Oral Pharmacokinetics of a Chitosan-Based Nano- Drug Delivery System of Interferon Alpha

 and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Interferon Alpha (IFNα)-Loaded Nanoparticles
2.3. Characterization of the Nanoparticles
2.3.1. Hydrodynamic Diameter, Polydispersity Index and Zeta-Potential
2.3.2. Nanoparticle-Tracking Analysis
2.3.3. Stability of Nanoparticles as a Function of pH
2.3.4. Drug-Encapsulation Efficiency
2.4. Cell Studies In Vitro
2.4.1. Cells
2.4.2. Cell Compatibility and Permeability Assays
2.4.3. Cell Uptake
2.5. Oral Pharmacokinetics
2.6. Statistical Analysis
3. Results and Discussions
3.1. Synthesis and Characterization of PEGylated Chitosan
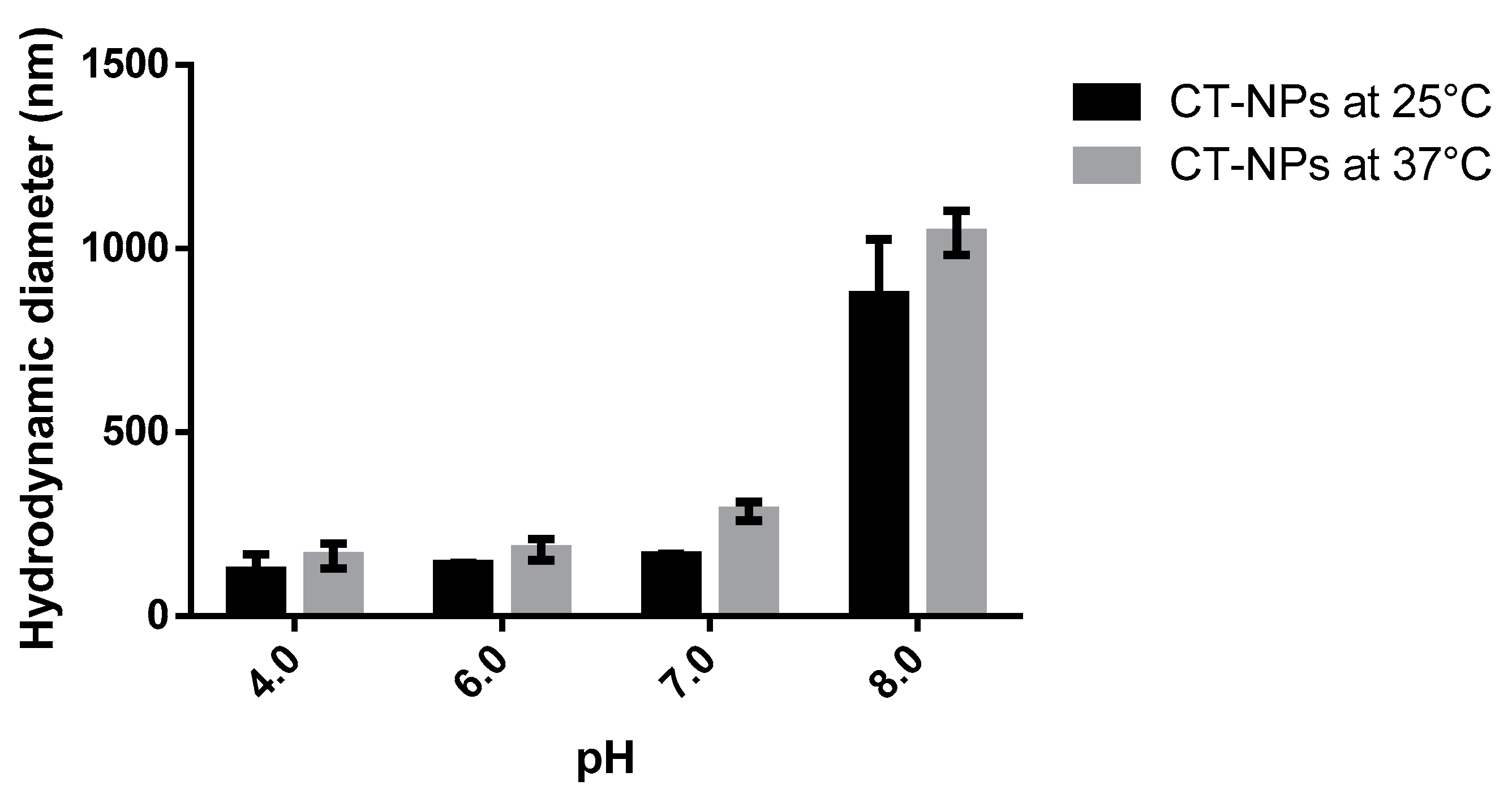
3.2. Preparation and Characterization of the Nanoparticles
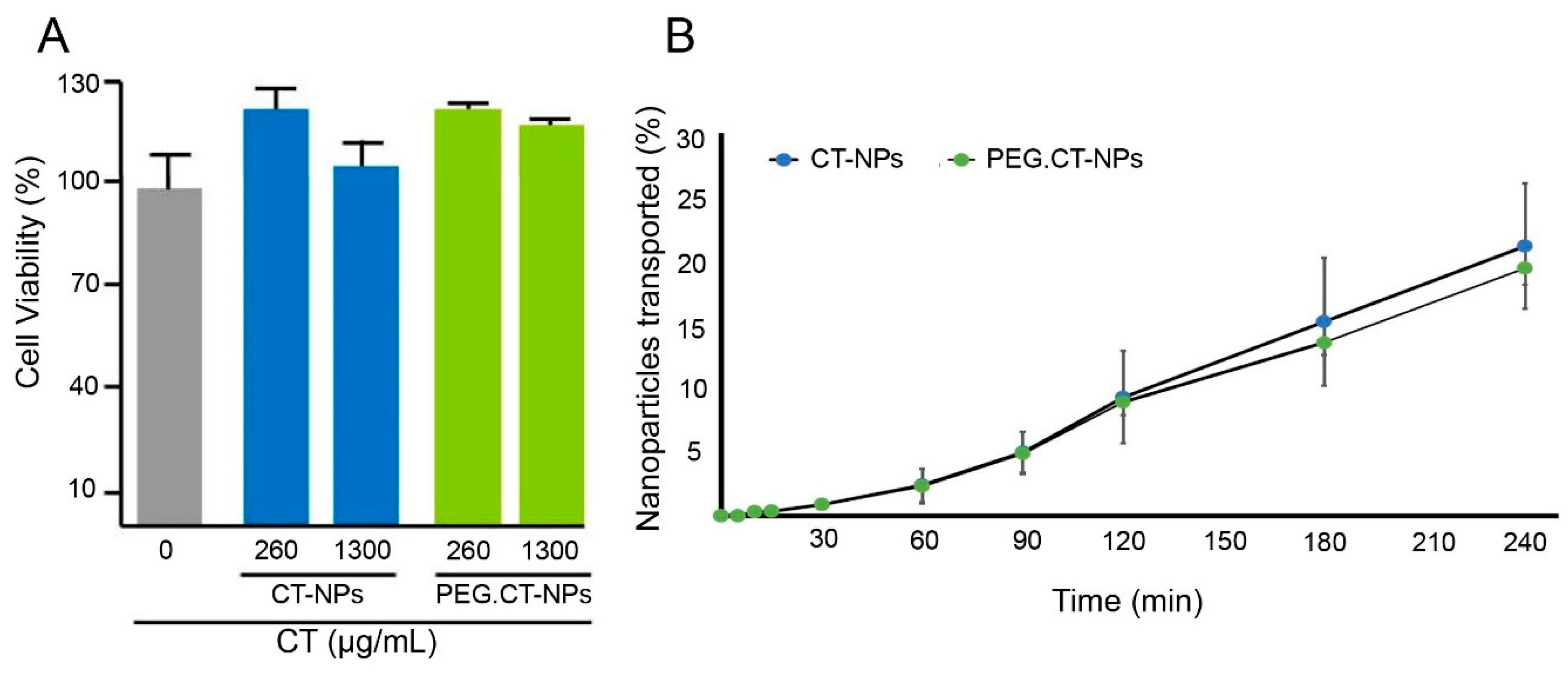
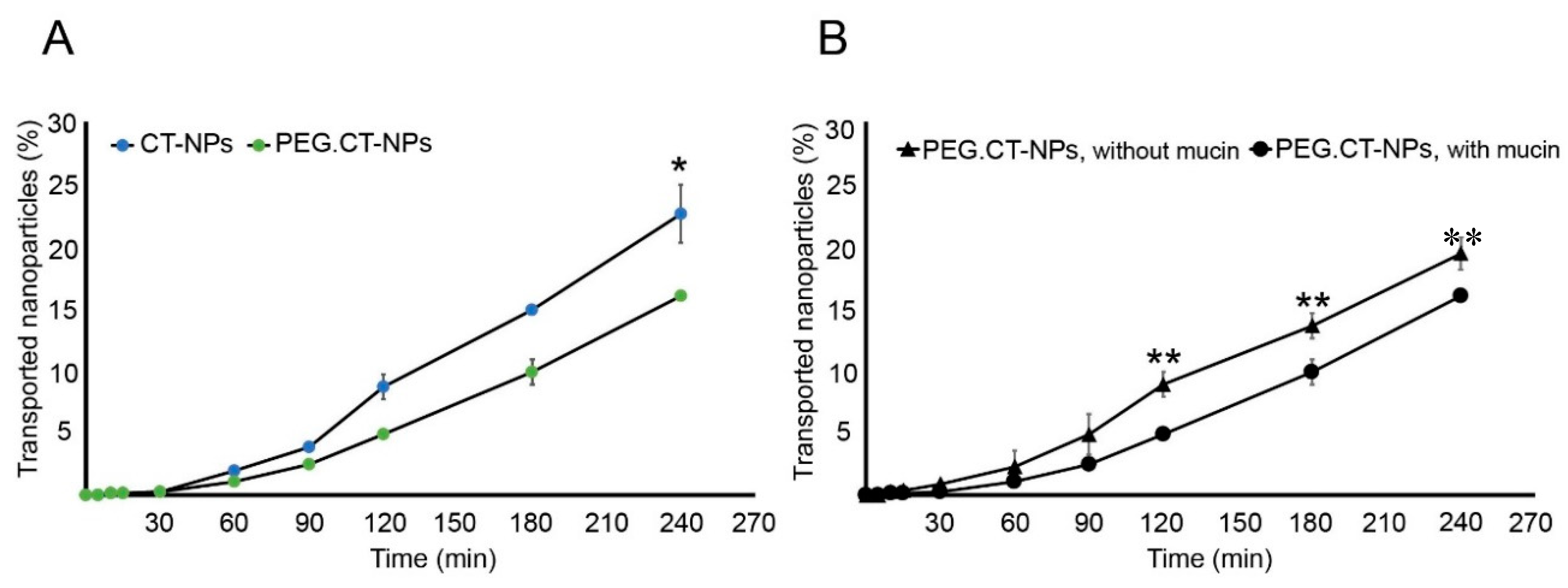
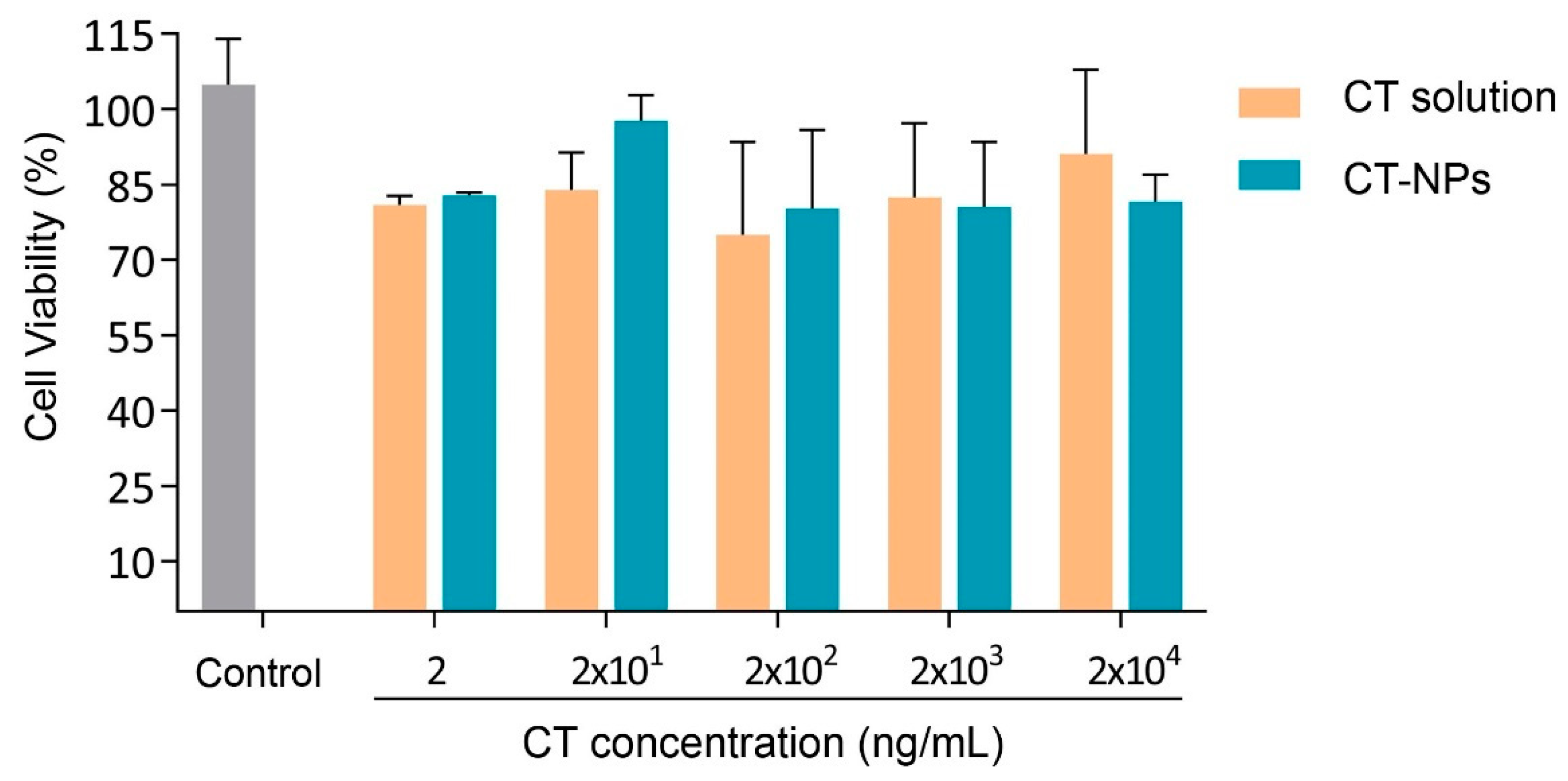
3.3. Cell Compatibility, Permeability and Uptake
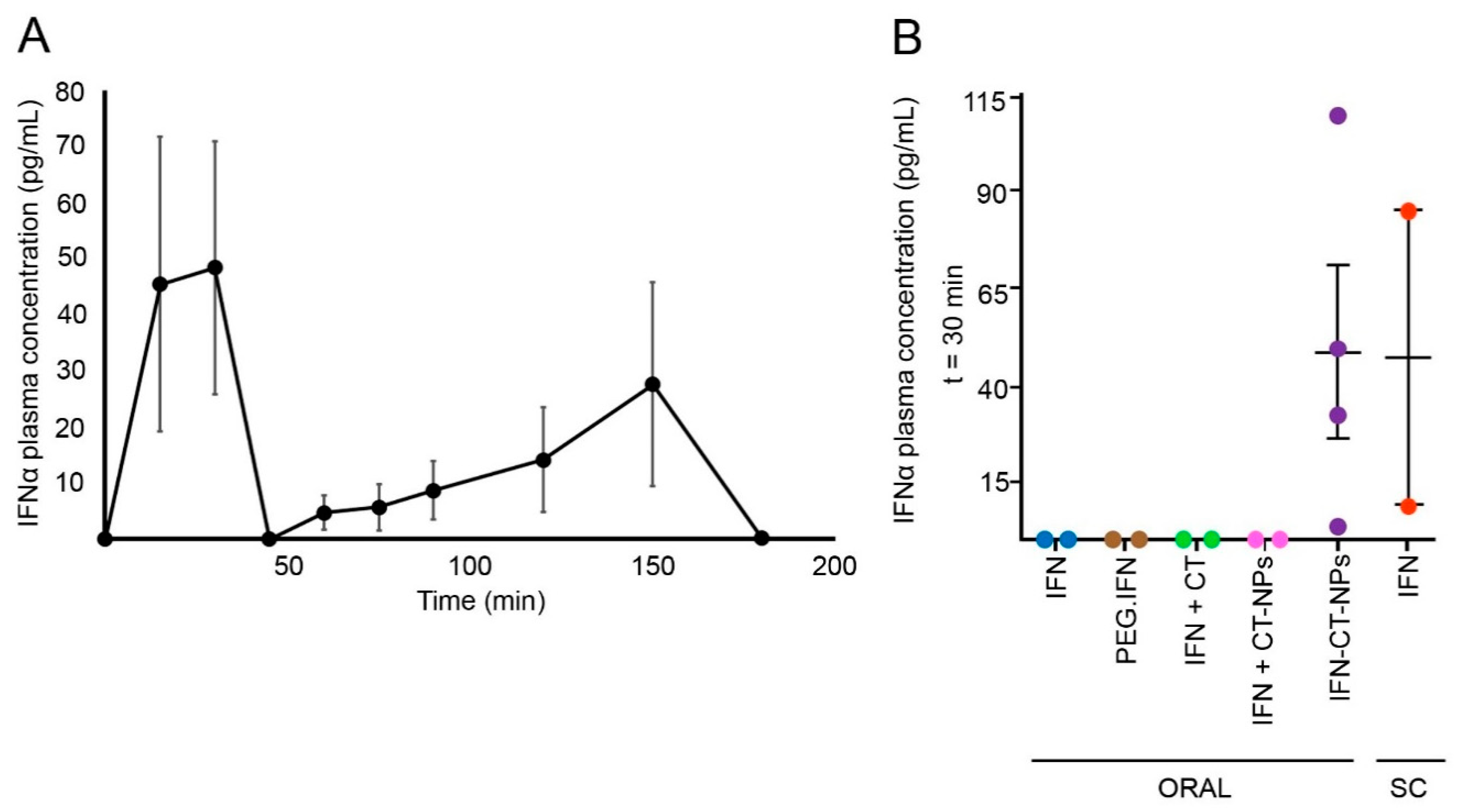
3.4. Oral Pharmacokinetics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Liu, C.; Kou, Y.; Zhang, X.; Cheng, H.; Chen, X.; Mao, S. Strategies and industrial perspectives to improve oral absorption of biological macromolecules. Expert Opin. Drug Deliv. 2018, 15, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Katze, M.G.; He, Y.; Gale, M. Viruses and interferon: A fight for supremacy. Nat. Rev. Immunol. 2002, 2, 675–687. [Google Scholar] [CrossRef] [PubMed]
- El-Baky, N.A.; Redwan, E.M. Therapeutic Alpha-Interferons Protein: Structure, Production, and Biosimilar. Prep. Biochem. Biotechnol. 2015, 45, 109–127. [Google Scholar] [CrossRef] [PubMed]
- Schandené, L.; Cogan, E.; Crusiaux, A.; Goldman, M. Interferon-α Upregulates Both Interleukin-10 and Interferon-γ Production by Human CD4+ T Cells. Blood 1997, 89, 1110–1111. [Google Scholar] [CrossRef] [PubMed]
- Hervas-Stubbs, S.; Perez-Gracia, J.L.; Rouzaut, A.; Sanmamed, M.F.; Le Bon, A.; Melero, I. Direct Effects of Type I Interferons on Cells of the Immune System. Clin. Cancer Res. 2011, 17, 2619–2627. [Google Scholar] [CrossRef] [PubMed]
- Pestka, S.; Krause, C.D.; Walter, M.R. Interferons, interferon-like cytokines, and their receptors. Immunol. Rev. 2004, 202, 8–32. [Google Scholar] [CrossRef] [PubMed]
- Radwanski, E.; Perentesis, G.; Jacobs, S.; Oden, E.; Affrime, M.; Symchowicz, S.; Zampaglione, N. Pharmacokinetics of interferon alpha-2b in healthy volunteers. J. Clin. Pharmacol. 1987, 27, 432–435. [Google Scholar] [CrossRef] [PubMed]
- Boulestin, A.; Kamar, N.; Sandres-Sauné, K.; Alric, L.; Vinel, J.-P.; Rostaing, L.; Izopet, J. Pegylation of IFN-alpha and antiviral activity. J. Interferon Cytokine Res. 2006, 26, 849–853. [Google Scholar] [CrossRef] [PubMed]
- Akash, M.S.H.; Rehman, K.; Chen, S. Polymeric-based particulate systems for delivery of therapeutic proteins. Pharm. Dev. Technol. 2016, 21, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Akash, M.S.H.; Rehman, K.; Chen, S. Natural and synthetic polymers as drug carriers for delivery of therapeutic proteins. Polym. Rev. 2015, 55, 371–406. [Google Scholar] [CrossRef]
- Mukhopadhyay, P.; Kundu, P.P. Chitosan-graft-PAMAM–alginate core–shell nanoparticles: A safe and promising oral insulin carrier in an animal model. RSC Adv. 2015, 5, 93995–94007. [Google Scholar] [CrossRef]
- Sheng, J.; He, H.; Han, L.; Qin, J.; Chen, S.; Ru, G.; Li, R.; Yang, P.; Wang, J.; Yang, V.C. Enhancing insulin oral absorption by using mucoadhesive nanoparticles loaded with LMWP-linked insulin conjugates. J. Control Release 2016, 233, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wei, W.; Lv, P.; Wang, L.; Ma, G. Preparation and evaluation of alginate-chitosan microspheres for oral delivery of insulin. Eur. J. Pharm. Biopharm. 2011, 77, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Sonaje, K.; Lin, Y.-H.; Juang, J.-H.; Wey, S.-P.; Chen, C.-T.; Sung, H.-W. In vivo evaluation of safety and efficacy of self-assembled nanoparticles for oral insulin delivery. Biomaterials 2009, 30, 2329–2339. [Google Scholar] [CrossRef] [PubMed]
- Cánepa, C.; Imperiale, J.; Berini, C.; Lewicki, M.; Sosnik, A.; Biglione, M. Development of a Drug Delivery System Based on Chitosan Nanoparticles for Oral Administration of Interferon-α. Biomacromolecules 2017, 18, 3302–3309. [Google Scholar] [CrossRef] [PubMed]
- Hirai, A.; Odani, H.; Nakajima, A. Determination of degree of deacetylation of chitosan by 1H NMR spectroscopy. Polym. Bull. 1991, 26, 87–94. [Google Scholar] [CrossRef]
- Schlachet, I.; Trousil, J.; Rak, D.; Knudsen, K.D.; Pavlova, E.; Nyström, B.; Sosnik, A. Chitosan-graft-poly(methyl methacrylate) amphiphilic nanoparticles: Self-association and physicochemical characterization. Carbohydr. Polym. 2019, 212, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Chang, F.-C.; Tsao, C.-T.; Lin, A.; Zhang, M.; Levengood, S.L.; Zhang, M. PEG-chitosan hydrogel with tunable stiffness for study of drug response of breast cancer cells. Polymers (Basel) 2016, 8, 112. [Google Scholar] [CrossRef] [PubMed]
- Sarmento, B.; Mazzaglia, D.; Bonferoni, M.C.; Neto, A.P.; do Céu Monteiro, M.; Seabra, V. Effect of chitosan coating in overcoming the phagocytosis of insulin loaded solid lipid nanoparticles by mononuclear phagocyte system. Carbohydr. Polym. 2011, 84, 919–925. [Google Scholar] [CrossRef]
- Xu, Q.; Ensign, L.M.; Boylan, N.J.; Schön, A.; Gong, X.; Yang, J.-C.; Lamb, N.W.; Cai, S.; Yu, T.; Freire, E.; et al. Impact of Surface Polyethylene Glycol (PEG) Density on Biodegradable Nanoparticle Transport in Mucus ex Vivo and Distribution in Vivo. ACS Nano 2015, 9, 9217–9227. [Google Scholar] [CrossRef] [PubMed]
- A Natural Source of Chitosan|ChitoClear|Primex. Available online: http://www.primex.is/products-services/chitoclear/ (accessed on 3 June 2019).
- Noi, I.; Schlachet, I.; Kumarasamy, M.; Sosnik, A. Permeability of Novel Chitosan-g-poly (Methyl Methacrylate) Amphiphilic Nanoparticles in a Model of Small Intestine in Vitro. Polymers (Basel) 2018, 10, 478. [Google Scholar] [CrossRef] [PubMed]
- Lea, T. Caco-2 Cell Line. In The Impact of Food Bioactives on Health; Springer International Publishing: Cham, Switzerland, 2015; pp. 103–111. [Google Scholar] [CrossRef]
- Yee, S. In vitro permeability across Caco-2 cells (colonic) can predict in vivo (small intestinal) absorption in man--fact or myth. Pharm. Res. 1997, 14, 763–766. [Google Scholar] [CrossRef] [PubMed]
- Slavin, J. Fibre-Rich and Wholegrain Foods. In Fibre-Rich and Wholegrain Foods, 1st ed.; Delcour, J., Poutanen, K., Eds.; Woodhead Publishing: Swaston, UK, 2013; pp. 61–75. [Google Scholar]
- Cummins, J.M.; Beilharz, M.W.; Krakowka, S. Oral use of interferon. J. Interferon Cytokine Res. 1999, 19, 853–857. [Google Scholar] [CrossRef] [PubMed]
- Coppi, G.; Sala, N.; Bondi, M.; Sergi, S.; Iannuccelli, V. Ex-vivo evaluation of alginate microparticles for Polymyxin B oral administration. J. Drug Target. 2006, 14, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Chatelut, E.; Rostaing, L.; Grégoire, N.; Payen, J.L.; Pujol, A.; Izopet, J.; Houin, G.; Canal, P. A pharmacokinetic model for alpha interferon administered subcutaneously. Br. J. Clin. Pharmacol. 1999, 47, 365–371. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | CT-NPs | PEG.CT-NPs |
|---|---|---|
| Dh (nm) | 47 | 93 |
| PDI | 0.47 | 0.32 |
| Concentration (particles/mL) | 3 × 1011 ± 6 × 107 | 3 × 1011 ± 3 × 1010 |
| Parameter | Value |
|---|---|
| tmax1 (h) | 0.5 |
| tmax2 (h) | 1.5 |
| Cmax1 (pg.mL−1) | 48.4 ± 22.5 |
| Cmax2 (pg.mL−1) | 27.6 ± 31.4 |
| t1/2 (h) | 0.07 ± 0.02 |
| AUC0-∞ (pg*h/mL) | 56.92 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Imperiale, J.C.; Schlachet, I.; Lewicki, M.; Sosnik, A.; Biglione, M.M. Oral Pharmacokinetics of a Chitosan-Based Nano- Drug Delivery System of Interferon Alpha. Polymers 2019, 11, 1862. https://doi.org/10.3390/polym11111862
Imperiale JC, Schlachet I, Lewicki M, Sosnik A, Biglione MM. Oral Pharmacokinetics of a Chitosan-Based Nano- Drug Delivery System of Interferon Alpha. Polymers. 2019; 11(11):1862. https://doi.org/10.3390/polym11111862
Chicago/Turabian StyleImperiale, Julieta C., Inbar Schlachet, Marianela Lewicki, Alejandro Sosnik, and Mirna M. Biglione. 2019. "Oral Pharmacokinetics of a Chitosan-Based Nano- Drug Delivery System of Interferon Alpha" Polymers 11, no. 11: 1862. https://doi.org/10.3390/polym11111862
APA StyleImperiale, J. C., Schlachet, I., Lewicki, M., Sosnik, A., & Biglione, M. M. (2019). Oral Pharmacokinetics of a Chitosan-Based Nano- Drug Delivery System of Interferon Alpha. Polymers, 11(11), 1862. https://doi.org/10.3390/polym11111862