-scale production, properties and commercial applications of polylactic acid polymers. Polym. Degrad. Stab. 1998, 59, 145–152. [Google Scholar] [CrossRef]
- Auras, R.; Harte, B.; Selke, S. An overview of polylactides as packaging materials. Macromol. Biosci. 2004, 4, 835–864. [Google Scholar] [CrossRef] [PubMed]
- Madhavan Nampoothiri, K.; Nair, N.R.; John, R.P. An overview of the recent developments in polylactide (PLA) research. Bioresour. Technol. 2010, 101, 8493–8501. [Google Scholar] [CrossRef]
- Hamad, K.; Kaseem, M.; Yang, H.W.; Deri, F.; Ko, Y.G. Properties and medical applications of polylactic acid: A review. Express Polym. Lett. 2015, 9, 435–455. [Google Scholar] [CrossRef]
- Vink, E.T.H.; Davies, S.; Kolstad, J.J. The eco-profile for current Ingeo polylactide production. Ind. Biotechnol. 2010, 6, 212–224. [Google Scholar] [CrossRef]
- Groot, W.J.; Borén, T. Life cycle assessment of the manufacture of lactide and PLA biopolymers from sugarcane in Thailand. Int. J. Life Cycle Assess. 2010, 15, 970–984. [Google Scholar] [CrossRef]
- Aeschelmann, F.; Carus, M. Bio-based Building Blocks and Polymers in the World Capacities, Production and Applications: Status Quo and Trends towards 2020. Ind. Biotechnol. 2015, 11, 154–159. [Google Scholar] [CrossRef]
- Sinclair, R.G. The case for polylactic acid as a commodity packaging plastic. J. Macromol. Sci. Pure Appl. Chem. 1996, 33, 585–597. [Google Scholar] [CrossRef]
- Dorgan, J.R.; Lehermeier, H.; Mang, M. Thermal and Rheological Properties of Commercial-Grade Poly (Lactic Acid)s. J. Polym. Environ. 2000, 8, 1–9. [Google Scholar] [CrossRef]
- Drumright, B.R.E.; Gruber, P.R.; Henton, D.E. Polylactic_Acid_Technology. Adv. Mater. 2000, 10, 1841–1846. [Google Scholar] [CrossRef]
- Abd Alsaheb, R.A.; Aladdin, A.; Othman, N.Z.; Abd Malek, R.; Leng, O.M.; Aziz, R.; El Enshasy, H.A. Recent applications of polylactic acid in pharmaceutical and medical industries. J. Chem. Pharm. Res. 2015, 7, 51–63. [Google Scholar]
- Mooney, D.J.; Baldwin, D.F.; Suht, N.P.; Vacantis, J.P.; Larger, R. Novel approach to fabricate porous sponges of poly (D, L-lactic-co-glycolic acid) without the use of organic solvents. Biomaterials 1996, 17, 1417–1422. [Google Scholar] [CrossRef]
- Fang, Q.; Milford, A. Hanna Functional Properties of Polylactic Acid Starch-Based Loose-Fill Packaging Foams. Cereal Chem. 2000, 77, 779–783. [Google Scholar] [CrossRef]
- Ramesh, N.S.; Nawaby, A.V.; Amrutiya, N. Polylactic Acid Foam Composition. U.S. Patent US 2011/0263732 A1, 27 October 2011. [Google Scholar]
- Britton, R.N.; Hendrikus, F.A.; Van Doormalen, C.; Noordegraaf, J.; Molenveld, K.; Schennink, G.G.J. Coated Particulate Expandable Polylactic Acid. U.S. Patent US 8268901 B2, 18 September 2012. [Google Scholar]
- Lohmann, J.; Sampath, B.D.S.; Gutmann, P.; Künkel, A.; Hahn, K.; Füßl, A. Process for Producing Expandable Pelletized Material which Comprises Polylactic Acid. U.S. Patent US 2013/0345327 A1, 26 December 2013. [Google Scholar]
- Lohmann, J.; Sampath, B.D.S.; Gutmann, P.; Künkel, A.; Hahn, K.; Füßl, A. Verfahren zur Herstellung von Expandierbaren Polymilchsäurehaltigen Granulaten. Patent WO 2014/001119 AI, 3 January 2014. [Google Scholar]
- Matuana, L.M. Solid state microcellular foamed poly(lactic acid): Morphology and property characterization. Bioresour. Technol. 2008, 99, 3643–3650. [Google Scholar] [CrossRef]
- Haraguchi, K.; Ohta, H. Expandable Polylactic Acid Resin Particles. European Patent EP 1683828 B1, 16 November 2011. [Google Scholar]
- Haraguchi, K.; Ohta, H. Expandable Polylactic Acid Resin Particles, Expanded Polylactic Acid Resin Beads and Molded Article Obtained from Expanded Polylactic Acid Resin Beads. U.S. Patent US 2006/0167122 A1, 27 July 2006. [Google Scholar]
- Maquet, V.; Boccaccini, A.R.; Pravata, L.; Notingher, I.; Jérôme, R. Porous poly(α-hydroxyacid)-bioglass composite scaffolds for bone tissue engineering. Biomaterials 2004, 25, 4185–4194. [Google Scholar] [CrossRef]
- Hirai, T.; Nishijima, K.; Ochiai, T. Polylactic Acid Resin Foam Particle for In-Mold Foam Forming, Process for Producing the Same, and Process for Producing Polylactic Acid Resin Foam Molding. Patent EP2135724 A1, 23 December 2009. [Google Scholar]
- Fujimoto, Y.; Sinha Ray, S.; Okamoto, M.; Ogami, A.; Yamada, K.; Ueda, K. Well-controlled biodegradable nanocomposite foams: From microcellular to nanocellular. Macromol. Rapid Commun. 2003, 24, 457–461. [Google Scholar] [CrossRef]
- Ray, S.S.; Okamoto, M. New Polylactide/Layered Silicate Nanocomposites, 6aMelt Rheology and Foam Processing. Macromol. Mater. Eng. 2003, 288, 936–944. [Google Scholar] [CrossRef]
- Ema, Y.; Ikeya, M.; Okamoto, M. Foam processing and cellular structure of polylactide-based nanocomposites. Polymer 2006, 47, 5350–5359. [Google Scholar] [CrossRef]
- Chen, J.W.; Liu, J.L. Batch-foamed biodegradable polylactide acid/organic modifi ed montmorillonite clays and polylactide/sericite powder nanocomposites. J. Polym. Eng. 2012, 32, 121–126. [Google Scholar] [CrossRef]
- Chen, P.; Wang, W.; Wang, Y.; Yu, K.; Zhou, H.; Wang, X.; Mi, J. Crystallization-induced microcellular foaming of poly (lactic acid) with high volume expansion ratio. Polym. Degrad. Stab. 2017, 144, 231–240. [Google Scholar] [CrossRef]
- Wang, J.; Zhai, W.; Ling, J.; Shen, B.; Zheng, W.; Park, C.B. Ultrasonic irradiation enhanced cell nucleation in microcellular poly(lactic acid): A novel approach to reduce cell size distribution and increase foam expansion. Ind. Eng. Chem. Res. 2011, 50, 13840–13847. [Google Scholar] [CrossRef]
- Zhai, W.; Ko, Y.; Zhu, W.; Wong, A.; Park, C.B. A study of the crystallization, melting, and foaming behaviors of polylactic acid in compressed CO2. Int. J. Mol. Sci. 2009, 10, 5381–5397. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Huang, H. Foaming of Poly(lactic acid) Using Supercritical Carbon Dioxide as Foaming Agent: Influence of Crystallinity and Spherulite Size on Cell Structure and Expansion Ratio. Ind. Eng. Chem. Res. 2014, 53, 2277–2286. [Google Scholar] [CrossRef]
- Wang, X.; Zhou, H.; Liu, B.; Du, Z.; Li, H. Chain Extension and Foaming Behavior of Poly(lactic acid) by Functionalized Multiwalled Carbon Nanotubes and Chain Extender. Adv. Polym. Technol. 2014, 33. [Google Scholar] [CrossRef]
- Liu, J.; Lou, L.; Yu, W.; Liao, R.; Li, R.; Zhou, C. Long chain branching polylactide: Structures and properties. Polymer 2010, 51, 5186–5197. [Google Scholar] [CrossRef]
- Zhang, W.; Chen, B.; Zhao, H.; Yu, P.; Fu, D.; Wen, J.; Peng, X. Processing and characterization of supercritical CO2batch foamed poly(lactic acid)/poly(ethylene glycol) scaffold for tissue engineering application. J. Appl. Polym. Sci. 2013, 130, 3066–3073. [Google Scholar] [CrossRef]
- Qiu, Y.; Lv, Q.; Wu, D.; Xie, W.; Peng, S.; Lan, R.; Xie, H. Cyclic tensile properties of the polylactide nanocomposite foams containing cellulose nanocrystals. Cellulose 2018, 25, 1795–1807. [Google Scholar] [CrossRef]
- Wu, D.; Lv, Q.; Feng, S.; Chen, J.; Chen, Y.; Qiu, Y.; Yao, X. Polylactide composite foams containing carbon nanotubes and carbon black: Synergistic effect of filler on electrical conductivity. Carbon N. Y. 2015, 95, 380–387. [Google Scholar] [CrossRef]
- Wei, L.; Shicheng, H.; Hongfu, Z. Effect of octa(epoxycyclohexyl) POSS on thermal, rheology property, and foaming behavior of PLA composites. J. Appl. Polym. Sci. 2018, 135, 1–10. [Google Scholar] [CrossRef]
- Huang, A.; Kharbas, H.; Ellingham, T.; Mi, H.; Turng, L.-S.; Peng, X. Mechanical properties, crystallization characteristics, and foaming behavior of polytetrafluoroethylene-reinforced poly(lactic acid) composites. Polym. Eng. Sci. 2017, 57, 570–580. [Google Scholar] [CrossRef]
- Zhao, H.; Cui, Z.; Sun, X.; Turng, L.S.; Peng, X. Morphology and properties of injection molded solid and microcellular polylactic acid/polyhydroxybutyrate-valerate (PLA/PHBV) blends. Ind. Eng. Chem. Res. 2013, 52, 2569–2581. [Google Scholar] [CrossRef]
- Tor-Świątek, A.; Garbacz, T.; Sedlarik, V.; Stloukal, P.; Kucharczyk, P. Influence of polylactide modifications with blowing agents on selected machanical properties. Adv. Sci. Technol. Res. J. 2017, 11, 206–214. [Google Scholar] [CrossRef]
- Xie, P.; Wu, G.; Cao, Z.; Han, Z.; Zhang, Y.; An, Y.; Yang, W. Effect of mold opening process on microporous structure and properties of microcellular polylactide-polylactide nanocomposites. Polymers 2018, 10, 554. [Google Scholar] [CrossRef]
- Zhao, H.; Zhao, G.; Turng, L.S.; Peng, X. Enhancing Nanofiller Dispersion Through Prefoaming and Its Effect on the Microstructure of Microcellular Injection Molded Polylactic Acid/Clay Nanocomposites. Ind. Eng. Chem. Res. 2015, 54, 7122–7130. [Google Scholar] [CrossRef]
- Larsen, A.; Neldin, C. Physical Extruder Foaming of Poly (lactic acid)—Processing and Foam Properties. Polym. Eng. Sci. 2013, 53, 941–949. [Google Scholar] [CrossRef]
- Mihai, M.; Huneault, M.A.; Favis, B.D.; Li, H. Extrusion foaming of semi-crystalline PLA and PLA/thermoplastic starch blends. Macromol. Biosci. 2007, 7, 907–920. [Google Scholar] [CrossRef] [PubMed]
- Mihai, M.; Huneault, M.A.; Favis, B.D. Crystallinity development in cellular poly(lactic acid) in the presence of supercritical carbon dioxide. J. Appl. Polym. Sci. 2009, 113, 2920–2932. [Google Scholar] [CrossRef]
- Wang, J.; Zhu, W.; Zhang, H.; Park, C.B. Continuous processing of low-density, microcellular poly(lactic acid) foams with controlled cell morphology and crystallinity. Chem. Eng. Sci. 2012, 75, 390–399. [Google Scholar] [CrossRef]
- Matuana, L.M.; Diaz, C.A. Study of cell nucleation in microcellular poly(lactic acid) foamed with supercritical CO2 through a continuous-extrusion process. Ind. Eng. Chem. Res. 2010, 49, 2186–2193. [Google Scholar] [CrossRef]
- Nofar, M. Effects of nano-/micro-sized additives and the corresponding induced crystallinity on the extrusion foaming behavior of PLA using supercritical CO2. Mater. Des. 2016, 101, 24–34. [Google Scholar] [CrossRef]
- Liu, W.; Wang, X.; Li, H.; Du, Z.; Zhang, C. Study on rheological and extrusion foaming behaviors of chain-extended poly (lactic acid)/clay nanocomposites. J. Cell. Plast. 2013, 49, 535–554. [Google Scholar] [CrossRef]
- Keshtkar, M.; Nofar, M.; Park, C.B.; Carreau, P.J. Extruded PLA/clay nanocomposite foams blown with supercritical CO2. Polymer 2014, 55, 4077–4090. [Google Scholar] [CrossRef]
- Vannini, C.; Fiordelisi, F.; Movilli, W.; Lanzani, F. PLA-Based Degradable Foams and Process for Their Production. Patent WO2005/042627 A1, 12 May 2005. [Google Scholar]
- Matuana, L.M.; Diaz, C.A. Strategy to produce microcellular foamed poly(lactic acid)/wood-flour composites in a continuous extrusion process. Ind. Eng. Chem. Res. 2013, 52, 12032–12040. [Google Scholar] [CrossRef]
- Gorrasi, G.; Pantani, R. Effect of PLA grades and morphologies on hydrolytic degradation at composting temperature: Assessment of structural modification and kinetic parameters. Polym. Degrad. Stab. 2013, 98, 1006–1014. [Google Scholar] [CrossRef]
- Zhou, H.; Zhao, M.; Qu, Z.; Mi, J.; Wang, X.; Deng, Y. Thermal and Rheological Properties of Poly(lactic acid)/Low-Density Polyethylene Blends and Their Supercritical CO2 Foaming Behavior. J. Polym. Environ. 2018, 26, 3564–3573. [Google Scholar] [CrossRef]
- Chu, R.K.M.; Mark, L.H.; Park, C.B. Cell nucleation in high-pressure foam injection molding. In Proceedings of the Annual Technical Conference SPE-ANTEC, Anaheim, CA, USA, 8–10 May 2017; pp. 2411–2415. [Google Scholar]
- Ludwiczak, J.; Kozlowski, M. Foaming of Polylactide in the Presence of Chain Extender. J. Polym. Environ. 2015, 23, 137–142. [Google Scholar] [CrossRef]
- Zhang, R.; Cai, C.; Liu, Q.; Hu, S. Enhancing the Melt Strength of Poly(Lactic Acid) via Micro-Crosslinking and Blending with Poly(Butylene Adipate-co-Butylene Terephthalate)for the Preparation of Foams. J. Polym. Environ. 2017, 25, 1335–1341. [Google Scholar] [CrossRef]
- Geissler, B.; Feuchter, M.; Laske, S.; Walluch, M.; Holzer, C.; Langecker, G.R. Tailor-Made High Density PLA Foam Sheets-Strategies to Improve the Mechanical Properties. Cell. Polym. 2014, 33, 249–257. [Google Scholar] [CrossRef]
- Tabatabaei, A.; Park, C.B. In-situ visualization of PLA crystallization and crystal effects on foaming in extrusion. Eur. Polym. J. 2017, 96, 505–519. [Google Scholar] [CrossRef]
- Zhou, M.; Zhou, P.; Xiong, P.; Qian, X.; Zheng, H. Crystallization, rheology and foam morphology of branched PLA prepared by novel type of chain extender. Macromol. Res. 2015, 23, 231–236. [Google Scholar] [CrossRef]
- Tiwary, P.; Park, C.B.; Kontopoulou, M. Transition from microcellular to nanocellular PLA foams by controlling viscosity, branching and crystallization. Eur. Polym. J. 2017, 91, 283–296. [Google Scholar] [CrossRef]
- Lee, R.E.; Guo, Y.; Tamber, H.; Planeta, M.; Leung, S.N.S. Thermoforming of Polylactic Acid Foam Sheets: Crystallization Behaviors and Thermal Stability. Ind. Eng. Chem. Res. 2016, 55, 560–567. [Google Scholar] [CrossRef]
- Di, Y.; Iannace, S.; Di Maio, E.; Nicolais, L. Poly(lactic acid)/organoclay nanocomposites: Thermal, rheological properties and foam processing. J. Polym. Sci. Part B Polym. Phys. 2005, 43, 689–698. [Google Scholar] [CrossRef]
- Di, Y.; Iannace, S.; Di Maio, E.; Nicolais, L. Reactively modified poly (lactic acid): Properties and foam processing. Macromol. Mater. Eng. 2005, 290, 1083–1090. [Google Scholar] [CrossRef]
- Najafi, N.; Heuzey, M.C.; Carreau, P.J.; Therriault, D.; Park, C.B. Rheological and foaming behavior of linear and branched polylactides. Rheol. Acta 2014, 53, 779–790. [Google Scholar] [CrossRef]
- Zafar, M.T.; Kumar, S.; Singla, R.K.; Maiti, S.N.; Ghosh, A.K. Surface Treated Jute Fiber Induced Foam Microstructure Development in Poly(lactic acid)/Jute Fiber Biocomposites and their Biodegradation Behavior. Fibers Polym. 2018, 19, 648–659. [Google Scholar] [CrossRef]
- Wang, Y.; Song, Y.; Du, J.; Xi, Z.; Wang, Q. Preparation of desirable porous cell structure polylactide/wood flour composite foams assisted by chain extender. Materials 2017, 10, 23. [Google Scholar] [CrossRef] [PubMed]
- Nofar, M. Rheological, thermal, and foaming behaviors of different polylactide grades. Int. J. Mater. Sci. Res. 2018, 1, 16–22. [Google Scholar] [CrossRef]
- Rizvi, R.; Cochrane, B.; Naguib, H.; Lee, P.C. Fabrication and characterization of melt-blended polylactide-chitin composites and their foams. J. Cell. Plast. 2011, 47, 283–300. [Google Scholar] [CrossRef]
- Witt, M.R.J.; Shah, S. Methods of manufacture of polylactic acid foams. U.S. Patent US 8283389 B2, 9 October 2012. [Google Scholar]
- Ameli, A.; Jahani, D.; Nofar, M.; Jung, P.U.; Park, C.B. Processing and characterization of solid and foamed injection-molded polylactide with talc. J. Cell. Plast. 2013, 49, 351–374. [Google Scholar] [CrossRef]
- Najafi, N.; Heuzey, M.C.; Carreau, P.J.; Therriault, D.; Park, C.B. Mechanical and morphological properties of injection molded linear and branched-polylactide (PLA) nanocomposite foams. Eur. Polym. J. 2015, 73, 455–465. [Google Scholar] [CrossRef]
- Kramschuster, A.; Turng, L.S. An injection molding process for manufacturing highly porous and interconnected biodegradable polymer matrices for use as tissue engineering scaffolds. J. Biomed. Mater. Res. Part B Appl. Biomater. 2010, 92, 366–376. [Google Scholar] [CrossRef]
- Pradeep, S.A.; Kharbas, H.; Turng, L.S.; Avalos, A.; Lawrence, J.G.; Pilla, S. Investigation of thermal and thermomechanical properties of biodegradable PLA/PBSA composites processed via supercritical fluid-assisted foam injection molding. Polymers 2017, 9, 22. [Google Scholar] [CrossRef]
- Ameli, A.; Nofar, M.; Jahani, D.; Rizvi, G.; Park, C.B. Development of high void fraction polylactide composite foams using injection molding: Crystallization and foaming behaviors. Chem. Eng. J. 2015, 262, 78–87. [Google Scholar] [CrossRef]
- Sun, X.; Kharbas, H.; Peng, J.; Turng, L.-S. A novel method of producing lightweight microcellular injection molded parts with improved ductility and toughness. Polymer 2015, 56, 102–110. [Google Scholar] [CrossRef]
- Pilla, S.; Kim, S.G.; Auer, G.K.; Gong, S.; Park, C.B. Microcellular extrusion-foaming of polylactide with chain-extender. Polym. Eng. Sci. 2009, 49, 1653–1660. [Google Scholar] [CrossRef]
- Corre, Y.-M.; Maazouz, A.; Duchet, J.; Reignier, J. Batch foaming of chain extended PLA with supercritical CO2: Influence of the rheological properties and the process parameters on the cellular structure. J. Supercrit. Fluids 2011, 58, 177–188. [Google Scholar] [CrossRef]
- Richards, E.; Rizvi, R.; Chow, A.; Naguib, H. Biodegradable composite foams of PLA and PHBV using subcritical CO2. J. Polym. Environ. 2008, 16, 258–266. [Google Scholar] [CrossRef]
- Mallet, B.; Lamnawar, K.; Maazouz, A. Compounding and Melt Strengthening of Poly(Lactic Acid): Shear and Elongation Rheological Investigations for Forming Process. Key Eng. Mater. 2013, 554–557, 1751–1756. [Google Scholar] [CrossRef]
- Ludwiczak, J.; Kozlowski, M. Dynamic mechanical properties of foamed polylactide and polylactide/wood flour composites. J. Biobased Mater. Bioenergy 2015, 9, 227–230. [Google Scholar] [CrossRef]
- Zhang, X.; Ding, W.; Zhao, N.; Chen, J.; Park, C.B. Effects of Compressed CO2 and Cotton Fibers on the Crystallization and Foaming Behaviors of Polylactide. Ind. Eng. Chem. Res. 2018, 57, 2094–2104. [Google Scholar] [CrossRef]
- Ludwiczak, J.; Frąckowiak, S.; Łużny, R. Effect of Recycling on the Cellular Structure of Polylactide in a Batch Process. Cell. Polym. 2018, 37, 69–79. [Google Scholar] [CrossRef]
- Bocz, K.; Tábi, T.; Vadas, D.; Sauceau, M.; Fages, J.; Marosi, G. Characterisation of natural fibre reinforced PLA foams prepared by supercritical CO2 assisted extrusion. Express Polym. Lett. 2016, 10, 771–779. [Google Scholar] [CrossRef]
- Vadas, D.; Igricz, T.; Sarazin, J.; Bourbigot, S.; Marosi, G.; Bocz, K. Flame retardancy of microcellular poly(lactic acid) foams prepared by supercritical CO2-assisted extrusion. Polym. Degrad. Stab. 2018, 153, 100–108. [Google Scholar] [CrossRef]
- Volpe, V.; De Filitto, M.; Klofacova, V.; De Santis, F.; Pantani, R. Effect of mold opening on the properties of PLA samples obtained by foam injection molding. Polym. Eng. Sci. 2018, 58, 475–484. [Google Scholar] [CrossRef]
- Volpe, V.; De Filitto, M.; Klofacova, V.; De Santis, F.; Pantani, R. Effect of processing conditions on the cell morphology distribution in foamed injection molded PLA samples. AIP Conf. Proc. 2017, 1914, 1–6. [Google Scholar] [CrossRef]
- Xue, S.; Jia, P.; Ren, Q.; Liu, X.; Lee, R.E.; Zhai, W. Improved expansion ratio and heat resistance of microcellular poly(L-lactide) foam via in-situ formation of stereocomplex crystallites. J. Cell. Plast. 2018, 54, 103–119. [Google Scholar] [CrossRef]
- Pantani, R.; Sorrentino, A.; Volpe, V.; Titomanlio, G. Foam injection molding of poly(lactic acid) with physical blowing agents. AIP Conf. Proc. 2014, 1593, 397–400. [Google Scholar] [CrossRef]
- Volpe, V.; Pantani, R. Foam injection molding of poly(lactic) acid: Effect of back pressure on morphology and mechanical properties. J. Appl. Polym. Sci. 2015, 132. [Google Scholar] [CrossRef]
- Wang, G.; Zhao, G.; Wang, S.; Zhang, L.; Park, C.B. Injection-molded microcellular PLA/graphite nanocomposites with dramatically enhanced mechanical and electrical properties for ultra-efficient EMI shielding applications. J. Mater. Chem. C 2018, 6, 6847–6859. [Google Scholar] [CrossRef]
- Pantani, R.; Volpe, V.; Titomanlio, G. Foam injection molding of poly(lactic acid) with environmentally friendly physical blowing agents. J. Mater. Process. Technol. 2014, 214, 3098–3107. [Google Scholar] [CrossRef]
- Seo, J.H.; Han, J.; Lee, K.S.; Cha, S.W. Combined Effects of Chemical and Microcellular Foaming on Foaming Characteristics of PLA (Poly Lactic Acid) in Injection Molding Process. Polym. Plast. Technol. Eng. 2012, 51, 455–460. [Google Scholar] [CrossRef]
- Mihai, M.; Huneault, M.A.; Favis, B.D. Rheology and extrusion foaming of chain-branched poly(lactic acid). Polym. Eng. Sci. 2010, 50, 629–642. [Google Scholar] [CrossRef]
- Shi, X.; Wang, L.; Kang, Y.; Qin, J.; Li, J.; Zhang, H.; Fan, X.; Liu, Y. Effect of poly ( butylenes succinate ) on the microcellular foaming of polylactide using supercritical carbon dioxide. J. Polym. Res. 2018, 25. [Google Scholar] [CrossRef]
- Julien, J.-M.; Bénézet, J.-C.; Lafranche, E.; Quantin, J.-C.; Bergeret, A.; Lacrampe, M.-F.; Krawczak, P. Development of poly(lactic acid) cellular materials: Physical and morphological characterizations. Polymer 2012, 53, 5885–5895. [Google Scholar] [CrossRef]
- Li, S.; He, G.; Liao, X.; Park, C.B.; Yang, Q.; Li, G. Introduction of a long-chain branching structure by ultraviolet-induced reactive extrusion to improve cell morphology and processing properties of polylactide foam. RSC Adv. 2017, 7, 6266–6277. [Google Scholar] [CrossRef]
- Meng, Q.; Heuzey, M.-C.; Carreau, P.J. Control of thermal degradation of polylactide/clay nanocomposites during melt processing by chain extension reaction. Polym. Degrad. Stab. 2012, 97, 2010–2020. [Google Scholar] [CrossRef]
- Ding, W.; Kuboki, T.; Wong, A.; Park, C.B.; Sain, M. Rheology, thermal properties, and foaming behavior of high d-content polylactic acid/cellulose nanofiber composites. RSC Adv. 2015, 5, 91544–91557. [Google Scholar] [CrossRef]
- Walallavita, A.; Verbeek, C.J.R.; Lay, M. Blending Novatein® thermoplastic protein with PLA for carbon dioxide assisted batch foaming. AIP Conf. Proc. 2016, 1713, 100006. [Google Scholar] [CrossRef]
- Dlouhá, J.; Suryanegara, L.; Yano, H. The role of cellulose nanofibres in supercritical foaming of polylactic acid and their effect on the foam morphology. Soft Matter 2012, 8, 8704–8713. [Google Scholar] [CrossRef]
- Witt, M.R.J.; Shah, S. Methods of Manufacture of Polylactic Acid Foams. Patent WO2008/093284 A1, 7 August 2008. [Google Scholar]
- Cink, K.; Smith, J.; Nangeroni, J.; Randall, J.R. Extruded Polylactide Foams Blown with Carbon Dioxide. Patent EP1735373 B1, 27 December 2006. [Google Scholar]
- Zhao, H.; Yan, X.; Zhao, G.; Guo, Z. Microcellular injection molded polylactic acid/poly (ε-caprolactone) blends with supercritical CO2: Correlation between rheological properties and their foaming behavior. Polym. Eng. Sci. 2016, 56, 939–946. [Google Scholar] [CrossRef]
- Lee, S.T.; Kareko, L.; Jun, J. Study of thermoplastic PLA foam extrusion. J. Cell. Plast. 2008, 44, 293–305. [Google Scholar] [CrossRef]
- Yu, L.; Toikka, G.; Dean, K.; Bateman, S.; Yuan, Q.; Filippou, C.; Nguyen, T. Foaming behaviour and cell structure of poly(lactic acid) after various modifications. Polym. Int. 2013, 62, 759–765. [Google Scholar] [CrossRef]
- Julien, J.M.; Quantin, J.C.; Bénézet, J.C.; Bergeret, A.; Lacrampe, M.F.; Krawczak, P. Chemical foaming extrusion of poly(lactic acid) with chain-extenders: Physical and morphological characterizations. Eur. Polym. J. 2015, 67, 40–49. [Google Scholar] [CrossRef]
- Göttermann, S.; Weinmann, S.; Bonten, C.; Standau, T.; Altstädt, V. Modifiziertes Polylactid für die Schaumextrusion. In Proceedings of the 24. Stuttgarter Kunststoffkolloquium, Stuttgart, Germany, 25–26 February 2015. [Google Scholar]
- Standau, T.; Murillo Castellón, S.; Delavoie, A.; Bonten, C.; Altstädt, V. Effects of Chemical Modifications on the Rheological and the Expansion Behavior of Polylactide (PLA) in Foam Extrusion. e-Polymers 2019. [Google Scholar] [CrossRef]
- Deroiné, M.; Le Duigou, A.; Corre, Y.M.; Le Gac, P.Y.; Davies, P.; César, G.; Bruzaud, S. Accelerated ageing of polylactide in aqueous environments: Comparative study between distilled water and seawater. Polym. Degrad. Stab. 2014, 108, 319–329. [Google Scholar] [CrossRef]
- Ren, Q.; Wang, J.; Zhai, W.; Su, S. Solid State Foaming of Poly(lactic acid) Blown with Compressed CO2: Influences of Long Chain Branching and Induced Crystallization on Foam Expansion and Cell Morphology. Ind. Eng. Chem. Res. 2013, 52, 13411–13421. [Google Scholar] [CrossRef]
- Nofar, M.; Ameli, A.; Park, C.B. A novel technology to manufacture biodegradable polylactide bead foam products. Mater. Des. 2015, 83, 413–421. [Google Scholar] [CrossRef]
- Nofar, M.; Ameli, A.; Park, C.B. Development of polylactide bead foams with double crystal melting peaks. Polymer 2015, 69, 83–94. [Google Scholar] [CrossRef]
- Boissard, C.; Bourban, P.-E.; Plummer, C.J.; Neagu, C.; Manson, J.-A.E. Cellular Biocomposites from Polylactide and Microfibrillated Cellulose. J. Cell. Plast. 2012. [Google Scholar] [CrossRef]
- Reignier, J.; Gendron, R.; Champagne, M.F. Extrusion foaming of poly(lactic acid) blown with CO2: Toward 100% green material. Cell. Polym. 2007, 26, 83–115. [Google Scholar] [CrossRef]
- Lim, L.-T.; Auras, R.; Rubino, M. Processing technologies for poly(lactic acid). Prog. Polym. Sci. 2008, 33, 820–852. [Google Scholar] [CrossRef]
- Dean, K.M.; Petinakis, E.; Meure, S.; Yu, L.; Chryss, A. Melt Strength and Rheological Properties of Biodegradable Poly(Lactic Aacid) Modified via Alkyl Radical-Based Reactive Extrusion Processes. J. Polym. Environ. 2012, 20, 741–747. [Google Scholar] [CrossRef]
- Göttermann, S.; Standau, T.; Weinmann, S.; Altstädt, V.; Bonten, C. Effect of chemical modification on the thermal and rheological properties of polylactide. Polym. Eng. Sci. 2017. [Google Scholar] [CrossRef]
- Nofar, M.; Park, C.B. Poly (lactic acid) foaming. Prog. Polym. Sci. 2014, 39, 1–21. [Google Scholar] [CrossRef]
- Corre, Y.-M.; Duchet, J.; Reignier, J.; Maazouz, A. Melt strengthening of poly (lactic acid) through reactive extrusion with epoxy-functionalized chains. Rheol. Acta 2011, 50, 613–629. [Google Scholar] [CrossRef]
- Huang, Y.; Zhang, C.; Pan, Y.; Wang, W.; Jiang, L.; Dan, Y. Study on the Effect of Dicumyl Peroxide on Structure and Properties of Poly(Lactic Acid)/Natural Rubber Blend. J. Polym. Environ. 2013, 21, 375–387. [Google Scholar] [CrossRef]
- Rytlewski, P.; Zenkiewicz, M.; Malinowski, R. Influence of dicumyl peroxide content on thermal and mechanical properties of polylactide. Int. Polym. Process. 2011, 26, 580–586. [Google Scholar] [CrossRef]
- Zhou, Z.F.; Huang, G.Q.; Xu, W.B.; Ren, F.M. Chain extension and branching of poly(L-lactic acid) produced by reaction with a DGEBA-based epoxy resin. Express Polym. Lett. 2007, 1, 734–739. [Google Scholar] [CrossRef]
- Zhong, W.; Ge, J.; Gu, Z.; Li, W.; Chen, X.; Zang, Y.; Yang, Y. Study on biodegradable polymer materials based on poly(lactic acid). I. Chain extending of low molecular weight poly(lactic acid) with methylenediphenyl diisocyanate. J. Appl. Polym. Sci. 1999, 74, 2546–2551. [Google Scholar] [CrossRef]
- Hiltunen, K.; Seppälä, J.V.; Härkönen, M. Lactic acid based poly(ester-urethane)s: The effects of different polymerization conditions on the polymer structure and properties. J. Appl. Polym. Sci. 1997, 64, 865–873. [Google Scholar] [CrossRef]
- Tuominen, J.; Seppälä, J.V. Synthesis and characterization of lactic acid based poly(ester-amide). Macromolecules 2000, 33, 3530–3535. [Google Scholar] [CrossRef]
- Södergård, A.; Stolt, M. Properties of lactic acid based polymers and their correlation with composition. Prog. Polym. Sci. 2002, 27, 1123–1163. [Google Scholar] [CrossRef]
- Formela, K.; Zedler; Hejna, A.; Tercjak, A. Reactive extrusion of bio-based polymer blends and composites–current trends and future developments. Express Polym. Lett. 2018, 12, 24–57. [Google Scholar] [CrossRef]
- Raquez, J.M.; Narayan, R.; Dubois, P. Recent advances in reactive extrusion processing of biodegradable polymer-based compositions. Macromol. Mater. Eng. 2008, 293, 447–470. [Google Scholar] [CrossRef]
- Khankrua, R.; Pivsa-Art, S.; Hiroyuki, H.; Suttiruengwong, S. Effect of chain extenders on thermal and mechanical properties of poly(lactic acid) at high processing temperatures: Potential application in PLA/Polyamide 6 blend. Polym. Degrad. Stab. 2014, 108, 232–240. [Google Scholar] [CrossRef]
- Li, H.; Huneault, M.A. Effect of chain extension on the properties of PLA/TPS blends. J. Appl. Polym. Sci. 2011, 122, 134–141. [Google Scholar] [CrossRef]
- Cailloux, J.; Santana, O.O.; Franco-Urquiza, E.; Bou, J.J.; Carrasco, F.; Gámez-Pérez, J.; Maspoch, M.L. Sheets of branched poly(lactic acid) obtained by one step reactive extrusion calendering process: Melt rheology analysis. Express Polym. Lett. 2012, 7, 304–318. [Google Scholar] [CrossRef]
- Eslami, H.; Kamal, M.R. Effect of a chain extender on the rheological and mechanical properties of biodegradable poly(lactic acid)/poly[(butylene succinate)-co-adipate] blends. J. Appl. Polym. Sci. 2013, 129, 2418–2428. [Google Scholar] [CrossRef]
- Carlson, D.; Dubois, P.; Nie, L.; Narayan, R. Free radical branching of polylactide by reactive extrusion. Polym. Eng. Sci. 1998, 38, 311–321. [Google Scholar] [CrossRef]
- Moad, G. Synthesis of Polyolefin Graft Copolymers by Reactive Extrusion. Prog. Polym. Sci. 1999, 24, 81–142. [Google Scholar] [CrossRef]
- Praphulla. Free Radical-Mediated Reactive Extrusion of Commodity Polymers. Ph.D. Thesis, Queen’s University, Kingston, ON, Canada, 2018. [Google Scholar]
- Corneillie, S.; Smet, M. PLA architectures: The role of branching. Stijn Corneillie Mario Smet 2015, 6, 850–867. [Google Scholar] [CrossRef]
- Al-Itry, R.; Lamnawar, K.; Maazouz, A. Improvement of thermal stability, rheological and mechanical properties of PLA, PBAT and their blends by reactive extrusion with functionalized epoxy. Polym. Degrad. Stab. 2012, 97, 1898–1914. [Google Scholar] [CrossRef]
- Frenz, V.; Scherzer, D.; Villalobos, M.; Awojulu, A.; Edison, M.; Van Der Meer, R. Multifunctional polymers as chain extenders and compatibilizers for polycondensates and biopolymers. Antec 2008, 3, 1682–1686. [Google Scholar]
- Gu, L.; Xu, Y.; Fahnhorst, G.W.; Macosko, C.W. Star vs long chain branching of poly(lactic acid) with multifunctional aziridine. J. Rheol. 2017, 61, 785–796. [Google Scholar] [CrossRef]
- Cherykhunthod, W.; Seadan, M.; Suttiruengwong, S. Effect of peroxide and chain extender on mechanical properties and morphology of poly (butylene succinate)/poly (lactic acid) blends. IOP Conf. Ser. Mater. Sci. Eng. 2015, 87. [Google Scholar] [CrossRef]
- Walha, F.; Lamnawar, K.; Maazouz, A.; Jaziri, M. Rheological, morphological and mechanical studies of sustainably sourced polymer blends based on poly(lactic acid) and polyamide 11. Polymers 2016, 8, 61. [Google Scholar] [CrossRef]
- Mallet, B.; Maazouz, K.; Abderrahim, L. Improvement of Blown Film Extrusion of Poly(Lactic Acid): Structure–Processing–Properties Relationships. Polym. Eng. Sci. 2014. [Google Scholar] [CrossRef]
- Hachana, N.; Wongwanchai, T.; Chaochanchaikul, K.; Harnnarongchai, W. Influence of Crosslinking Agent and Chain Extender on Properties of Gamma-Irradiated PLA. J. Polym. Environ. 2017, 25, 323–333. [Google Scholar] [CrossRef]
- Najafi, N.; Heuzey, M.C.; Carreau, P.J. Polylactide (PLA)-clay nanocomposites prepared by melt compounding in the presence of a chain extender. Compos. Sci. Technol. 2012, 72, 608–615. [Google Scholar] [CrossRef]
- Najafi, N.; Heuzey, M.C.; Carreau, P.J.; Wood-Adams, P.M. Control of thermal degradation of polylactide (PLA)-clay nanocomposites using chain extenders. Polym. Degrad. Stab. 2012, 97, 554–565. [Google Scholar] [CrossRef]
- Al-Itry, R.; Lamnawar, K.; Maazouz, A. Reactive extrusion of PLA, PBAT with a multi-functional epoxide: Physico-chemical and rheological properties. Eur. Polym. J. 2014, 58, 90–102. [Google Scholar] [CrossRef]
- Wang, J. Rheology of Foaming Polymers and Its Influence on Microcellular Processing; University of Toronto: Toronto, ON, USA, 2009; p. 136. ISBN 9780494608807. [Google Scholar]
- Jaszkiewicz, A.; Bledzki, A.K.; Van Der Meer, R.; Franciszczak, P.; Meljon, A. How does a chain-extended polylactide behave?: A comprehensive analysis of the material, structural and mechanical properties. Polym. Bull. 2014, 71, 1675–1690. [Google Scholar] [CrossRef]
- Jaszkiewicz, A.; Meljon, A.; Bledzki, A.K. Mechanical and thermomechanical properties of PLA/Man-made cellulose green composites modified with functional chain extenders—A comprehensive study. Polym. Compos. 2018, 39, 1716–1723. [Google Scholar] [CrossRef]
- Chaiwutthinan, P.; Leejarkpai, T.; Kashima, D.P.; Chuayjuljit, S. Poly(Lactic Acid)/Poly(Butylene Succinate) Blends Filled with Epoxy Functionalised Polymeric Chain Extender. Adv. Mater. Res. 2013, 664, 644–648. [Google Scholar] [CrossRef]
- Yousfi, M.; Dadouche, T.; Chomat, D.; Samuel, C.; Soulestin, J.; Lacrampe, M.F.; Krawczak, P. Development of nanofibrillar morphologies in poly(l-lactide)/poly(amide) blends: Role of the matrix elasticity and identification of the critical shear rate for the nodular/fibrillar transition. RSC Adv. 2018, 8, 22023–22041. [Google Scholar] [CrossRef]
- Cailloux, J.; Abt, T.; García-Masabet, V.; Santana, O.; Sánchez-Soto, M.; Carrasco, F.; Maspoch, M.L. Effect of the viscosity ratio on the PLA/PA10.10 bioblends morphology and mechanical properties. Express Polym. Lett. 2018, 12, 569–582. [Google Scholar] [CrossRef]
- Cailloux, J.; Santana, O.O.; Maspoch, M.L.; Bou, J.J.; Carrasco, F. Using viscoelastic properties to quantitatively estimate the amount of modified poly(lactic acid) chains through reactive extrusion. J. Rheol. 2015, 59, 1191–1227. [Google Scholar] [CrossRef]
- Södergård, A.; Stolt, M. Industrial Production of High Molecular Weight Poly(lactic acid). In Poly(Lactic Acid): Synthesis, Structures, Properties, Processing, and Applications; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2010; pp. 27–41. [Google Scholar]
- Bo-Hsin, L.; Ming-Chien, Y. Improvement of thermal and mechanical properties of poly(L-lactic acid) with 4,4-methylene diphenyl diisocyanate. Pharmacovigil. Rev. 2018, 10, 8–11. [Google Scholar] [CrossRef]
- Vachon, A.; Pépin, K.; Balampanis, E.; Veilleux, J.; Vuillaume, P.Y. Compatibilization of PLA/PEBA Blends via Reactive Extrusion: A Comparison of Different Coupling Agents. J. Polym. Environ. 2017, 25, 812–827. [Google Scholar] [CrossRef]
- Sungsanit, K. Rheological and Mechanical Behaviour of Poly (Lactic Acid)/Polyethylene Glycol Blends. Ph.D. Thesis, RMIT University, Melbourne, Australia, 2011; pp. 1–305. [Google Scholar]
- Liu, X.; Yu, L.; Dean, K.; Toikka, G.; Bateman, S.; Nguyen, T.; Yuan, Q.; Filippou, C. Improving Melt Strength of Polylactic Acid. Int. Polym. Process. 2013, 28, 64–71. [Google Scholar] [CrossRef]
- You, J.; Lou, L.; Yu, W.; Zhou, C. The preparation and crystallization of long chain branching polylactide made by melt radicals reaction. J. Appl. Polym. Sci. 2013, 129, 1959–1970. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, S.; Zhang, L.; Bai, Y. Preparation and rheological characterization of long chain branching polylactide. Polymer 2014, 55, 2472–2480. [Google Scholar] [CrossRef]
- Quanxiao, D.; Chow, L.C.; Wang, T.; Frukhtbeyn, S.A.; Wang, F.; Yang, M.; Mitchell, J.W. A New Bioactive Polylactide-based Composite with High Mechanical Strength. Acc. Chem. Res. 2008, 45, 788–802. [Google Scholar] [CrossRef]
- Stloukal, P.; Jandikova, G.; Koutny, M.; Sedlařík, V. Carbodiimide additive to control hydrolytic stability and biodegradability of PLA. Polym. Test. 2016, 54, 19–28. [Google Scholar] [CrossRef]
- Stloukal, P.; Kalendova, A.; Mattausch, H.; Laske, S.; Holzer, C.; Koutny, M. The influence of a hydrolysis-inhibiting additive on the degradation and biodegradation of PLA and its nanocomposites. Polym. Test. 2015, 41, 124–132. [Google Scholar] [CrossRef]
- Yang, L.; Chen, X.; Jing, X. Stabilization of poly(lactic acid) by polycarbodiimide. Polym. Degrad. Stab. 2008, 93, 1923–1929. [Google Scholar] [CrossRef]
- Bousfield, G. Effect of Chain Extension on Rheology and Tensile Properties of PHB and PHB-PLA Blends. Master’s Thesis, Universite de Montreal, Montreal, QC, Canada, 2014. [Google Scholar]
- Holcapkova, P.; Stloukal, P.; Kucharczyk, P.; Omastova, M.; Kovalcik, A. Anti-hydrolysis effect of aromatic carbodiimide in poly(lactic acid)/wood flour composites. Compos. Part A Appl. Sci. Manuf. 2017, 103, 283–291. [Google Scholar] [CrossRef]
- Cicero, J.A.; Dorgan, J.R.; Dec, S.F.; Knauss, D.M. Phosphite stabilization effects on two-step melt-spun fibers of polylactide. Polym. Degrad. Stab. 2002, 78, 95–105. [Google Scholar] [CrossRef]
- Dorgan, J.R.; Janzen, J.; Clayton, M.P.; Hait, S.B.; Knauss, D.M. Melt rheology of variable L-content poly(lactic acid). J. Rheol. 2005, 49, 607–619. [Google Scholar] [CrossRef]
- Lehermeier, H.J.; Dorgan, J.R. Melt rheology of poly(lactic acid): Consequences of blending chain architectures. Polym. Eng. Sci. 2001, 41, 2172–2184. [Google Scholar] [CrossRef]
- Palade, L.I.; Lehermeier, H.J.; Dorgan, J.R. Melt rheology of high L-content poly(lactic acid). Macromolecules 2001, 34, 1384–1390. [Google Scholar] [CrossRef]
- Meng, X.; Shi, G.; Chen, W.; Wu, C.; Xin, Z.; Han, T. Structure effect of phosphite on the chain extension in PLA. Polym. Degrad. Stab. 2015, 120, 283–289. [Google Scholar] [CrossRef]
- Meng, X.; Shi, G.; Wu, C.; Chen, W.; Xin, Z.; Shi, Y.; Sheng, Y. Chain extension and oxidation stabilization of Triphenyl Phosphite (TPP) in PLA. Polym. Degrad. Stab. 2016, 124, 112–118. [Google Scholar] [CrossRef]
- Gu, L. Modification of Poly (lactic acid) by Melt Blending. Ph.D. Thesis, University of Minnesota, Minneapolis, MN, USA, 2017. [Google Scholar]
- Liu, C.; Jia, Y.; He, A. Preparation of higher molecular weight poly (L-lactic acid) by chain extension. Int. J. Polym. Sci. 2013, 2013, 1–7. [Google Scholar] [CrossRef]
- Wang, H.; Sun, X.; Seib, P. Strengthening blends of poly(lactic acid) and starch with methylenediphenyl diisocyanate. J. Appl. Polym. Sci. 2001, 82, 1761–1767. [Google Scholar] [CrossRef]
- Jun, C.L. Reactive blending of biodegradable polymers: PLA and starch. J. Polym. Environ. 2000, 8, 33–37. [Google Scholar] [CrossRef]
- Kylmä, J.; Tuominen, J.; Helminen, A.; Seppälä, J. Chain extending of lactic acid oligomers. Effect of 2,2′-bis(2-oxazoline) on 1,6-hexamethylene diisocyanate linking reaction. Polymer 2001, 42, 3333–3343. [Google Scholar] [CrossRef]
- Tuominen, J.; Kylmä, J.; Seppälä, J. Chain extending of lactic acid oligomers. 2. Increase of molecular weight with 1,6-hexamethylene diisocyanate and 2,2′-bis(2-oxazoline). Polymer 2002, 43, 3–10. [Google Scholar] [CrossRef]
- Wei, L.; McDonald, A.G. Peroxide induced cross-linking by reactive melt processing of two biopolyesters: Poly(3-hydroxybutyrate) and poly(l -lactic acid) to improve their melting processability. J. Appl. Polym. Sci. 2015, 132. [Google Scholar] [CrossRef]
- Takamura, M.; Nakamura, T.; Takahashi, T.; Koyama, K. Effect of type of peroxide on cross-linking of poly(l-lactide). Polym. Degrad. Stab. 2008, 93, 1909–1916. [Google Scholar] [CrossRef]
- Thitithammawong, A.; Nakason, C.; Sahakaro, K.; Noordermeer, J. Effect of different types of peroxides on rheological, mechanical, and morphological properties of thermoplastic vulcanizates based on natural rubber/polypropylene blends. Polym. Test. 2007, 26, 537–546. [Google Scholar] [CrossRef]
- Signori, F.; Boggioni, A.; Righetti, M.C.; Rondán, C.E.; Bronco, S.; Ciardelli, F. Evidences of transesterification, chain branching and cross-linking in a biopolyester commercial blend upon reaction with dicumyl peroxide in the melt. Macromol. Mater. Eng. 2015, 300, 153–160. [Google Scholar] [CrossRef]
- Södergård, A.; Niemi, M.; Selin, J.F.; Näsman, J.H. Changes in Peroxide Melt-Modified Poly(L-lactide). Ind. Eng. Chem. Res. 1995, 34, 1203–1207. [Google Scholar] [CrossRef]
- Takamura, M.; Nakamura, T.; Kawaguchi, S.; Takahashi, T.; Koyama, K. Molecular characterization and crystallization behavior of peroxide-induced slightly crosslinked poly(L-lactide) during extrusion. Polym. J. 2010, 42, 600–608. [Google Scholar] [CrossRef]
- Takamura, M.; Sugimoto, M.; Kawaguchi, S.; Takahashi, T.; Koyama, K. Influence of extrusion temperature on molecular architecture and crystallization behavior of peroxide-induced slightly crosslinked poly(L-lactide) by reactive extrusion. J. Appl. Polym. Sci. 2012, 123, 1468–1478. [Google Scholar] [CrossRef]
- Södergård, A. Modification of polylactide. Polym. Sci. 1998, 2, 263–275. [Google Scholar]
- Nijenhuis, A.J.; GrijpmaA, D.W.; Pennings, J. Crosslinked poly(l-lactide) and poly(ε-caprolactone). Polymer 1996, 37, 2783–2791. [Google Scholar] [CrossRef]
- Yang, S.; Wu, Z.H.; Yang, W.; Yang, M.B. Thermal and mechanical properties of chemical crosslinked polylactide (PLA). Polym. Test. 2008, 27, 957–963. [Google Scholar] [CrossRef]
- Dawidziuk, K.; Simmons, H.; Kontopoulou, M.; ScottParent, J. Peroxide-initiated graft modification of thermoplastic BioPolyesters: Introduction of long-chain branching. Polymer 2018. [Google Scholar] [CrossRef]
- Dong, W.; Ma, P.; Wang, S.; Chen, M.; Cai, X.; Zhang, Y. Effect of partial crosslinking on morphology and properties of the poly(β-hydroxybutyrate)/poly(d,l-lactic acid) blends. Polym. Degrad. Stab. 2013, 98, 1549–1555. [Google Scholar] [CrossRef]
- Zhang, J.F.; Sun, X. Mechanical properties of poly(lactic acid)/starch composites compatibilized by maleic anhydride. Biomacromolecules 2004, 5, 1446–1451. [Google Scholar] [CrossRef]
- Carlson, D.; Nie, L.; Narayan, R.; Dubois, P. Maleation of Polylactide (PLA) by Reactive Extrusion. J. Appl. Polym. Sci. 1999, 72, 477–485. [Google Scholar] [CrossRef]
- Detyothin, S.; Selke, S.E.M.; Narayan, R.; Rubino, M.; Auras, R. Reactive functionalization of poly(lactic acid), PLA: Effects of the reactive modifier, initiator and processing conditions on the final grafted maleic anhydride content and molecular weight of PLA. Polym. Degrad. Stab. 2013, 98, 2697–2708. [Google Scholar] [CrossRef]
- Huneault, M.A.; Li, H. Morphology and properties of compatibilized polylactide/thermoplastic starch blends. Polymer 2007, 48, 270–280. [Google Scholar] [CrossRef]
- Ma, P.; Jiang, L.; Ye, T.; Dong, W.; Chen, M. Melt free-radical grafting of maleic anhydride onto biodegradable poly(lactic acid) by using styrene as a comonomer. Polymers 2014, 6, 1528–1543. [Google Scholar] [CrossRef]
- Hwang, S.W.; Lee, S.B.; Lee, C.K.; Lee, J.Y.; Shim, J.K.; Selke, S.E.M.; Soto-Valdez, H.; Matuana, L.; Rubino, M.; Auras, R. Grafting of maleic anhydride on poly(L-lactic acid). Effects on physical and mechanical properties. Polym. Test. 2012, 31, 333–344. [Google Scholar] [CrossRef]
- Avella, M.; Bogoeva-Gaceva, G.; Bužarovska, A.; Errico, M.E.; Gentile, G.; Grozdanov, A. Poly(lactic acid)-based biocomposites reinforced with kenaf fibers. J. Appl. Polym. Sci. 2008, 108, 3542–3551. [Google Scholar] [CrossRef]
- Tang, H.; Dai, W.; Chen, B. A New Method for Producing High Melt Strength Polypropylene With Reactive Extrusion. Polym. Eng. Sci. 2008, 48, 1339–1344. [Google Scholar] [CrossRef]
- Dorgan, J.R.; Williams, J.S.; Lewis, D.N. Melt rheology of poly(lactic acid): Entanglement and chain architecture effects. J. Rheol. 1999, 43, 1141–1155. [Google Scholar] [CrossRef]
- Locati, G.; Pegoraro, M.; Iti, D.N.; Milan, V.M.B. A Model for the Zero Shear Viscosity. Polym. Eng. Sci. 1999, 39, 741–748. [Google Scholar] [CrossRef]
- Cooper-White, J.J.; Mackay, M.E. Rheological properties of poly(lactides). Effect of molecular weight and temperature on the viscoelasticity of poly(l-lactic acid). J. Polym. Sci. Part B Polym. Phys. 1999, 37, 1803–1814. [Google Scholar] [CrossRef]
- Cox, W.; Merz, E. Correlation of dynamic and steady flow viscosities. J. Polym. Sci. Part A Polym. Chem. 1958, 28, 619–622. [Google Scholar]
- Spitael, P.; Macosko, C.W. Strain hardening in polypropylenes and its role in extrusion foaming. Polym. Eng. Sci. 2004, 44, 2090–2100. [Google Scholar] [CrossRef]
- Wagner, M.H.; Bastian, H.; Hachmann, P.; Meissner, J.; Kurzbeck, S.; Münstedt, H.; Langouche, F. The strain-hardening behaviour of linear and long-chain-branched polyolefin melts in extensional flows. Rheol. Acta 2000, 39, 97–109. [Google Scholar] [CrossRef]
- Saeidlou, S.; Huneault, M.A.; Li, H.; Park, C.B. Poly(lactic acid) crystallization. Prog. Polym. Sci. 2012, 37, 1657–1677. [Google Scholar] [CrossRef]
- Hartmann, M.H. High Molecular Weight Polylactic Acid Polymers. In Biopolymers from Renewable Resources; Kaplan, D.L., Ed.; Springer: Berlin/Heidelberg, Germany, 1998; pp. 367–411. ISBN 978-3-662-03680-8. [Google Scholar]
- Jamshidi, K.; Hyon, S.-H.; Ikada, Y. Thermal Characterization of polylactides. Polymer 1988, 29, 2229–2234. [Google Scholar] [CrossRef]
- Ahmed, J.; Zhang, J.X.; Song, Z.; Varshney, S.K. Thermal properties of polylactides: Effect of molecular mass and nature of lactide isomer. J. Therm. Anal. Calorim. 2009, 95, 957–964. [Google Scholar] [CrossRef]
- Wang, Y.; Li, M.; Wang, K.; Shao, C.; Li, Q.; Shen, C. Unusual structural evolution of poly(lactic acid) upon annealing in the presence of an initially oriented mesophase. Soft Matter 2014, 10, 1512–1518. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Duan, Y.; Sato, H.; Tsuji, H.; Noda, I.; Yan, S.; Ozaki, Y. Crystal modifications and thermal behavior of poly(L-lactic acid) revealed by infrared spectroscopy. Macromolecules 2005, 38, 8012–8021. [Google Scholar] [CrossRef]
- Zhang, J.; Tashiro, K.; Tsuji, H.; Domb, A.J. Disorder-to-order phase transition and multiple melting behavior of poly(L-lactide) investigated by simultaneous measurements of WAXD and DSC. Macromolecules 2008, 41, 1352–1357. [Google Scholar] [CrossRef]
- Pan, P.; Kai, W.; Zhu, B.; Dong, T.; Inoue, Y. Polymorphous crystallization and multiple melting behavior of poly(L-lactide): Molecular weight dependence. Macromolecules 2007, 40, 6898–6905. [Google Scholar] [CrossRef]
- Stoclet, G.; Seguela, R.; Vanmansart, C.; Rochas, C.; Lefebvre, J.M. WAXS study of the structural reorganization of semi-crystalline polylactide under tensile drawing. Polymer 2012, 53, 519–528. [Google Scholar] [CrossRef]
- Puchalski, M.; Kwolek, S.; Szparaga, G.; Chrzanowski, M.; Krucinska, I. Investigation of the influence of PLA molecular structure on the crystalline forms (α’’ and α) and Mechanical Properties ofWet Spinning Fibres. Polymers 2017, 9, 18. [Google Scholar] [CrossRef]
- Eling, B.; Gogolewski, S.; Pennings, A.J. Biodegradable materials of poly ( L-lactic acid ): 1. Melt-spun and solution-spun fibres. Polymer 1982, 23, 1587–1593. [Google Scholar] [CrossRef]
- Hoogsteen, W.; Postema, A.R.; Pennings, A.J.; Brinke, G.T.; Zugenmaier, P. Crystal Structure, Conformation, and Morphology of Solution-Spun Poly(L-Lactide) Fibers. Macromolecules 1990, 23, 634–642. [Google Scholar] [CrossRef]
- Cartier, L.; Okihara, T.; Ikada, Y.; Tsuji, H.; Puiggali, J.; Lotz, B. Epitaxial crystallization and crystalline polymorphism of polylactides. Polymer 2000, 41, 8909–8919. [Google Scholar] [CrossRef]
- Nofar, M.; Zhu, W.; Park, C.B.; Randall, J. Crystallization kinetics of linear and long-chain-branched polylactide. Ind. Eng. Chem. Res. 2011, 50, 13789–13798. [Google Scholar] [CrossRef]
- Nofar, M.; Zhu, W.; Park, C.B. Effect of dissolved CO2 on the crystallization behavior of linear and branched PLA. Polymer 2012, 53, 3341–3353. [Google Scholar] [CrossRef]
- De Santis, F.; Pantani, R. Melt compounding of poly (Lactic Acid) and talc: Assessment of material behavior during processing and resulting crystallization. J. Polym. Res. 2015, 22, 1–9. [Google Scholar] [CrossRef]
- Battegazzore, D.; Bocchini, S.; Frache, A. Crystallization kinetics of poly(lactic acid)-talc composites. Express Polym. Lett. 2011, 5, 849–858. [Google Scholar] [CrossRef]
- Papageorgiou, G.Z.; Achilias, D.S.; Nanaki, S.; Beslikas, T.; Bikiaris, D. PLA nanocomposites: Effect of filler type on non-isothermal crystallization. Thermochim. Acta 2010, 511, 129–139. [Google Scholar] [CrossRef]
- Shi, X.; Zhang, G.; Phuong, T.V.; Lazzeri, A. Synergistic effects of nucleating agents and plasticizers on the crystallization behavior of Poly(lactic acid). Molecules 2015, 20, 1579–1593. [Google Scholar] [CrossRef] [PubMed]
- Ikada, Y.; Jamshidi, K.; Tsuji, H.; Hyon, S.H. Stereocomplex formation between enantiomeric poly(lactides). Macromolecules 1987, 20, 904–906. [Google Scholar] [CrossRef]
- Tsuji, H.; Takai, H.; Saha, S.K. Isothermal and non-isothermal crystallization behavior of poly(l-lactic acid): Effects of stereocomplex as nucleating agent. Polymer 2006, 47, 3826–3837. [Google Scholar] [CrossRef]
- Brzeziński, M.; Biela, T. Polylactide nanocomposites with functionalized carbon nanotubes and their stereocomplexes: A focused review. Mater. Lett. 2014, 121, 244–250. [Google Scholar] [CrossRef]
- Nofar, M.; Tabatabaei, A.; Ameli, A.; Park, C.B. Comparison of melting and crystallization behaviors of polylactide under high-pressure CO2, N2, and He. Polymer 2013, 54, 6471–6478. [Google Scholar] [CrossRef]
- Takada, M.; Hasegawa, S.; Ohshima, M. Crystallization kinetics of poly(L-lactide) in contact with pressurized CO2. Polym. Eng. Sci. 2004, 44, 186–196. [Google Scholar] [CrossRef]
- Kokturk, G.; Piskin, E.; Serhatkulu, T.F.; Cakmak, M. Evolution of phase behavior and orientation in uniaxially deformed polylactic acid films. Polym. Eng. Sci. 2002, 42, 1619–1628. [Google Scholar] [CrossRef]
- Wong, A.; Guo, Y.; Parka, C.B. Fundamental mechanisms of cell nucleation in polypropylene foaming with supercritical carbon dioxide—Effects of extensional stresses and crystals. J. Supercrit. Fluids 2013, 79, 142–151. [Google Scholar] [CrossRef]
- Yasuniwa, M.; Tsubakihara, S.; Sugimoto, Y.; Nakafuku, C. Thermal analysis of the double-melting behavior of poly(L-lactic acid). J. Polym. Sci. Part B Polym. Phys. 2004, 42, 25–32. [Google Scholar] [CrossRef]
- Okolieocha, C.; Raps, D.; Subramaniam, K.; Altstädt, V. Microcellular to nanocellular polymer foams: Progress (2004–2015) and future directions—A review. Eur. Polym. J. 2015, 73, 500–519. [Google Scholar] [CrossRef]
- Raps, D.; Hossieny, N.; Park, C.B.; Altstädt, V. Past and present developments in polymer bead foams and bead foaming technology. Polymer 2015, 56, 5–19. [Google Scholar] [CrossRef]
- Sauceau, M.; Fages, J.; Common, A.; Nikitine, C.; Rodier, E. New challenges in polymer foaming: A review of extrusion processes assisted by supercritical carbon dioxide. Prog. Polym. Sci. 2011, 36, 749–766. [Google Scholar] [CrossRef]
- Colton, J.S.; Suh, N.P. Nucleation of microcellular foam: Theory and practice. Polym. Eng. Sci. 1987, 27, 500–503. [Google Scholar] [CrossRef]
- Goswami, J.; Bhatnagar, N.; Mohanty, S.; Ghosh, A.K. Processing and characterization of poly(lactic acid) based bioactive composites for biomedical scaffold application. Express Polym. Lett. 2013, 7, 767–777. [Google Scholar] [CrossRef]
- Juntunen, R.P.; Kumar, V.; Weller, J.E.; Bezubic, W.P.; Mi, J. Impact Strength of High Density Microcellular Poly(Vinyl Chloride) Foams. J. Vinyl Addit. Technol. 2000, 6, 93–99. [Google Scholar] [CrossRef]
- Sun, H.; Sur, G.S.; Mark, J.E. Microcellular foams from polyethersulfone and polyphenylsulfone: Preparation and mechanical properties. Eur. Polym. J. 2002, 38, 2373–2381. [Google Scholar] [CrossRef]
- Urbanczyk, L.; Calberg, C.; Detrembleur, C.; Jérôme, C.; Alexandre, M. Batch foaming of SAN/clay nanocomposites with scCO2: A very tunable way of controlling the cellular morphology. Polymer 2010, 51, 3520–3531. [Google Scholar] [CrossRef]
- Saiz-Arroyo, C.; Saja, J.A.; Velasco, J.I.; Rodríguez-Pérez, M.Á. Moulded polypropylene foams produced using chemical or physical blowing agents: Structure–properties relationship. J. Mater. Sci. 2012, 47, 5680–5692. [Google Scholar] [CrossRef]
- Guanghong, H.; Yue, W. Microcellular Foam Injection Molding Process. In Some Critical Issues for Injection Molding; Wang, J., Ed.; Intech: London, UK, 2012; pp. 175–202. ISBN 9789537619992. [Google Scholar]
- Hwang, S.-S.; Hsu, P.P.; Yeh, J.-M.; Chang, K.-C.; Lai, Y.-Z. The mechanical/thermal properties of microcellular injection-molded poly-lactic-acid nanocomposites. Polym. Compos. 2009, 30, 1625–1630. [Google Scholar] [CrossRef]
- Standau, T.; Hädelt, B.; Schreier, P.; Altstädt, V. Development of a Bead Foam from an Engineering Polymer with Addition of Chain Extender: Expanded Polybutylene Terephthalate. Ind. Eng. Chem. Res. 2018, 57, 17170–17176. [Google Scholar] [CrossRef]
- Stastny, F.; Gäth, R. Verfahren zur Herstellung Poröser Massen Oder Poröser Formkörper aus Polymerisaten. Patent DE845264, 14 August 1952. [Google Scholar]
- Lee, E.K. Novel Manufacturing Processes for Polymer Bead Foams. Ph.D. Thesis, University of Toronto, Toronto, ON, Canada, 2010. [Google Scholar]
- Shen, J.; Cao, X.; Lee, L.J. Synthesis and foaming of water expandable polystyrene-clay nanocomposites. Polymer 2006, 47, 6303–6310. [Google Scholar] [CrossRef]
- Parker, K.; Garancher, J.-P.; Shah, S.; Fernyhough, A. Expanded polylactic acid—An eco-friendly alternative to polystyrene foam. J. Cell. Plast. 2011, 47, 233–243. [Google Scholar] [CrossRef]
- Harrison, I.R. Modelling ‘melting’ in macromolecules. Polymer 1985, 26, 3–7. [Google Scholar] [CrossRef]
- Samuels, R.J. Quantitative structural characterization of the melting behavior of isotactic polypropylene. J. Polym. Sci. Polym. Phys. Ed. 1975, 13, 1417–1446. [Google Scholar] [CrossRef]
- Kardos, J.L.; Christiansen, A.W.; Baer, E. Structure of pressure-crystallized polypropylene. J. Polym. Sci. Part A-2 Polym. Phys. 1966, 4, 777–788. [Google Scholar] [CrossRef]
- Padden, F.J.; Keith, H.D. Spherulitic crystallization in polypropylene. J. Appl. Phys. 1959, 30, 1479–1484. [Google Scholar] [CrossRef]
- Pae, K.D. Solid-Solid Transition of Isotactic Polypropylene. Polymer 1968, 6, 657–663. [Google Scholar] [CrossRef]
- Zhang, R.; Luo, X.; Wang, Q.; Ma, D. Melting Behavior of Low Ethylene Content Polypropylene Copolymers with and without Nucleating Agents. Chin. J. Polym. Sci. 1994, 12, 246–255. [Google Scholar]
- Hingmann, R.; Rieger, J.; Kersting, M. Rheological Properties of a Partially Molten Polypropylene Random Coplymer during Annealing. Macromolecules 1995, 28, 3801–3806. [Google Scholar] [CrossRef]
- Nofar, M.; Guo, Y.; Park, C.B. Double crystal melting peak generation for expanded polypropylene bead foam manufacturing. Ind. Eng. Chem. Res. 2013, 52, 2297–2303. [Google Scholar] [CrossRef]
- Park, C.B.; Nofar, M. A Method for the Preparation of PLA Bead Foams. Patent WO 2014158014A1, 2 October 2014. [Google Scholar]
- Tang, L.; Zhai, W.; Zheng, W. Autoclave preparation of expanded polypropylene/poly(lactic acid) blend bead foams with a batch foaming process. J. Cell. Plast. 2011, 47, 429–446. [Google Scholar] [CrossRef]
- Fossey, D.J.; Smith, C.H. A New Potting Material—Expandable Polystyrene Bead Foam. J. Cell. Plast. 1977, 13, 347–353. [Google Scholar] [CrossRef]
- Järvelä, P.; Sarlin, J.; Järvelä, P.; Törmälä, P. A method to measure the fusion strength between expanded polystyrene (EPS) beads. J. Mater. Sci. 1986, 21, 3139–3142. [Google Scholar] [CrossRef]
- Stupak, P.; Frye, W.; Donovan, J. The Effect of Bead Fusion on the Energy Absorption of Polystyrene Foam. Part I: 9 Fracture Toughness. J. Cell. Plast. 1991, 27, 484–505. [Google Scholar] [CrossRef]
- Rossacci, J.; Shivkumar, S. Bead fusion in polystyrene foams. J. Mater. Sci. 2003, 38, 201–206. [Google Scholar] [CrossRef]
- Rossacci, J.; Shivkumar, S. Influence of EPS bead fusion on pattern degradation and casting formation in the lost foam process. J. Mater. Sci. 2003, 38, 2321–2330. [Google Scholar] [CrossRef]
- Standau, T.; Schreier, P.; Görl, J.; Mühlbacher, M.; Neumeyer, T.; Altstädt, V. Partikelschäume—Neue Materialien und Verarbeitungstechnologien Eröffnen neue Anwendungen. DGM-diALOG 2018, 2, 26–33. [Google Scholar]
- Endres, H.-J.; Siebert-Raths, A. Technische Biopolymere—Rahmenbedingungen, Marksituation, Herstellung, Aufbau und Eigenschaften; Carl Hanser Verlag: München, Germany, 2009; ISBN 978-3-446-41683-3. [Google Scholar]
- Tokiwa, Y.; Calabia, B.P. Biodegradability and biodegradation of poly(lactide). Appl. Microbiol. Biotechnol. 2006, 72, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, C.M.; Athanasiou, K.A.; Heckman, J.D. Biodegradable PLA-PGA Polymers for Tissue Engineering in Orthopaedics. Mater. Sci. Forum 1997, 250, 115–128. [Google Scholar] [CrossRef]
- Agrawal, C.M.; Bay, R.B. Biodegradable polymeric scaffolds for musculoskeletal tissue engineering. J. Biomed. Mater. Res. 2001, 55, 141–150. [Google Scholar] [CrossRef]
- Shao, J.; Xiang, S.; Bian, X.; Sun, J.; Li, G.; Chen, X. Remarkable melting behavior of PLA stereocomplex in linear PLLA/PDLA blends. Ind. Eng. Chem. Res. 2015, 54, 2246–2253. [Google Scholar] [CrossRef]
- Tsuji, H.; Ikada, Y. Stereocomplex formation between enantiomeric poly(lactic acid)s. XI. Mechanical properties and morphology of solution-cast films. Polymer 1999, 40, 6699–6708. [Google Scholar] [CrossRef]
- Srithep, Y.; Nealey, P.; Turng, L.-S. Effects of annealing time and temperature on the crystallinity and heat resistance behavior of injection-molded poly(lactic acid). Polym. Eng. Sci. 2013, 53, 580–588. [Google Scholar] [CrossRef]
- Harris, A.M.; Lee, E.C. Improving mechanical performance of injection molded PLA by controlling crystallinity. J. Appl. Polym. Sci. 2008, 107, 2246–2255. [Google Scholar] [CrossRef]
- Tang, Z.; Zhang, C.; Liu, X.; Zhu, J. The crystallization behavior and mechanical properties of polylactic acid in the presence of a crystal nucleating agent. J. Appl. Polym. Sci. 2012, 125, 1108–1115. [Google Scholar] [CrossRef]
- Zhu, H.; Zhu, Q.; Li, J.; Tao, K.; Xue, L.; Yan, Q. Synergistic effect between expandable graphite and ammonium polyphosphate on flame retarded polylactide. Polym. Degrad. Stab. 2011, 96, 183–189. [Google Scholar] [CrossRef]
- Ke, C.H.; Li, J.; Fang, K.Y.; Zhu, Q.L.; Zhu, J.; Yan, Q.; Wang, Y.Z. Synergistic effect between a novel hyperbranched charring agent and ammonium polyphosphate on the flame retardant and anti-dripping properties of polylactide. Polym. Degrad. Stab. 2010, 95, 763–770. [Google Scholar] [CrossRef]
- Tang, G.; Zhang, R.; Wang, X.; Wang, B.; Song, L.; Hu, Y.; Gong, X. Enhancement of flame retardant performance of bio-based polylactic acid composites with the incorporation of aluminum hypophosphite and expanded graphite. J. Macromol. Sci. Part A Pure Appl. Chem. 2013, 50, 255–269. [Google Scholar] [CrossRef]
- Wang, J.; Ren, Q.; Zheng, W.; Zhai, W. Improved Flame-Retardant Properties of Poly(lactic acid) Foams Using Starch as a Natural Charring Agent. Ind. Eng. Chem. Res. 2014, 53, 1422–1430. [Google Scholar] [CrossRef]
- Wang, K.; Wang, J.; Zhao, D.; Zhai, W. Preparation of microcellular poly(lactic acid) composites foams with improved flame retardancy. J. Cell. Plast. 2017, 53, 45–63. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PLA Grade (NatureWorks) | Foamed | d-Content | |
|---|---|---|---|
| Neat | Chemically Modified | (%) | |
| Extrusion and thermoforming | |||
| 2002 D | A [30,31,32,33,34,35,36,37,38,39,40], F [41,42,43,44,45], X [43,46,47,48,49,50,51,52,53,54,55] | A [31,35,36], X [46,47,52,55], | 4.0–4.3 [32,37,41,44,48,49,55,56] |
| 2003 D | A [57], F [58], X [59,60,61,62] | A [63], X [59,60] | 4.3 [57] |
| 2500 HP | A [64] | A [64] | 0.4 [65] |
| Injection molding | |||
| 3000 D | A [66,67] | A [67] | N/A |
| 3001 D | A [68,69,70,71,72], B [73], F [74,75,76,77,78,79], X [80] | A [68,70], F [75], X [80] | 1.4–1.5 [71,76,78] |
| 3051 D | A [81,82,83], X [84] | A [81,83], X [18] | 4–4.15 [18,81] |
| 3052 D | A [85,86], X [59,87] | X [59,87,88] | 4 [85,87] |
| 3251 D | A [64], F [89,90], X [46] | A [64] | 1.4 [89] |
| Films and cards | |||
| 4032 D | A [91], F [89,90,92,93,94,95,96], X [48,97,98,99] | X [18,97,100] | 1.4–2.0 [18,48,56,89,101] |
| 4060 D | A [102,103,104], B [73,105], X [106], F [107] | B [19], X [106] | 12–12.3 [56,106] |
| Fibers and nonwovens | |||
| 6300 D | X [106,108] | 9.5 [106], 9.85 [108] | |
| Blow molding | |||
| 7000 D | A [109], X [99,110] | A [109], X [110] | 6.4 [110] |
| 7001 D | A [111], X [111,112] | A [111], X [111,112] | 4.4 +/− 0.5 [113] |
| Foaming | |||
| 8051 D | X [49], A [71,114] | A [71,114], B [20,21,115,116], X [49] | 4.2–4.6 [49,71,114,115] |
| 8052 D | A [117], X [46] | A [117], X [46,65] | 4.7 [65] |
| 8300 D | X [106] | 11 [106] | |
| 8302 D | A [71], X [48,50,97,118] | X [97] | 9.85–10.1 [48,71,118] |
| Type | Functional Group | Chemical Modifier | Reference |
|---|---|---|---|
| Epoxide | 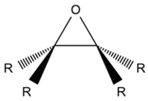 | Multifunctional epoxy-based oligomer | [18,31,35,46,49,52,59,64,65,68,70,71,75,80,81,83,87,88,97,101,110,112,114,115,116,117,121,123,134,135,136,142,143,144,145,146,147,148,149,150,151,152,153,154,155,156,157] |
| Isocyanate |  | 1,4-butane diisocyanate (BDI) | [67] |
| 1,6-hexamethylene diisocyanate (HDI) | [101,121,158] | ||
| 4,4-methylene diphenyl diisocyanate (MDI) | [127,159,160] | ||
| Anhydride | 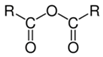 | Pyromellitic dianhydride (PMDA) | [36,101,109,143,161,162,163,164,165] |
| Oxazoline | 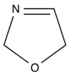 | 1,3-bisoxazoline | [121] |
| 1,4-phenylene-bis-oxazoline | [164] | ||
| 2,2-bis(2-oxazoline) | [162] | ||
| Not specified | [109] | ||
| Carbodiimide (CDI) | 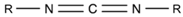 | CDI | [166,167] |
| Polycarbodiimide (PCDI) | [149,168,169] | ||
| Bis(2,6-diisopropylphenyl) carbodiimide (BDICDI) | [166,170] | ||
| Phosphite | 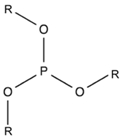 | Tris(nonyl-phenyl) phosphite (TNPP) | [149,171,172,173,174] |
| Triphenylphosphite (TPP) | [175,176] |
| Volume Expansion Rate (VER) (-) | Void Fraction (), Degree of Foaming (-) | Density Reduction (DR), Foaming Ratio (%) | Relative Density (RD), Specific Gravity (-) |
|---|---|---|---|
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Standau, T.; Zhao, C.; Murillo Castellón, S.; Bonten, C.; Altstädt, V. Chemical Modification and Foam Processing of Polylactide (PLA). Polymers 2019, 11, 306. https://doi.org/10.3390/polym11020306
Standau T, Zhao C, Murillo Castellón S, Bonten C, Altstädt V. Chemical Modification and Foam Processing of Polylactide (PLA). Polymers. 2019; 11(2):306. https://doi.org/10.3390/polym11020306
Chicago/Turabian StyleStandau, Tobias, Chunjing Zhao, Svenja Murillo Castellón, Christian Bonten, and Volker Altstädt. 2019. "Chemical Modification and Foam Processing of Polylactide (PLA)" Polymers 11, no. 2: 306. https://doi.org/10.3390/polym11020306
APA StyleStandau, T., Zhao, C., Murillo Castellón, S., Bonten, C., & Altstädt, V. (2019). Chemical Modification and Foam Processing of Polylactide (PLA). Polymers, 11(2), 306. https://doi.org/10.3390/polym11020306