Understanding the Polymerization of Polyfurfuryl Alcohol: Ring Opening and Diels-Alder Reactions

 ,
, 
 , , , ,
, , , ,  and
and 
Abstract
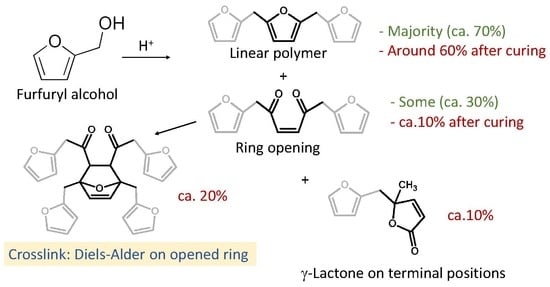
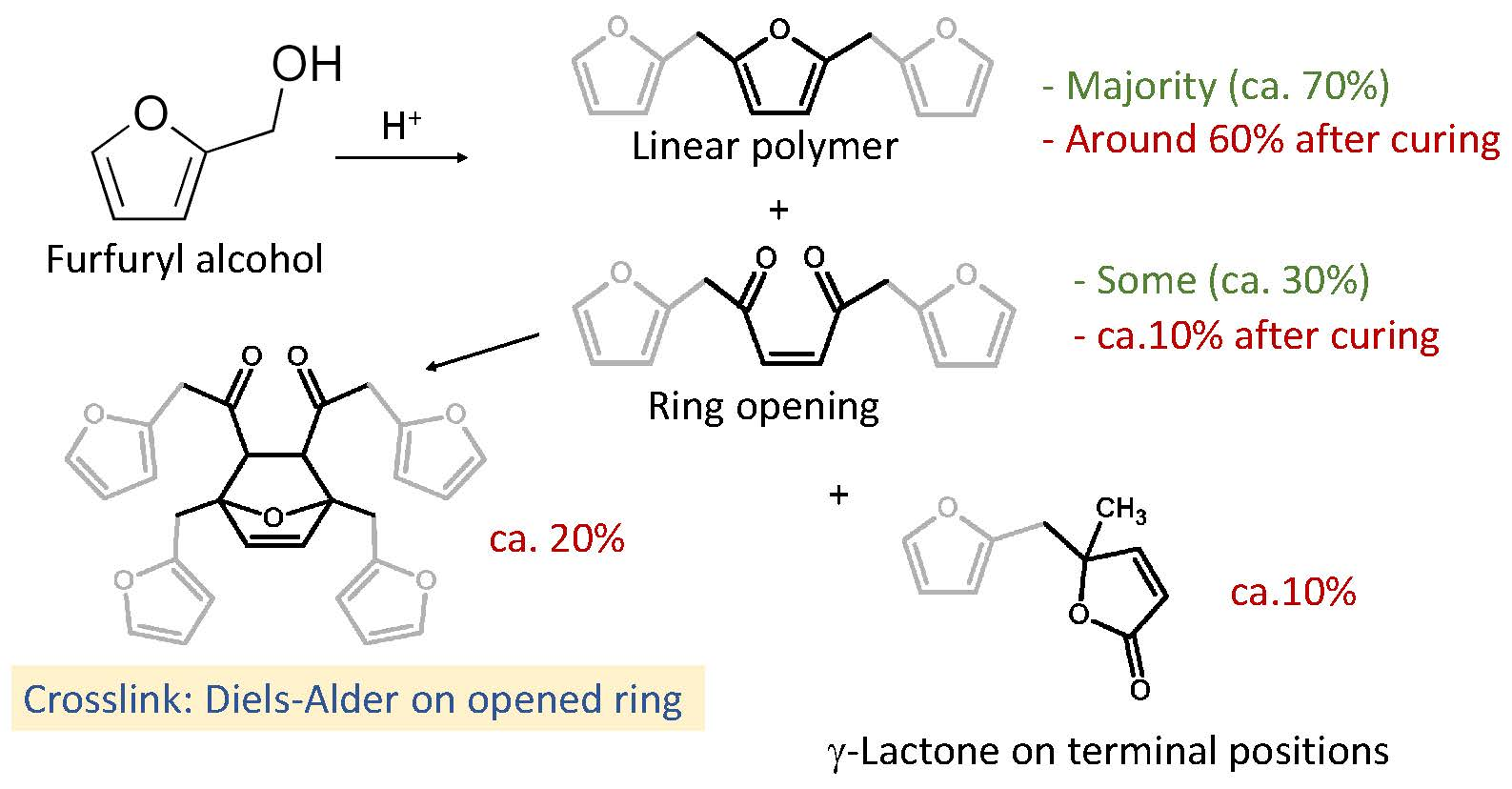
1. Introduction
2. Materials and Methods
2.1. Sample Preparation
2.2. Solid-State 13C-NMR
2.3. ATR-FTIR Analysis
2.4. Raman Investigation
2.5. Computational Details
3. Results and Discussion
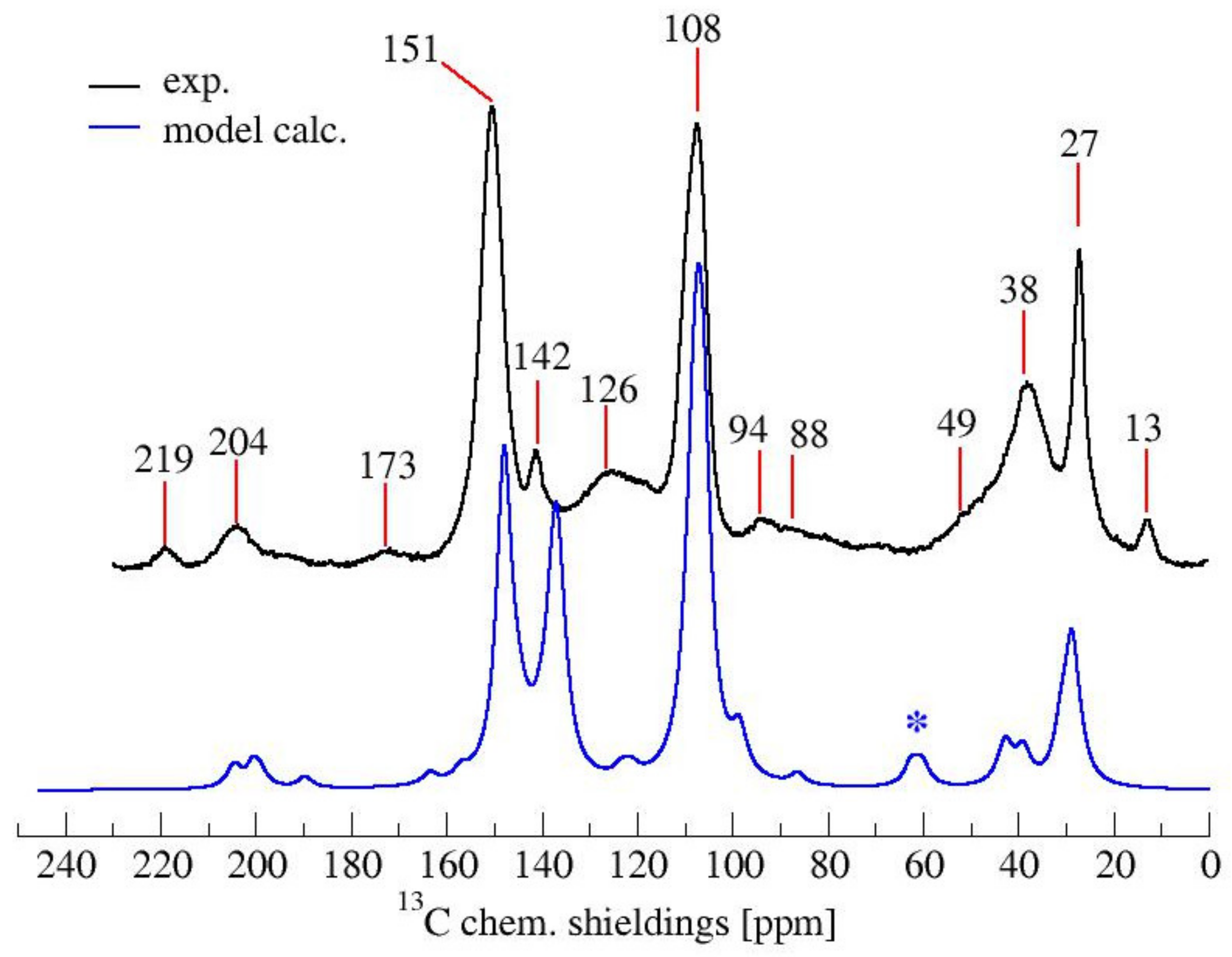
3.1. Solid-State 13C-NMR
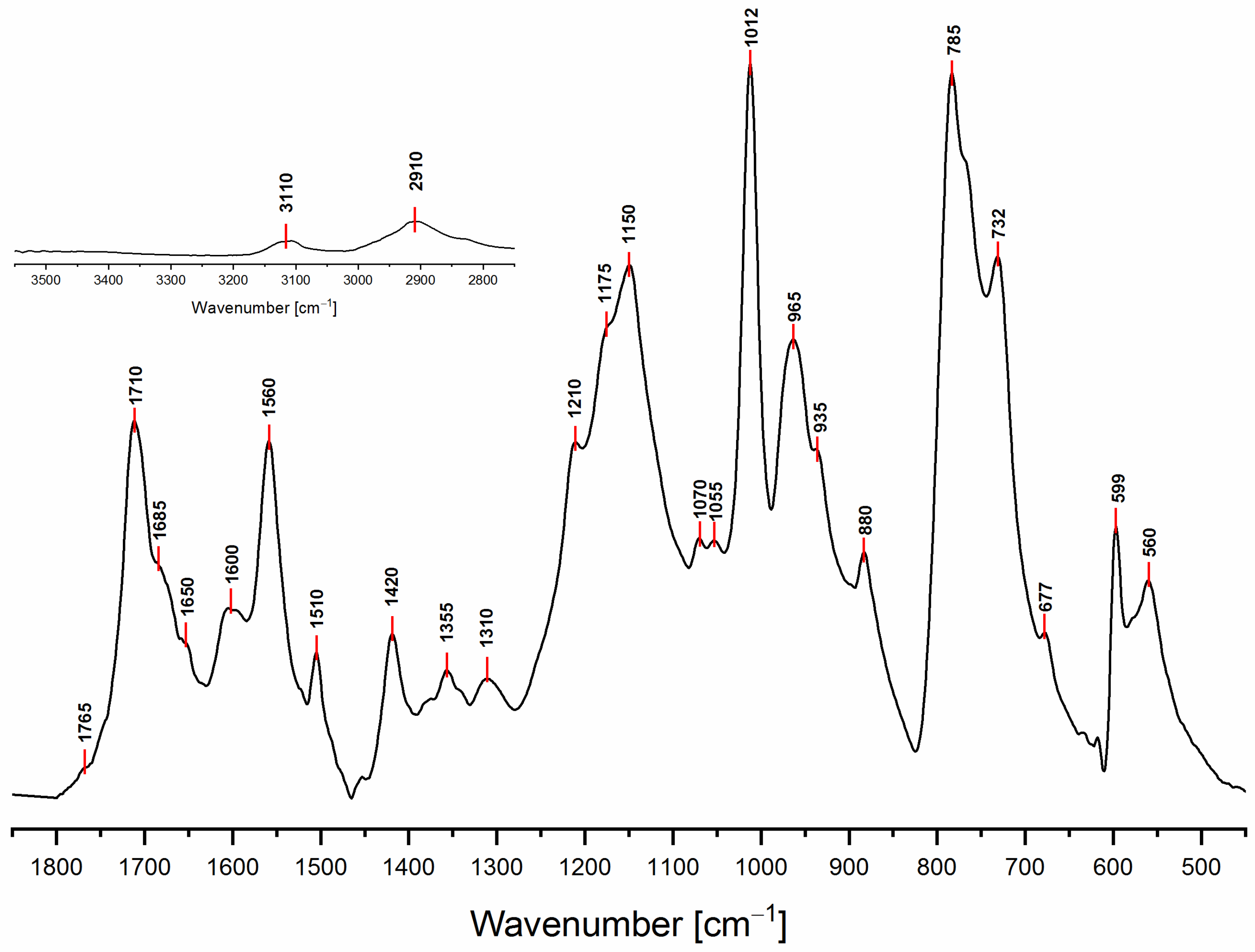
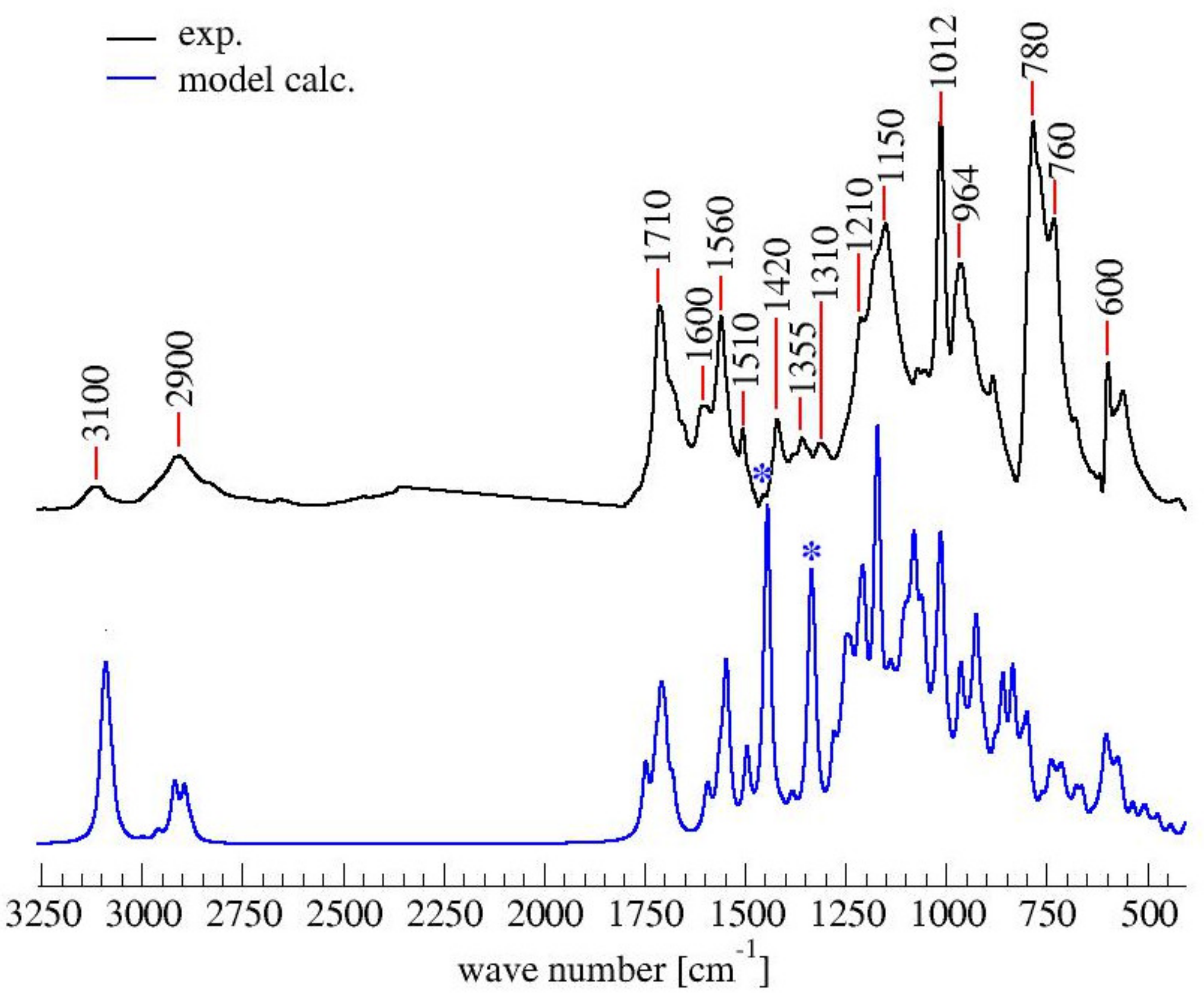
3.2. ATR-FTIR Spectroscopy
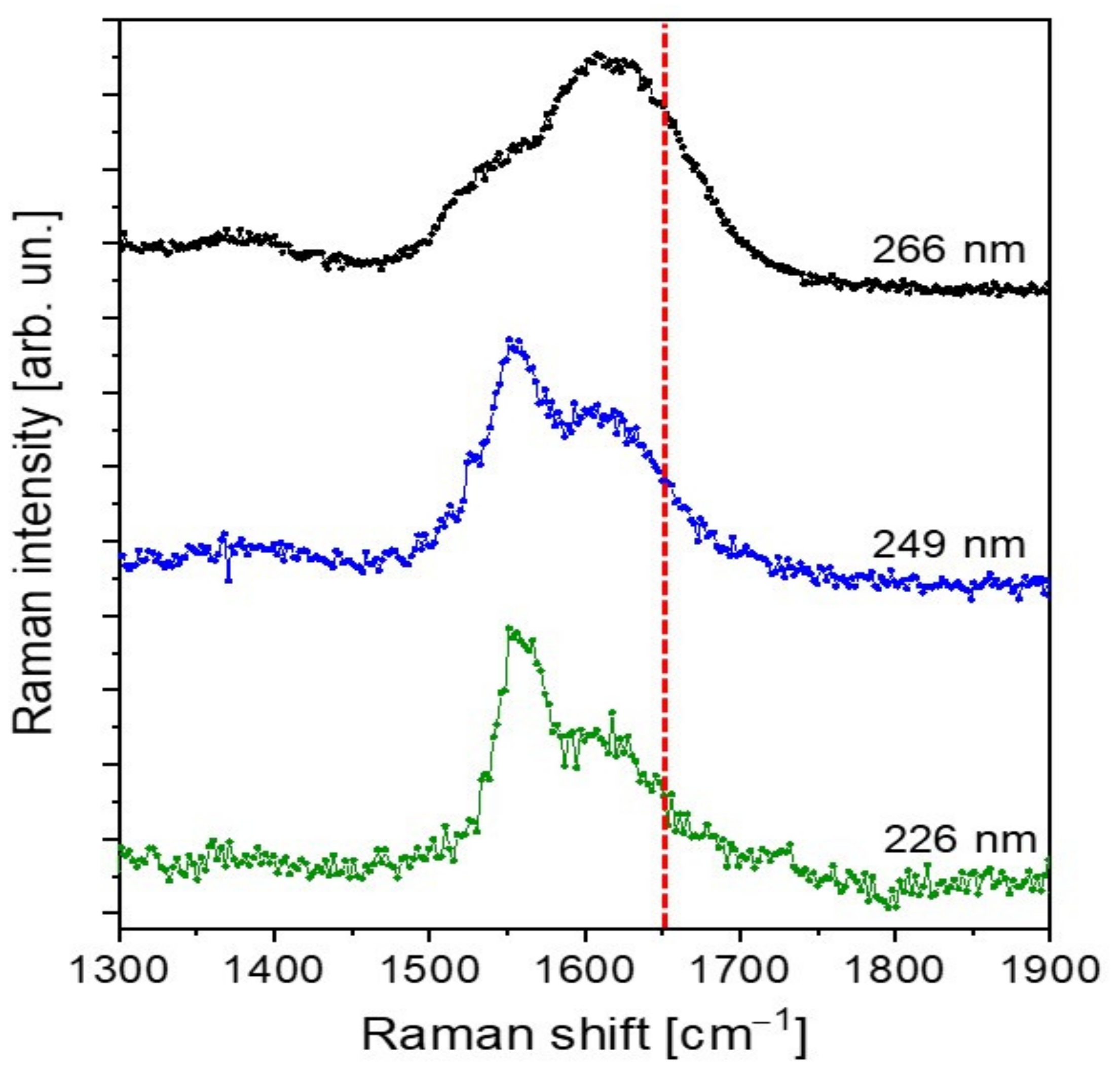
3.3. UV-Raman Spectroscopy
3.4. Computational Results
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dunlop, A.P.; Peters, F.N. The Furans; Reinhold Pub. Corp: New York, NY, USA, 1953; Volume 1. [Google Scholar]
- Conley, R.T.; Metil, I. An investigation of the structure of furfuryl alcohol polycondensates with infrared spectroscopy. J. Appl. Polym. Sci. 1963, 7, 37–52. [Google Scholar] [CrossRef]
- Wewerka, E.M. Study of the γ-alumina polymerization of furfuryl alcohol. J. Polym. Sci. Part A-1 Polym. Chem. 1971, 9, 2703–2715. [Google Scholar] [CrossRef]
- Chuang, I.S.; Maciel, G.E.; Myers, G.E. Carbon-13 NMR study of curing in furfuryl alcohol resins. Macromolecules 1984, 17, 1087–1090. [Google Scholar] [CrossRef]
- Buchwalter, S.L. The polymerization of furfuryl acetate in acetonitrile. J. Polym. Sci. Polym. Chem. Ed. 1985, 23, 2897–2911. [Google Scholar] [CrossRef]
- Choura, M.; Belgacem, N.M.; Gandini, A. Acid-catalyzed polycondensation of furfuryl alcohol: Mechanisms of chromophore formation and cross-linking. Macromolecules 1996, 29, 3839–3850. [Google Scholar] [CrossRef]
- Choura, M.; Belgacem, N.M.; Gandini, A. The acid-catalyzed polycondensation of furfuryl alcohol: Old puzzles unravelled. In Macromolecular Symposia; Hüthig & Wepf Verlag: Basel, Switzerland, 1997; Volume 122, pp. 263–268. [Google Scholar]
- Bertarione, S.; Bonino, F.; Cesano, F.; Damin, A.; Scarano, D.; Zecchina, A. Furfuryl Alcohol Polymerization in H−Y Confined Spaces: Reaction Mechanism and Structure of Carbocationic Intermediates. J. Phys. Chem. B 2008, 112, 2580–2589. [Google Scholar] [CrossRef]
- Guigo, N.; Mija, A.; Vincent, L.; Sbirrazzuoli, N. Chemorheological analysis and model-free kinetics of acid catalysed furfuryl alcohol polymerization. Phys. Chem. Chem. Phys. 2007, 9, 5359–5366. [Google Scholar] [CrossRef]
- Guigo, N.; Mija, A.; Zavaglia, R.; Vincent, L.; Sbirrazzuoli, N. New insights on the thermal degradation pathways of neat poly (furfuryl alcohol) and poly (furfuryl alcohol)/SiO2 hybrid materials. Polym. Degrad. Stab. 2009, 94, 908–913. [Google Scholar] [CrossRef]
- Barsberg, S.; Thygesen, L.G. Poly (furfuryl alcohol) formation in neat furfuryl alcohol and in cymene studied by ATR-IR spectroscopy and density functional theory (B3LYP) prediction of vibrational bands. Vib. Spectrosc. 2009, 49, 52–63. [Google Scholar] [CrossRef]
- Kim, T.; Assary, R.S.; Marshall, C.L.; Gosztola, D.J.; Curtiss, L.A.; Stair, P.C. Acid Catalyzed Furfuryl Alcohol Polymerization: Characterizations of Molecular Structure and Thermodynamic Properties. ChemCatChem 2011, 3, 1451–1458. [Google Scholar] [CrossRef]
- Kim, T.; Assary, R.S.; Kim, H.; Marshall, C.L.; Gosztola, D.J.; Curtiss, L.A.; Stair, P.C. Effects of solvent on the furfuryl alcohol polymerization reaction: UV Raman spectroscopy study. Catal. Today 2013, 205, 60–66. [Google Scholar] [CrossRef]
- Falco, G.; Guigo, N.; Vincent, L.; Sbirrazzuoli, N. Opening Furan for Tailoring Properties of Bio-based Poly (Furfuryl Alcohol) Thermoset. ChemSusChem 2018, 11, 1805–1812. [Google Scholar] [CrossRef] [PubMed]
- Banfi, D.; Patiny, L. www.nmrdb.org: Resurrecting and Processing NMR Spectra On-line. Chim. Int. J. Chem. 2008, 62, 280–281. [Google Scholar] [CrossRef]
- Steinbeck, C.; Krause, S.; Kuhn, S. NMRShiftDB constructing a Free Chemical Information System with Open-Source Components. J. Chem. Inf. Comput. Sci. 2003, 43, 1733–1739. [Google Scholar] [CrossRef] [PubMed]
- Castillo, A.M.; Patiny, L.; Wist, J. Fast and accurate algorithm for the simulation of NMR spectra of large spin systems. J. Magn. Reson. 2011, 209, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Lupi, S.; Nucara, A.; Perucchi, A.; Calvani, P.; Ortolani, M.; Quaroni, L.; Kiskinova, M. Performance of SISSI, the infrared beamline of the ELETTRA storage ring. J. Opt. Soc. Am. B 2007, 24, 4. [Google Scholar] [CrossRef]
- D’Amico, F.; Saito, M.; Bencivenga, F.; Marsi, M.; Gessini, A.; Camisasca, G.; Principi, E.; Cucini, R.; Di Fonzo, S.; Battistoni, A.; et al. UV resonant Raman scattering facility at Elettra. Nucl. Instrum. Methods Phys. Res. Sect. A Accel. Spectrom. Detect. Assoc. Equip. 2013, 703, 33–37. [Google Scholar] [CrossRef]
- Richard, L. McCreery, Photometric Standards for Raman Spectroscopy. In Handbook of Vibrational Spectroscopy; Chalmers, J.M., Griffiths, P.R., Eds.; Wiley: Hoboken, NJ, USA, 2006; Volume 1. [Google Scholar]
- ChemAxon. MarvinSketch, Version 15.2.16.0. 2014. Available online: http://www.chemaxon.com (accessed on 1 June 2019).
- Turbomole V7.0 2015, a Development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989-2007, TURBOMOLE GmbH, since 2007. Available online: http://www.turbomole.com (accessed on 1 June 2019).
- Sierka, M.; Hogekamp, A.; Ahlrichs, R. Fast evaluation of the Coulomb potential for electron densities using multipole accelerated resolution of identity approximation. J. Chem. Phys. 2003, 118, 9136–9148. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098. [Google Scholar] [CrossRef]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822. [Google Scholar] [CrossRef]
- Weigend, F.; Häser, M.; Patzelt, H.; Ahlrichs, R. RI-MP2: Optimized auxiliary basis sets and demonstration of efficiency. Chem. Phys. Lett. 1998, 294, 143–152. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Kollwitz, M.; Gauss, J. A direct implementation of the GIAO-MBPT (2) method for calculating NMR chemical shifts. Application to the naphthalenium and anthracenium ions. Chem. Phys. Lett. 1996, 260, 639–646. [Google Scholar] [CrossRef]
- Grimme, S.; Bannwarth, C.; Shushkov, P. A robust and accurate tight-binding quantum chemical method for structures, vibrational frequencies, and noncovalent interactions of large molecular systems parametrized for all spd-block elements (Z = 1–86). J. Chem. Theory Comput. 2017, 13, 1989–2009. [Google Scholar] [CrossRef] [PubMed]
- Socrates, G. Infrared and Raman Characteristic Group Frequencies: Tables and Charts; Wiley: Hoboken, NJ, USA, 2004. [Google Scholar]
- Kim, T.; Jeong, J.; Rahman, M.; Zhu, E.; Mahajan, D. Characterizations of furfuryl alcohol oligomer/polymerization catalyzed by homogeneous and heterogeneous acid catalysts. Korean J. Chem. Eng. 2014, 31, 2124–2129. [Google Scholar] [CrossRef]
- Asher, S.A. Ultraviolet resonance Raman spectrometry for detection and speciation of trace polycyclic aromatic hydrocarbons. Anal. Chem. 1984, 56, 720–724. [Google Scholar] [CrossRef]
- Asher, S.A.; Johnson, C.R. Raman Spectroscopy of a Coal Liquid Shows That Fluorescence Interference Is Minimized with Ultraviolet Excitation. Science 1984, 225, 311–313. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | Arrangement | Chemical Formula | Reference(s) |
|---|---|---|---|
| 1 | Linear |  | Dunlop & Peters—1953 [1] |
| 2 | Ring opening |  | Conley & Metil—1963 [2] |
| 3 | α,β-unsaturated γ-lactons |  | Wewerka—1971 [3] |
| 4 | Methylene bridge |  | Chuang et al.—1984 [4] |
| 5 | Conjugated |  | Buchwalter—1985 [5] |
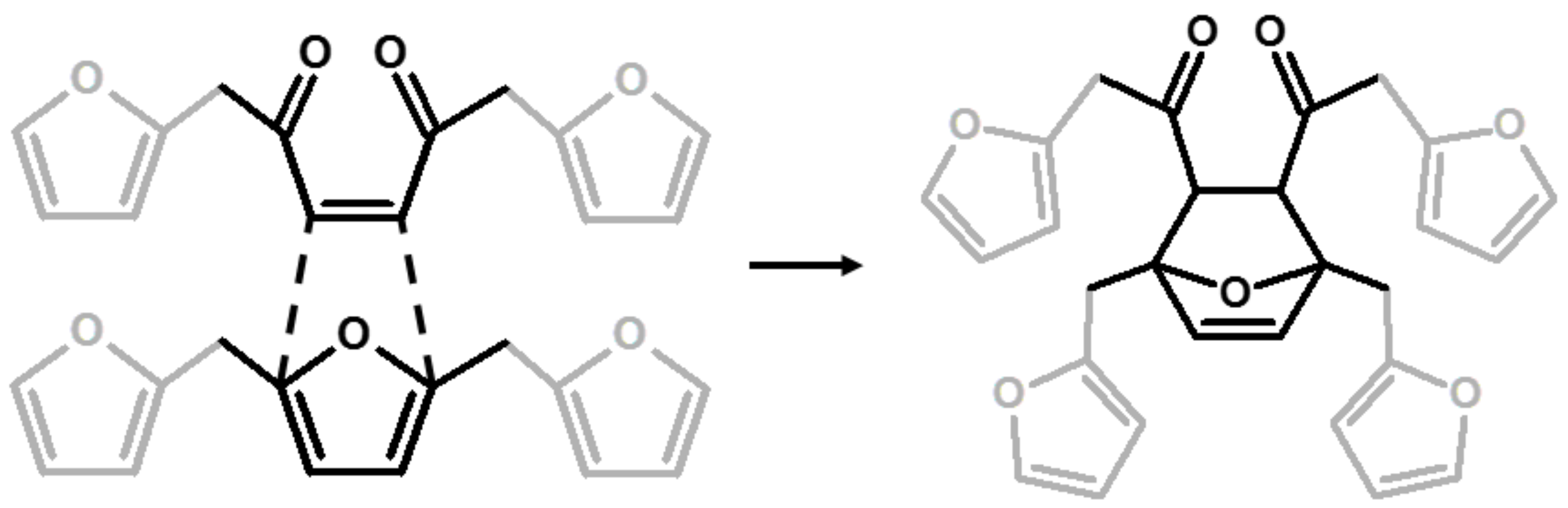
| 6 | Diels-Alder |  | Choura et al.—1996 [6] |
| 7 | Ring-Opening + Diels-Alder |  | Present paper |
| Chem. Shift | Chemical Structures from Table 1 | Attributions | ||||||
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | ||
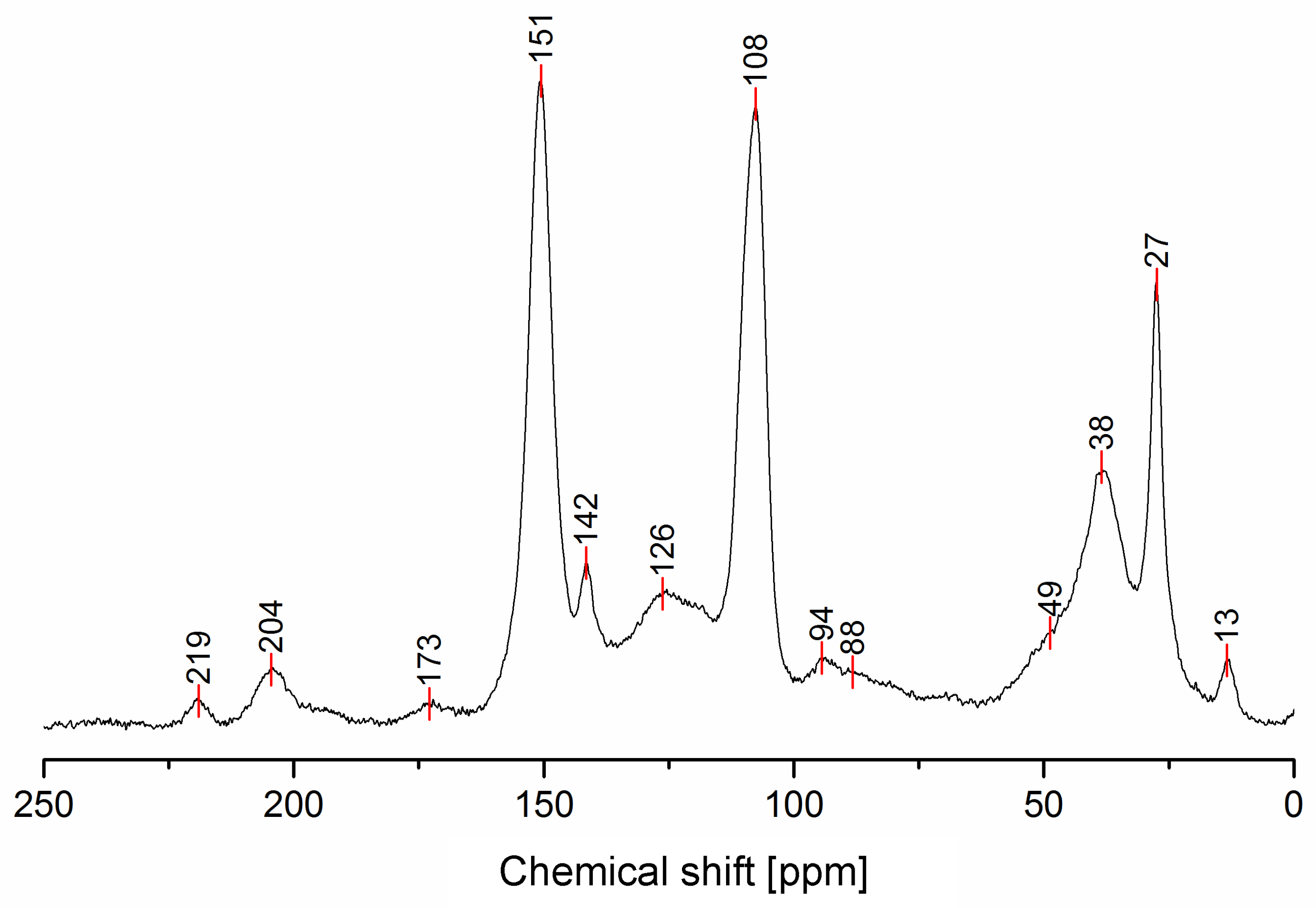
| 219 | No | Yes | No | No | No | No | Yes | C=O diketones vicinal dienophyle |
| 204 | No | Yes | No | No | No | No | Yes | C=O diketones |
| 173 | No | No | Yes | No | No | No | No | γ-lactones or Levulinic acid/ester |
| 151 | Yes | No | No | No | Yes | Yes | No | C2, C5 Furan |
| 142 | No | No | Yes | No | No | Yes | Yes | C=C in Diels-Alder bicycle or lactones |
| 126 | No | Yes | Yes | No | Yes | No | No | C=C in β between ketones or ester, C=C in the conjugated furan ring. |
| 108 | Yes | No | No | No | No | No | No | C3, C4 Furan |
| 94 | No | No | No | No | Yes | Yes | No | -CH= Bridge in conjugated systems |
| 88 | No | No | Yes | No | No | Yes | Yes | Quaternary C in Diels-Alder or lactones |
| 49 | No | Yes | No | No | No | No | Yes | -CH2- Bridge between furans & C=O |
| 38 | No | No | No | Yes | No | Yes | Yes | Tertiary C in Diels-Alder, -CH- bridge in methylene bridge (4) |
| 27 | Yes | No | Yes | Yes | No | Yes | Yes | -CH2- bridge in between furans, -CH2- bridge (4) |
| 13 | No | No | Yes | No | No | No | No | -CH3 in lactone structures |
| Wavenumber (cm−1) | Chemical Structures from Table 1 | Attributions [11,30,31] | ||||||
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | ||
| 3150–3050 | Yes | Yes | Yes | No | Yes | Yes | Yes | C-H stretching aromatic and vinyl |
| 2950–2850 | Yes | Yes | Yes | Yes | No | Yes | Yes | C-H stretching aliphatic |
| 1790–1740 | No | No | Yes | No | No | No | No | C=O stretching α,β-unsat γ -lactone |
| 1720–1700 | No | No | No | No | No | No | Yes | C=O stretching (isolated) |
| 1690–1670 | No | Yes | No | No | No | No | No | C=O vicinal to C=C (α,β-unsat. ketone) |
| 1650 | No | No | No | No | No | Yes | Yes | C=C stretching in D.A. (isolated) or conjugated diene |
| 1615–1590 | No | Yes | No | No | No | No | No | C=C vicinal to C=O (α,β-unsat. ketone) or conjugated diene |
| 1560 | Yes | No | No | No | Yes | No | No | C=C stretching (ring vibr. 2,5-disubstituted furans) |
| 1510 | Yes | No | No | No | Yes | No | No | C=C stretching (ring vibr. 2,5-disubstituted furans) |
| 1450–1345 | Yes | Yes | Yes | Yes | No | Yes | Yes | -CH2 scissoring and wagging |
| 1345–1290 | No | No | Yes | No | No | No | No | C-O stretching γ-lactone |
| 1230–1100 | No | No | No | Yes | No | Yes | Yes | Complex network of several vibrational modes associated with C-O ring stretching, C-C furan stretching,-CH2 in plane wagging.The peak at 1175 might be due to the C-O-C stretching of the D.A. (difficult to assign) |
| 1100–1040 | Yes | No | No | No | Yes | No | No | =C-O-C= ring vibration (associated with another peak in the range 1200–1120) |
| 1012 | Yes | No | No | No | No | No | No | -CH in plane wagging 2,5-disubstituted furan (Barsberg simulation) |
| 980–900 | No | Yes | No | No | No | No | Yes | -CH out of plane deformation vibration of alkenes –CH=CH– (usually 2 peaks and they are both present) |
| 880–860 | Yes | No | No | No | Yes | No | No | Furan ring C-H out-of-plane deform. vibration |
| 810–745 | Yes | No | No | No | Yes | No | No | Wagging/twisting -CH-ring structure |
| 745–700 | Yes | No | No | No | Yes | No | No | Furan ring -CH out of plane bend |
| 677 | No | Yes | Yes | No | No | Yes | Yes | -CH out of plane bending, cis -CH=CH- |
| 599 | Yes | No | No | No | Yes | No | No | Ring deformation vibration |
| 550 | No | Yes | Yes | No | No | Yes | Yes | -CH out of plane bending, cis -CH=CH- |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tondi, G.; Cefarin, N.; Sepperer, T.; D’Amico, F.; Berger, R.J.F.; Musso, M.; Birarda, G.; Reyer, A.; Schnabel, T.; Vaccari, L. Understanding the Polymerization of Polyfurfuryl Alcohol: Ring Opening and Diels-Alder Reactions. Polymers 2019, 11, 2126. https://doi.org/10.3390/polym11122126
Tondi G, Cefarin N, Sepperer T, D’Amico F, Berger RJF, Musso M, Birarda G, Reyer A, Schnabel T, Vaccari L. Understanding the Polymerization of Polyfurfuryl Alcohol: Ring Opening and Diels-Alder Reactions. Polymers. 2019; 11(12):2126. https://doi.org/10.3390/polym11122126
Chicago/Turabian StyleTondi, Gianluca, Nicola Cefarin, Thomas Sepperer, Francesco D’Amico, Raphael J. F. Berger, Maurizio Musso, Giovanni Birarda, Andreas Reyer, Thomas Schnabel, and Lisa Vaccari. 2019. "Understanding the Polymerization of Polyfurfuryl Alcohol: Ring Opening and Diels-Alder Reactions" Polymers 11, no. 12: 2126. https://doi.org/10.3390/polym11122126
APA StyleTondi, G., Cefarin, N., Sepperer, T., D’Amico, F., Berger, R. J. F., Musso, M., Birarda, G., Reyer, A., Schnabel, T., & Vaccari, L. (2019). Understanding the Polymerization of Polyfurfuryl Alcohol: Ring Opening and Diels-Alder Reactions. Polymers, 11(12), 2126. https://doi.org/10.3390/polym11122126