Fully Biobased Shape Memory Thermoplastic Vulcanizates from Poly(Lactic Acid) and Modified Natural Eucommia Ulmoides Gum with Co-Continuous Structure and Super Toughness

 ,
,
Abstract

1. Introduction
2. Experimental Section
2.1. Materials
2.2. Preparation of Modified EUG
2.3. Preparation of the TPVs
2.4. Characterizations
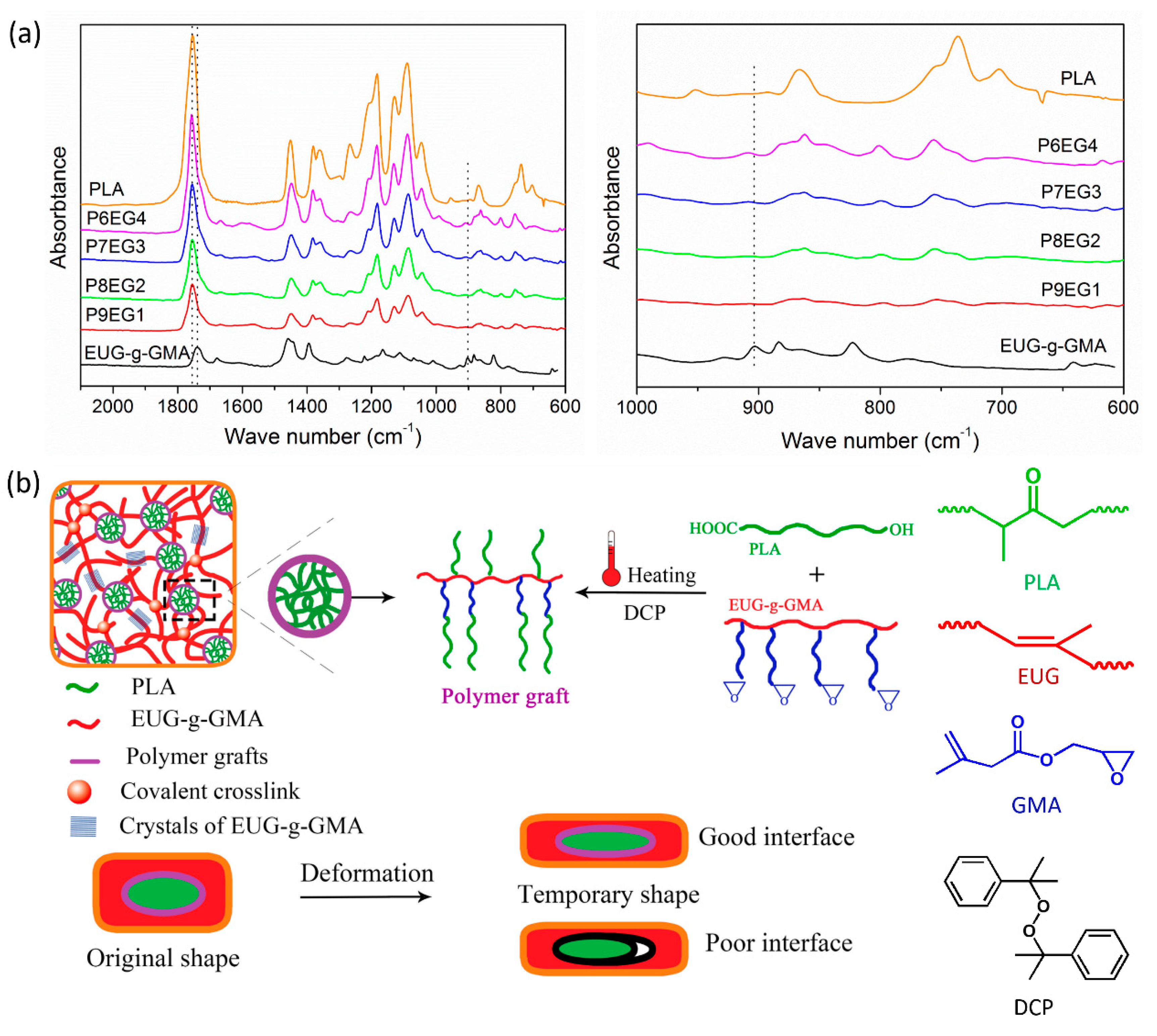
2.4.1. Fourier Transform Infrared Spectroscopy (FTIR)
2.4.2. Scanning Electron Microscopy (SEM)
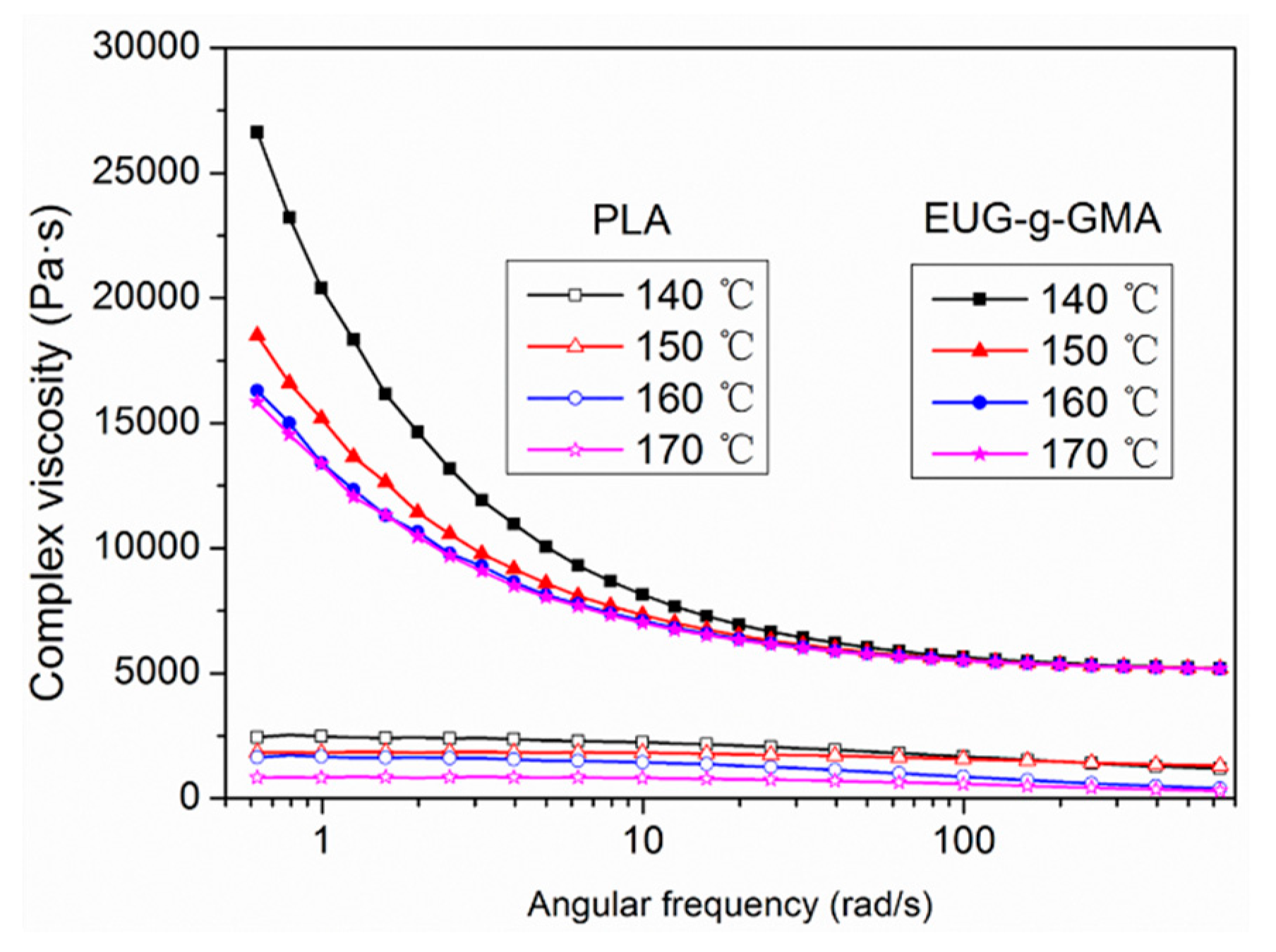
2.4.3. Rheological Measurement
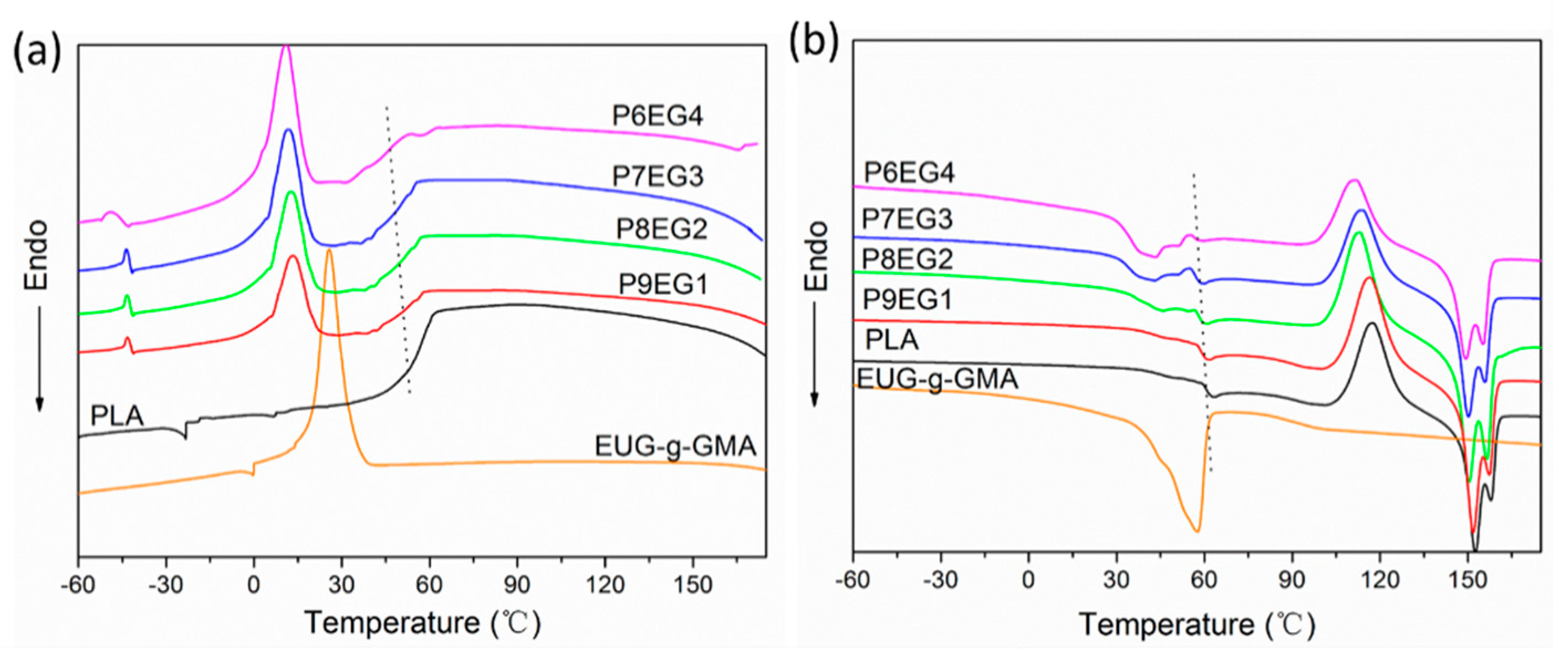
2.4.4. Different Scanning Calorimetry (DSC)
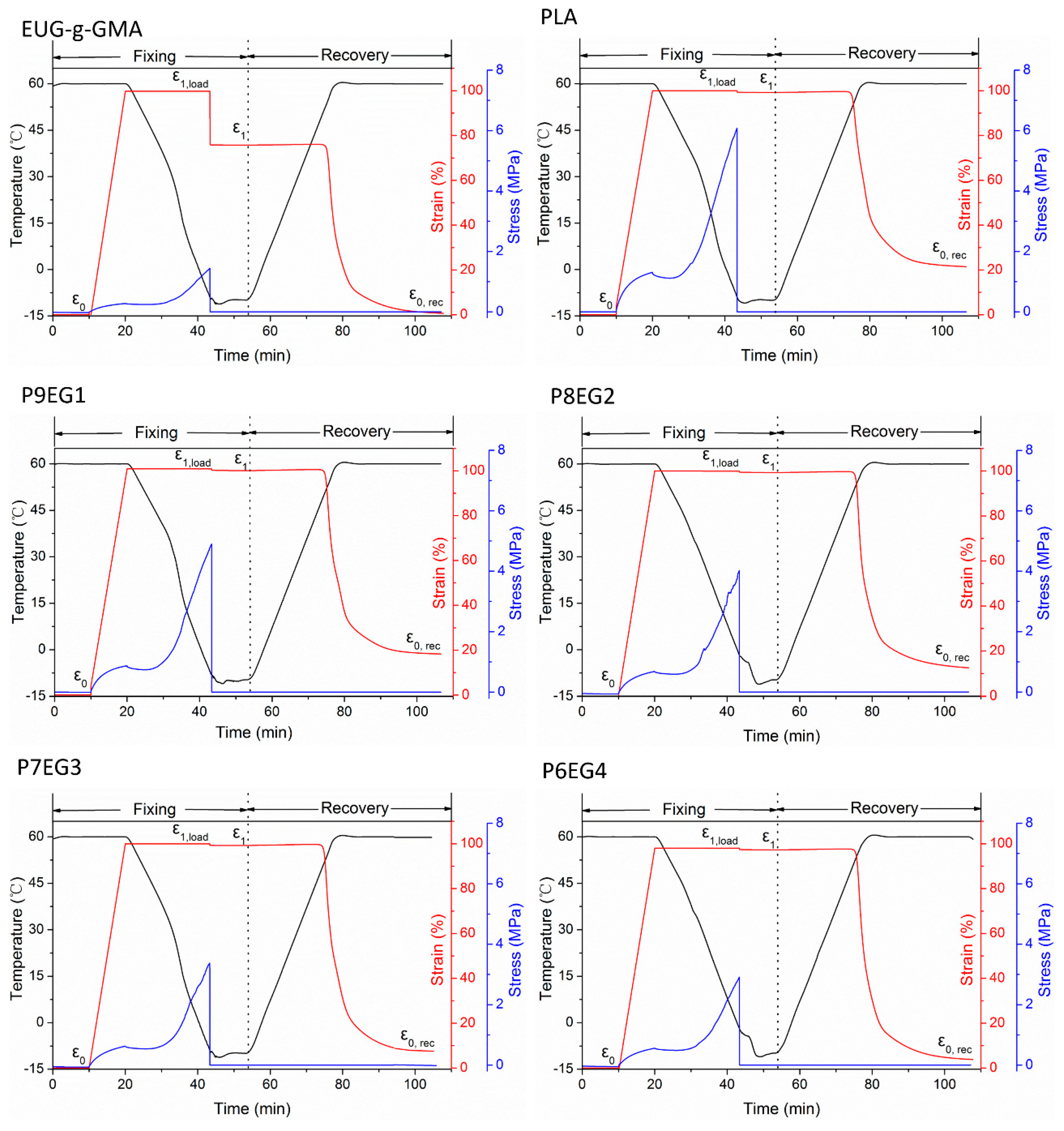
2.4.5. Shape Memory Analysis
2.4.6. Dynamic Mechanical Thermal Analysis (DMTA)
2.4.7. Mechanical Property Measurements
3. Results and Discussion
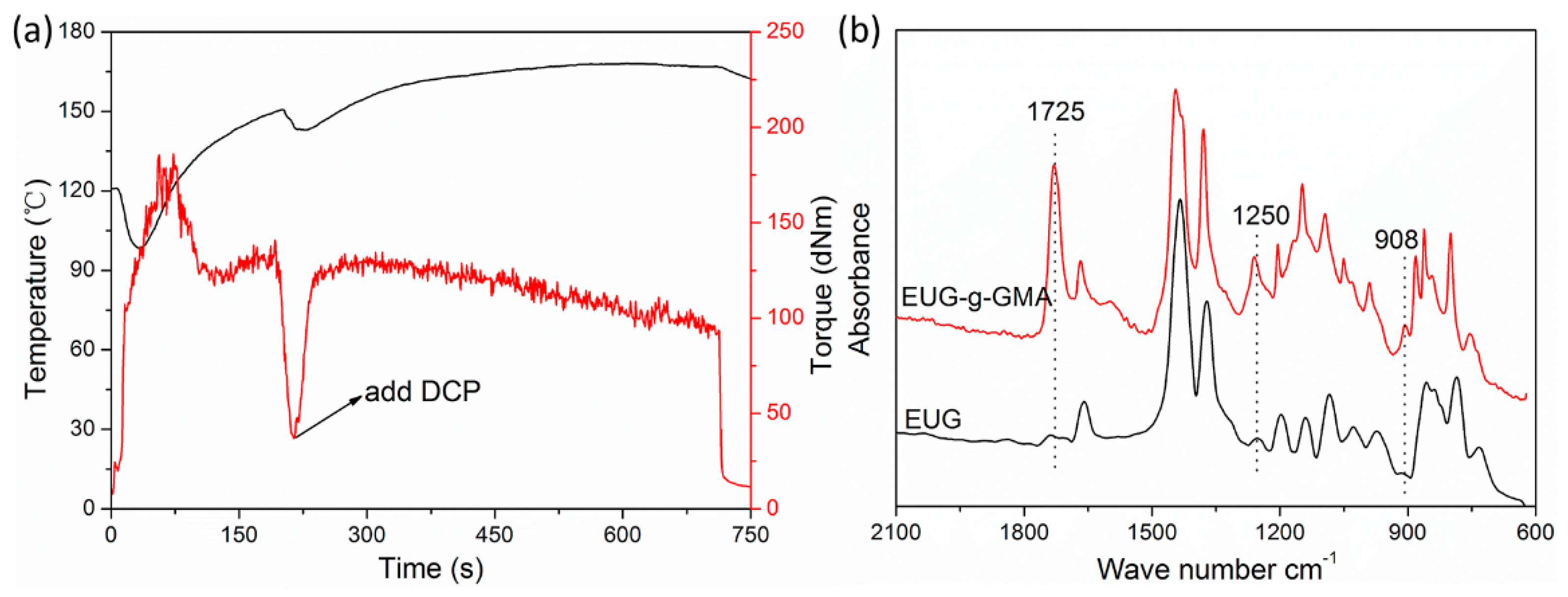
3.1. Preparation of Modified EUG
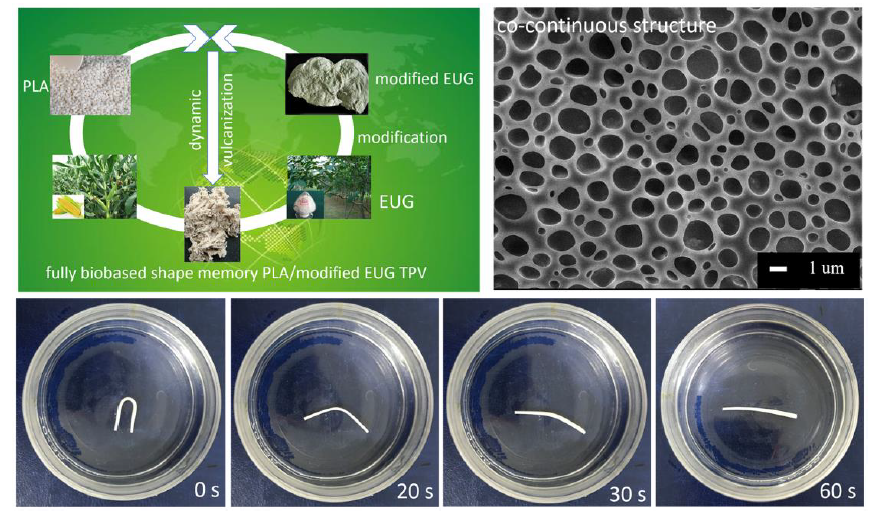
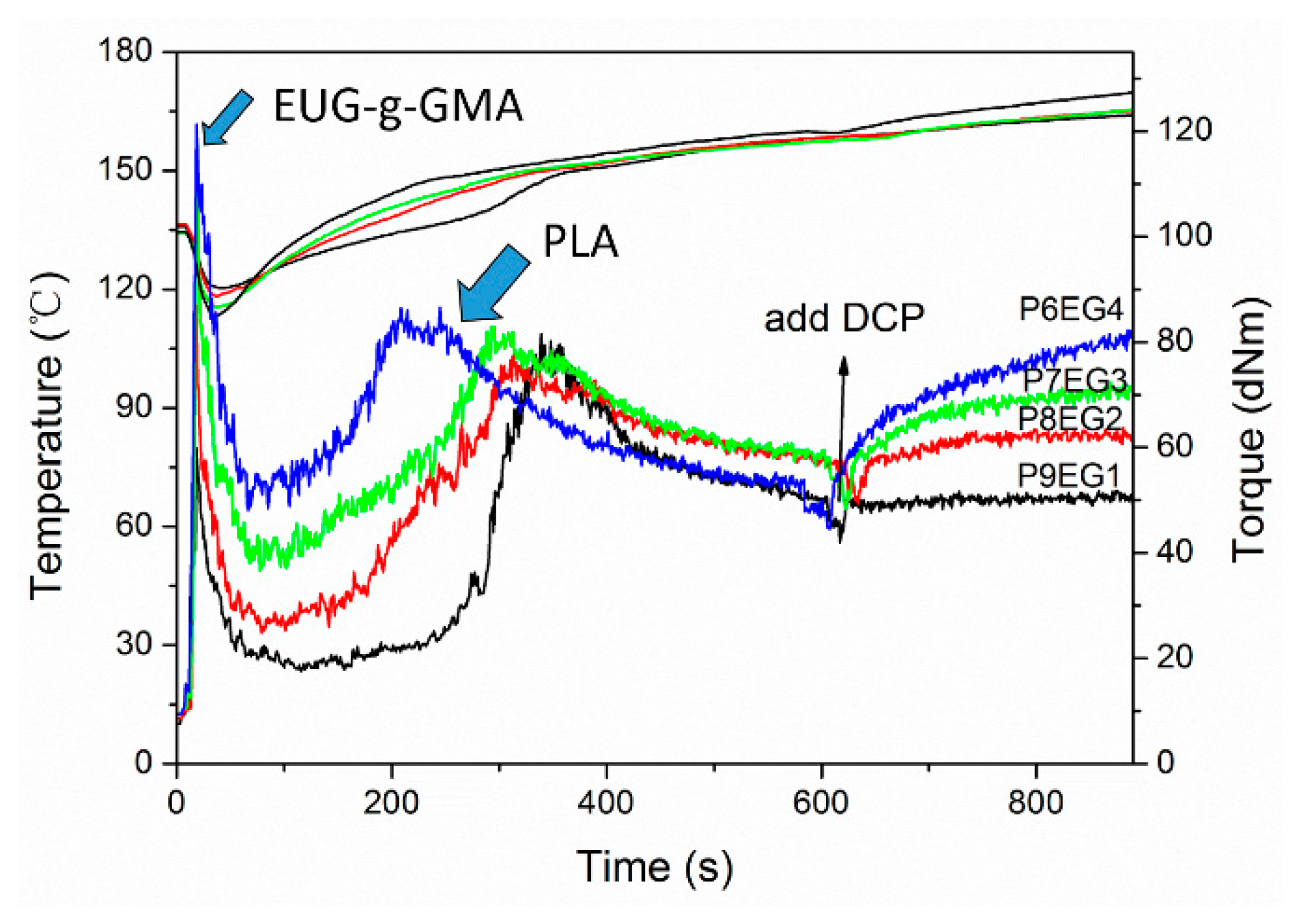
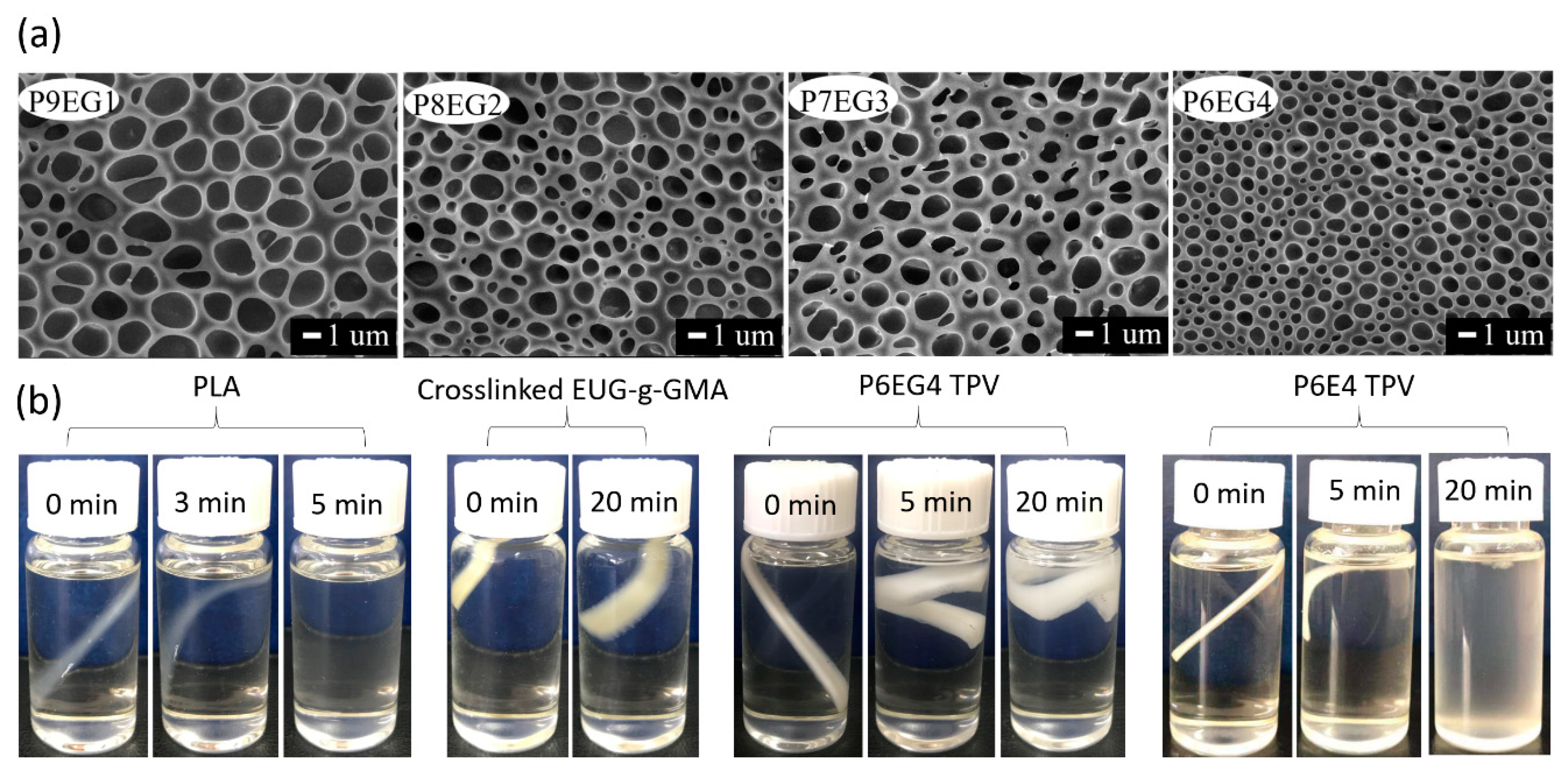
3.2. Preparation and Co-Continuous Structure in PLA/EUG-g-GMA TPVs
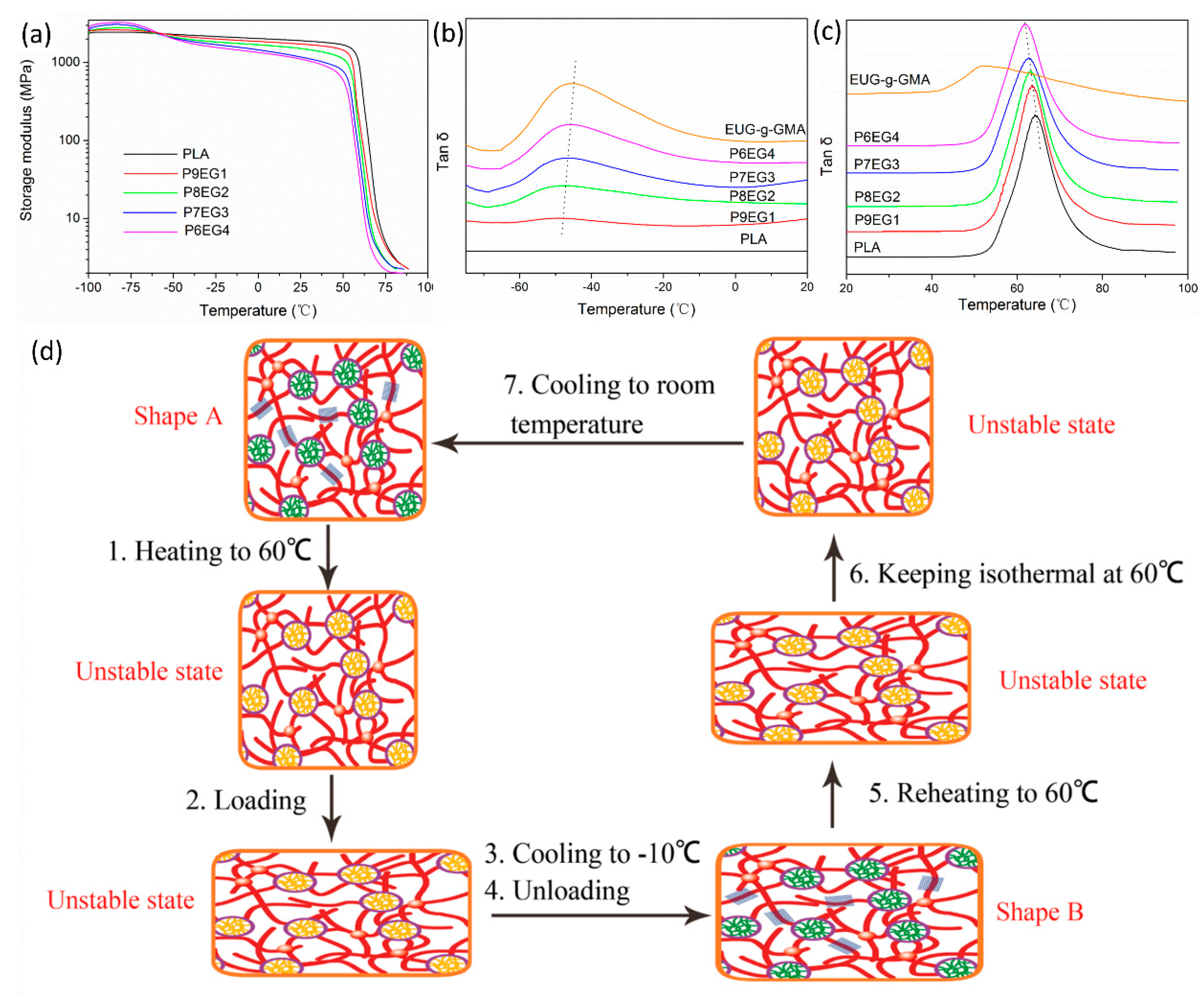
3.3. Improved Interface and Thermal Properties of the PLA/EUG-g-GMA TPVs
3.4. Shape Memory Analysis
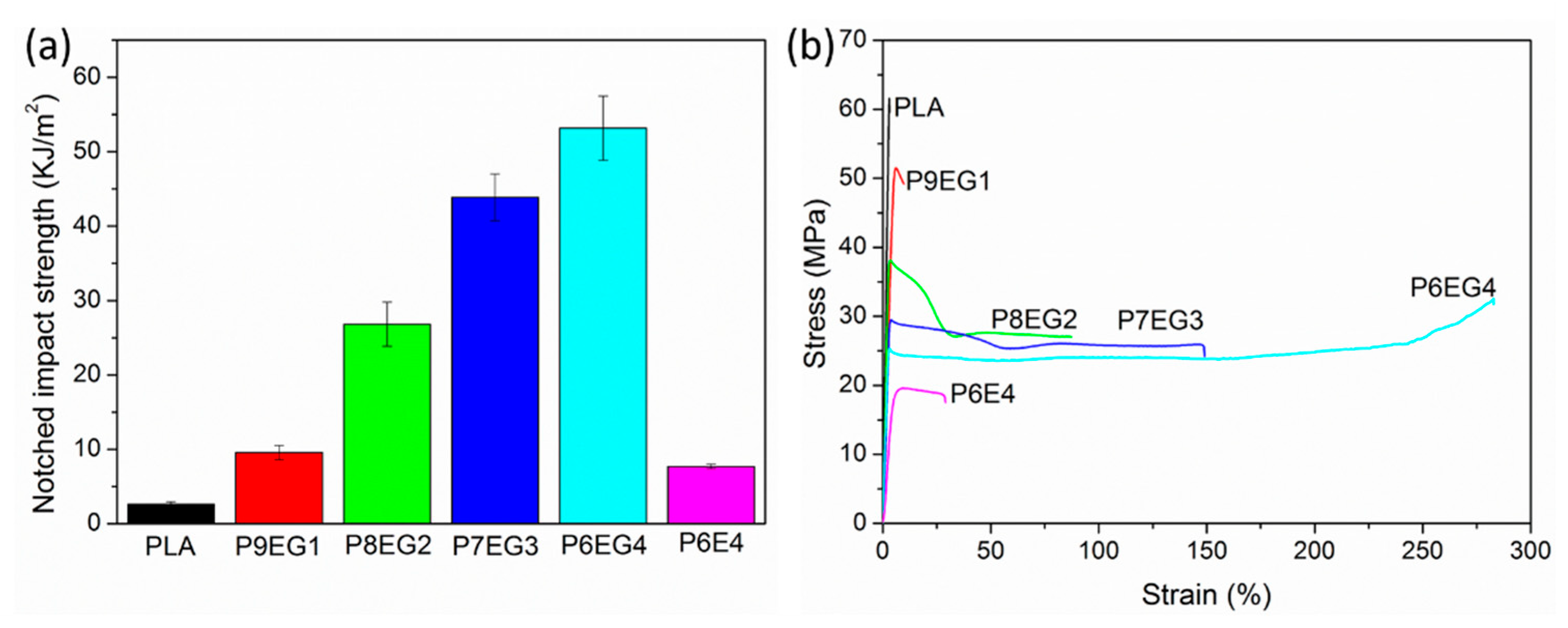
3.5. Mechanical Characterization
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Yang, Z.; Wang, Q.; Wang, T. Dual-triggered and thermally reconfigurable shape memory graphene-vitrimer composites. ACS Appl. Mater. Inter. 2016, 8, 21691–21699. [Google Scholar] [CrossRef] [PubMed]
- Andreas, L.; Hongyan, J.; Oliver, J.; Robert, L. Light-induced shape-memory polymers. Nature 2005, 434, 879–882. [Google Scholar]
- Schmidt, A.M. Electromagnetic activation of shape memory polymer networks containing magnetic nanoparticles. Macromol. Rapid Comm. 2006, 27, 1168–1172. [Google Scholar] [CrossRef]
- Yang, B.; Huang, W.M.; Li, C.; Li, L. Effects of moisture on the thermomechanical properties of a polyurethane shape memory polymer. Polymer 2006, 47, 1348–1356. [Google Scholar] [CrossRef]
- Xie, Y.; Lei, D.; Wang, S.; Liu, Z.; Sun, L.; Zhang, J.; Qing, F.L.; He, C.; You, Z. A biocompatible, biodegradable, and functionalizable copolyester and its application in water-responsive shape memory scaffold. ACS Biomater. Sci. Eng. 2019, 5, 1668–1676. [Google Scholar] [CrossRef]
- Guo, W.; Lu, C.; Ron, O.; Fuan, W.; Qi, X.; Alessandro, C.; Dror, S.; Itamar, W. pH-stimulated DNA hydrogels exhibiting shape-memory properties. Adv. Mater. 2015, 27, 73–78. [Google Scholar] [CrossRef]
- Liu, Y.; Du, H.; Liu, L.; Leng, J. Shape memory polymers and their composites in aerospace applications: A review. Smart Mater. Struct. 2014, 23, 23001–23022. [Google Scholar] [CrossRef]
- Chan, B.Q.Y.; Low, Z.W.K.; Heng, S.J.W.; Chan, S.Y.; Owh, C.; Loh, X.J. Recent advances in shape memory soft materials for biomedical applications. ACS Appl. Mater. Inter. 2016, 8, 10070–10087. [Google Scholar] [CrossRef]
- Weber, C. Shape memory polymers: Past, present and future developments. Prog. Polym. Sci. 2015, 49–50, 3–33. [Google Scholar]
- Andreas, L.; Robert, L. Biodegradable, elastic shape-memory polymers for potential biomedical applications. Science 2002, 296, 1673–1676. [Google Scholar]
- Xie, T. Tunable polymer multi-shape memory effect. Nature 2010, 464, 267–270. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Chen, M.; Wang, X.; Zhao, X.; Wang, Z.; Dang, Z.M.; Ma, L.; Hu, G.H.; Chen, F. Triple shape memory effects of cross-linked polyethylene/polypropylene blends with cocontinuous architecture. ACS Appl. Mater. Inter. 2013, 5, 5550–5556. [Google Scholar] [CrossRef] [PubMed]
- Shirole, A.; Perotto, C.U.; Balog, S.; Weder, C. Tailoring the shape memory properties of segmented poly(ester urethanes) via blending. ACS Appl. Mater. Inter. 2018, 10, 24829–24839. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Chen, S.; Fu, W.; Qiu, G. Shape memory behavior of natural Eucommia ulmoides gum and low-density polyethylene blends with two response temperatures. Polymers 2019, 11, 580. [Google Scholar] [CrossRef]
- Hu, J.; Zhu, Y.; Huang, H.; Lu, J. Recent advances in shape-memory polymers: Structure, mechanism, functionality, modeling and applications. Prog. Polym. Sci. 2012, 37, 1720–1763. [Google Scholar] [CrossRef]
- Xie, T. Recent advances in polymer shape memory. Polymer 2011, 52, 4985–5000. [Google Scholar] [CrossRef]
- Luo, X.; Mather, P.T. Triple-shape polymeric composites (TSPCs). Adv. Funct. Mater. 2010, 20, 2649–2656. [Google Scholar] [CrossRef]
- Meyer, A. Synthesis and characterization of polyurethane-based shape-memory polymers for tailored Tg around body temperature for medical applications. Macromol. Chem. Phys. 2011, 212, 592–602. [Google Scholar]
- Kolesov, I.; Dolynchuk, O.; Radusch, H.J. Modeling of shape-memory recovery in crosslinked semicrystalline polymers. Adv. Sci. Technol. 2012, 77, 319–324. [Google Scholar] [CrossRef]
- Behl, M.; Lendlein, A. Actively moving polymers. Soft Matter. 2006, 3, 58–67. [Google Scholar] [CrossRef]
- Yakacki, C.M.; Shandas, R.; Safranski, D.; Ortega, A.M.; Sassaman, K.; Gall, K. Strong, tailored, biocompatible shape-memory polymer networks. Adv. Funct. Mater. 2010, 18, 2428–2435. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.K.; Eugene, L.; Jiyeon, C.; Ki, J.Y.; Jun, A.D.; Keun, H.D. Shape-memory effect by specific biodegradable polymer blending for biomedical applications. Macromol. Biosci. 2014, 14, 667–678. [Google Scholar]
- Zhang, Z.X.; Fei, L.; He, Z.Z.; Yang, J.H.; Huang, T.; Nan, Z.; Yong, W.; Gao, X.L. Tunable shape memory behaviors of poly(ethylene vinyl acetate) achieved by adding poly(L-lactide). Smart Mater. Struct. 2015, 24, 125002–125014. [Google Scholar] [CrossRef]
- Zhang, Z.; He, Z.; Yang, J.; Huang, T.; Zhang, N.; Wang, Y. Crystallization controlled shape memory behaviors of dynamically vulcanized poly(l-lactide)/poly(ethylene vinyl acetate) blends. Polym. Test. 2016, 51, 82–92. [Google Scholar] [CrossRef]
- Dell’Erba, R.; Groeninckx, G.; Maglio, G.; Malinconico, M.; Migliozzi, A. Immiscible polymer blends of semicrystalline biocompatible components: Thermal properties and phase morphology analysis of PLLA/PCL blends. Polymer 2001, 42, 7831–7840. [Google Scholar] [CrossRef]
- Samuel, C.; Barrau, S.; Lefebvre, J.M.; Raquez, J.M.; Dubois, P. Designing multiple-shape memory polymers with miscible polymer blends: Evidence and origins of a triple-shape memory effect for miscible PLLA/PMMA blends. Macromolecules 2014, 47, 6791–6803. [Google Scholar] [CrossRef]
- Xu, C.; Lin, B.; Liang, X.; Chen, Y. Zinc dimethacrylate induced in situ interfacial compatibilization turns EPDM/PP TPVs into a shape memory material. Ind. Eng. Chem. Res. 2016, 55, 4539–4548. [Google Scholar] [CrossRef]
- Yuan, D.; Chen, Z.; Xu, C.; Chen, K.; Chen, Y. Fully biobased shape memory material based on novel cocontinuous structure in poly(lactic acid)/natural rubber TPVs fabricated via peroxide-induced dynamic vulcanization and in situ interfacial compatibilization. ACS Sustain. Chem. Eng. 2015, 3, 2856–2865. [Google Scholar] [CrossRef]
- Ning, N.; Li, S.; Wu, H.; Tian, H.; Yao, P.; Hu, G.H.; Tian, M.; Zhang, L. Preparation, microstructure, and microstructure-properties relationship of thermoplastic vulcanizates (TPVs): A review. Prog. Polym. Sci. 2018, 79, 61–97. [Google Scholar] [CrossRef]
- Wu, N.; Hong, Z.; Fu, G. Super-tough poly(lactide) thermoplastic vulcanizates based on modified natural rubber. ACS Sustain. Chem. Eng. 2016, 5, 78–84. [Google Scholar] [CrossRef]
- Liu, G.; He, Y.; Zeng, J.; Li, Q.; Wang, Y. Fully biobased and supertough polylactide-based thermoplastic vulcanizates fabricated by peroxide-induced dynamic vulcanization and interfacial compatibilization. Biomacromolecules 2014, 15, 4260–4271. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Chen, L.; Zhang, Y. Surprising shape-memory effect of polylactide resulted from toughening by polyamide elastomer. Polymer 2009, 50, 1311–1315. [Google Scholar] [CrossRef]
- Wang, Y.; Xia, L.; Xin, Z. Triple shape memory effect of foamed natural Eucommia ulmoides gum/high-density polyethylene composites. Polym. Advan. Technol. 2018, 29, 190–197. [Google Scholar] [CrossRef]
- Xia, L.; Wang, Y.; Lu, N.; Xin, Z. Facile fabrication of shape memory composites from natural Eucommia rubber and high density polyethylene. Polym. Inter. 2017, 66, 653–658. [Google Scholar] [CrossRef]
- Wu, S. Formation of dispersed phase in incompatible polymer blends: Interfacial and rheological effects. Polym. Eng. Sci. 1987, 27, 335–343. [Google Scholar] [CrossRef]
- Kang, H.; Gong, M.; Xu, M.; Wang, H.; Li, Y.; Fang, Q.; Zhang, L. Fabricated biobased Eucommia ulmoides gum/polyolefin elastomer thermoplastic vulcanizates into a shape memory material. Ind. Eng. Chem. Res. 2019, 58, 6375–6384. [Google Scholar] [CrossRef]
- Xia, L.; Wang, Y.; Ma, Z.; Du, A.; Qiu, G.; Xin, Z. Preparation of epoxidized Eucommia ulmoides gum and its application in styrene-butadiene rubber (SBR)/silica composites. Polym. Advan. Technol. 2017, 28, 94–101. [Google Scholar] [CrossRef]
- Meng, X.; Bocharova, V.; Tekinalp, H.; Cheng, S.; Kisliuk, A.; Sokolov, A.P.; Kunc, V.; Peter, W.H.; Ozcan, S. Toughening of nanocelluose/PLA composites via bio-epoxy interaction: Mechanistic study. Mater. Des. 2018, 139, 188–197. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, K.; Xu, C.; Chen, Y. Supertoughened biobased poly(lactic acid)–epoxidized natural rubber thermoplastic vulcanizates: Fabrication, co-continuous phase structure, interfacial in situ compatibilization, and toughening mechanism. J. Phys. Chem. B 2015, 119, 12138–12146. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemicals | Weight Ratio | |||||
|---|---|---|---|---|---|---|
| Crosslinked EUG-g-GMA | P9EG1 | P8EG2 | P7EG3 | P6EG4 | P6E4 | |
| PLA | 0 | 90 | 80 | 70 | 60 | 60 |
| EUG-g-GMA | 100 | 10 | 20 | 30 | 40 | 0 |
| EUG | 0 | 0 | 0 | 0 | 0 | 40 |
| Antioxidant1010 | 0.2 | 0.2 | 0.2 | 0.2 | 0.2 | 0.2 |
| DCP | 0.15 | 0.15 | 0.3 | 0.45 | 0.6 | 0.6 |
| Properties | Crosslinked EUG-g-GMA | Neat PLA | P9EG1 | P8EG2 | P7EG3 | P6EG4 |
|---|---|---|---|---|---|---|
| Rf (%) | 77.34 | 99.54 | 99.64 | 99.63 | 99.63 | 99.83 |
| Rr (%) | 99.48 | 70.47 | 83.28 | 86.57 | 90.97 | 93.74 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Liu, J.; Xia, L.; Shen, M.; Wei, L.; Xin, Z.; Kim, J. Fully Biobased Shape Memory Thermoplastic Vulcanizates from Poly(Lactic Acid) and Modified Natural Eucommia Ulmoides Gum with Co-Continuous Structure and Super Toughness. Polymers 2019, 11, 2040. https://doi.org/10.3390/polym11122040
Wang Y, Liu J, Xia L, Shen M, Wei L, Xin Z, Kim J. Fully Biobased Shape Memory Thermoplastic Vulcanizates from Poly(Lactic Acid) and Modified Natural Eucommia Ulmoides Gum with Co-Continuous Structure and Super Toughness. Polymers. 2019; 11(12):2040. https://doi.org/10.3390/polym11122040
Chicago/Turabian StyleWang, Yan, Jinhui Liu, Lin Xia, Mei Shen, Liping Wei, Zhenxiang Xin, and Jinkuk Kim. 2019. "Fully Biobased Shape Memory Thermoplastic Vulcanizates from Poly(Lactic Acid) and Modified Natural Eucommia Ulmoides Gum with Co-Continuous Structure and Super Toughness" Polymers 11, no. 12: 2040. https://doi.org/10.3390/polym11122040
APA StyleWang, Y., Liu, J., Xia, L., Shen, M., Wei, L., Xin, Z., & Kim, J. (2019). Fully Biobased Shape Memory Thermoplastic Vulcanizates from Poly(Lactic Acid) and Modified Natural Eucommia Ulmoides Gum with Co-Continuous Structure and Super Toughness. Polymers, 11(12), 2040. https://doi.org/10.3390/polym11122040