Studying the Ring-Opening Polymerization of 1,5-Dioxepan-2-one with Organocatalysts

Abstract
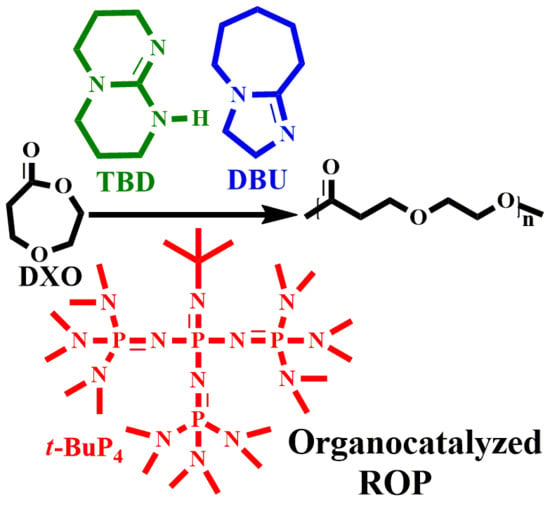
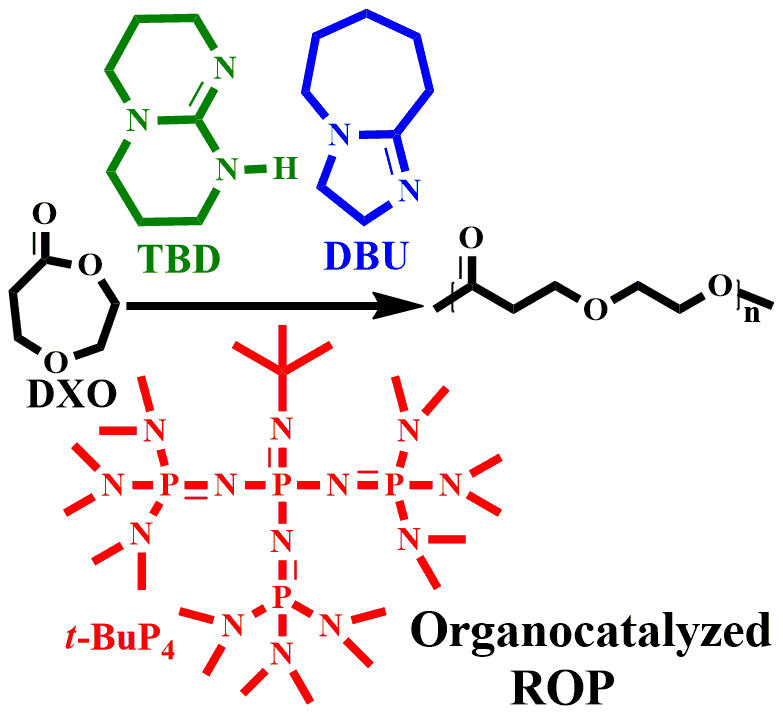
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Characterizations
2.3. Synthesis of PDXO Homopolymer
2.4. Synthesis of Copolymer
3. Results and Discussion
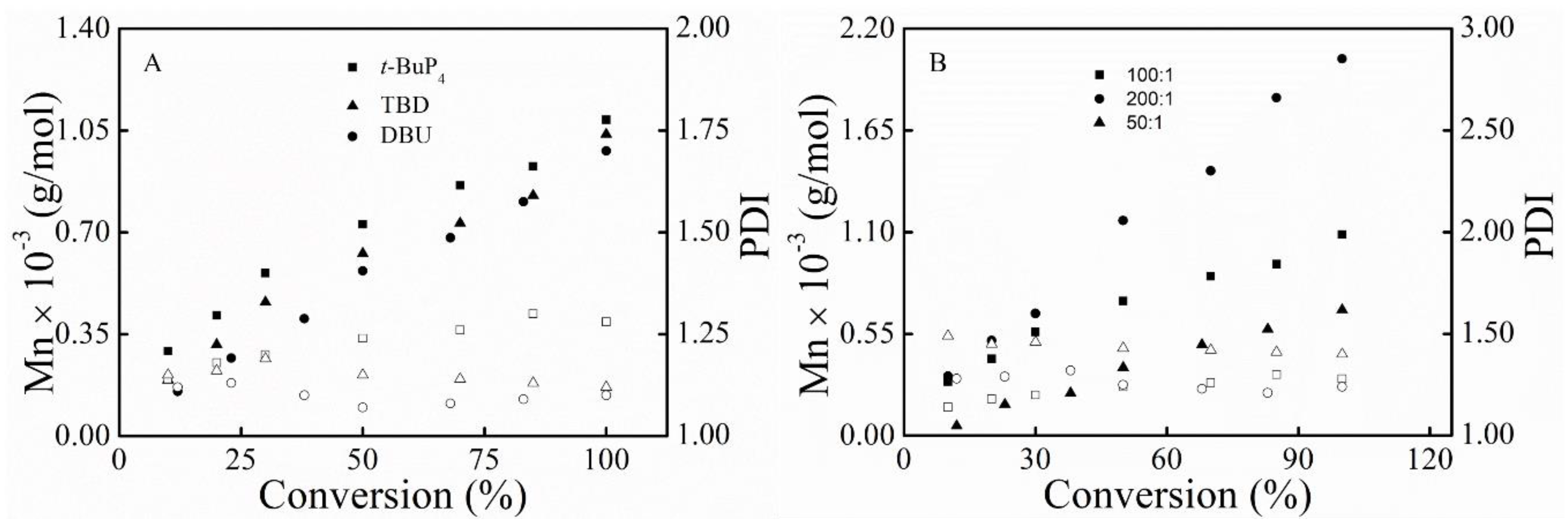
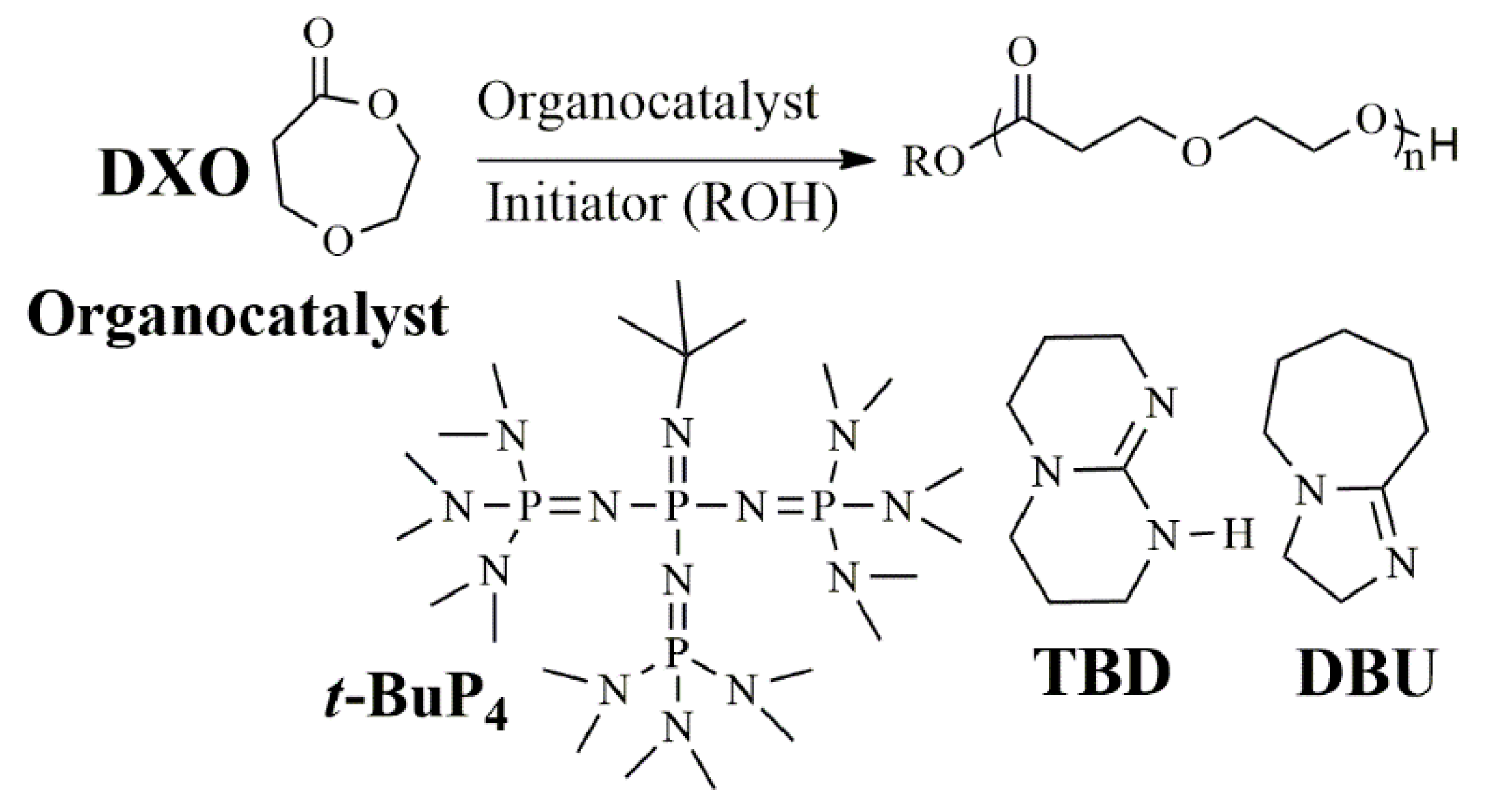
3.1. Polymerization
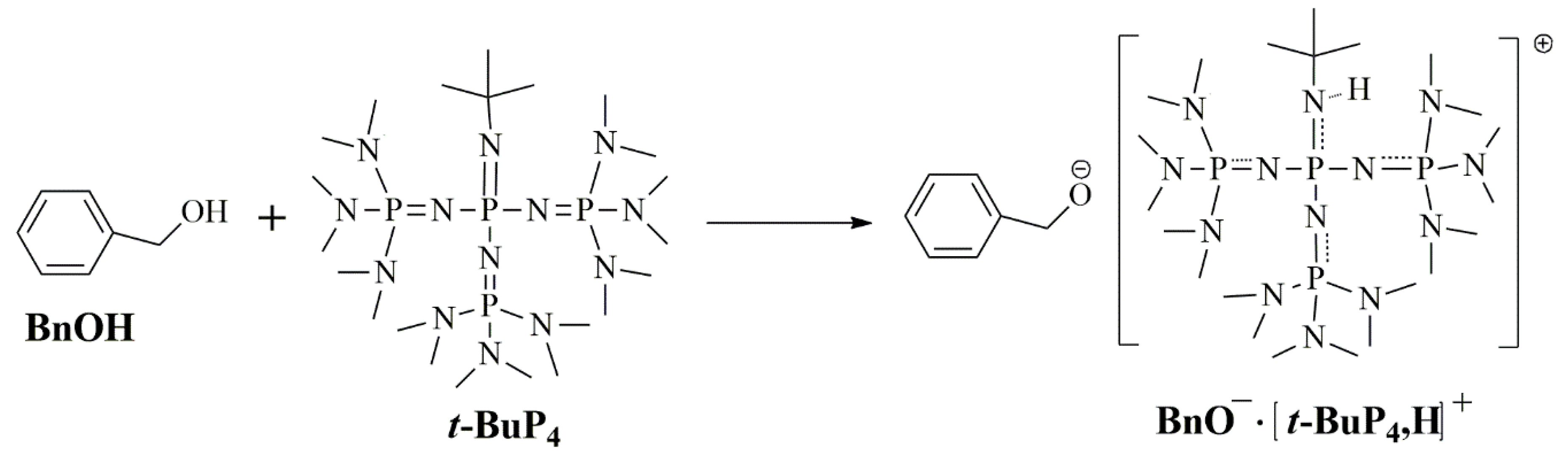
3.2. Ether Bond Fragmentation
3.3. Kinetic Study
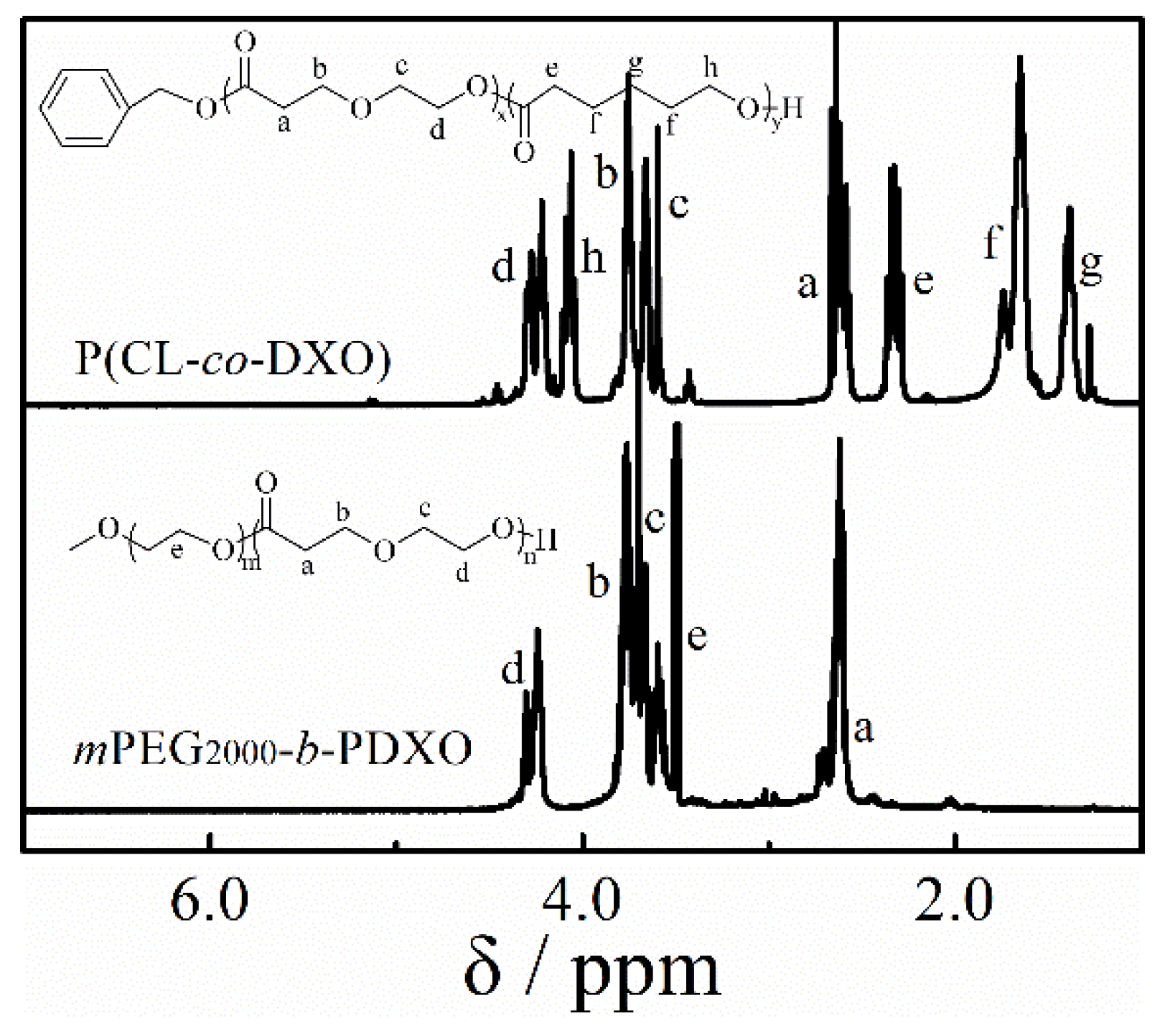
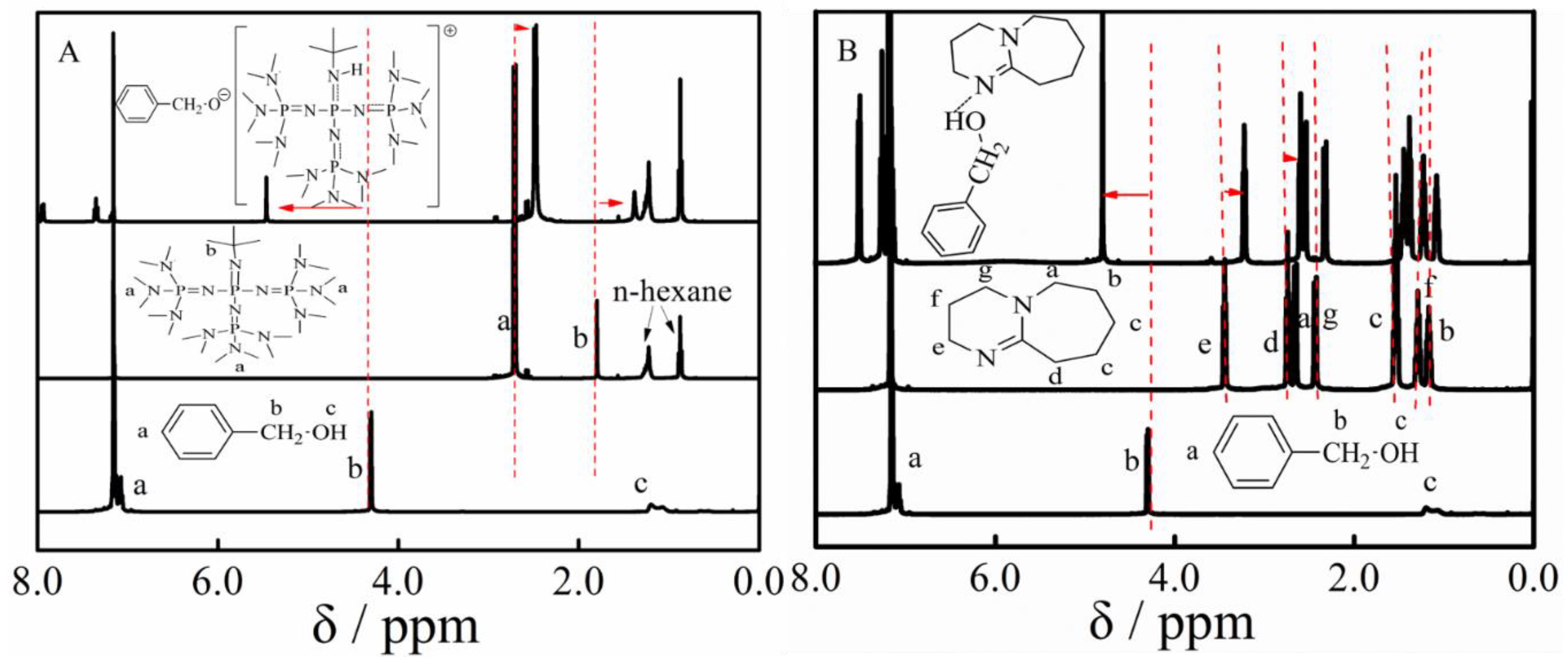
3.4. Copolymerization and Macroinitiator Initiation
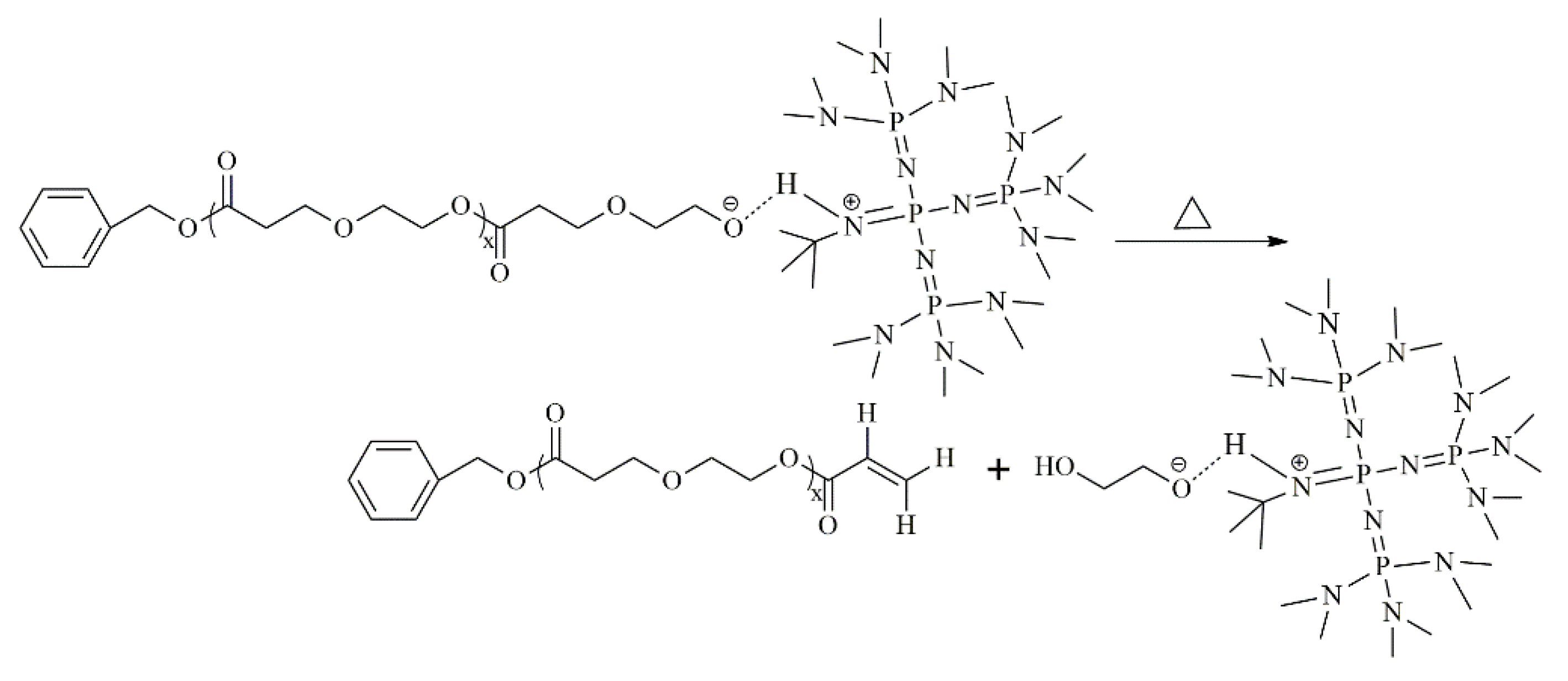
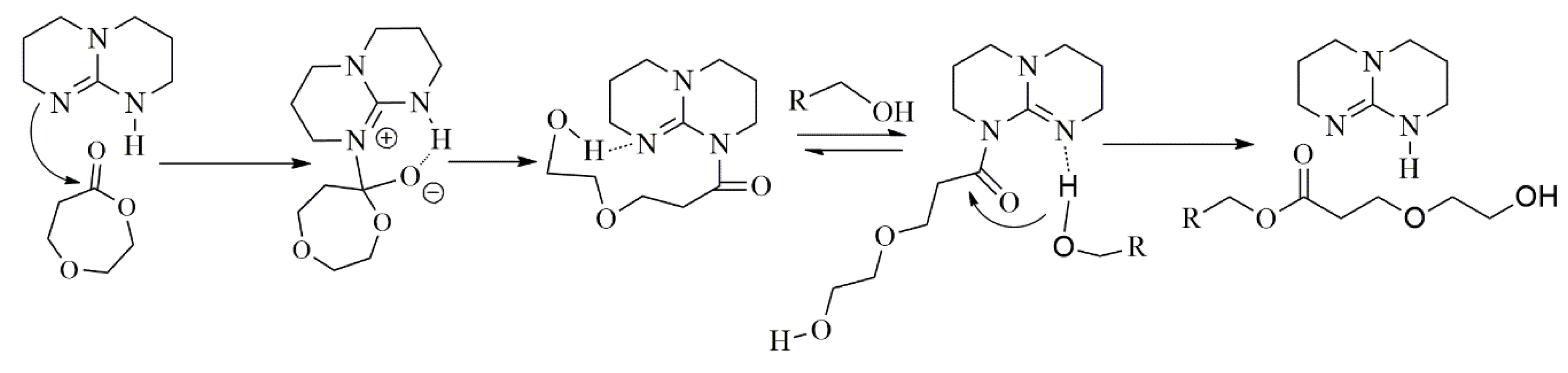
3.5. Proposed Mechanism
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Jerome, C.; Lecomte, P. Recent Advances in the Synthesis of Aliphatic Polyesters by Ring-Opening Polymerization. Adv. Drug Del. Rev. 2008, 60, 1056–1076. [Google Scholar] [CrossRef] [PubMed]
- Hadjichristidis, N.; Iatrou, H.; Pitsikalis, M.; Sakellariou, G. Synthesis of Well-Defined Polypeptide-based Materials via the Ring-Opening Polymerization of α-Amino Acid N-Carboxyanhydrides. Chem. Rev. 2009, 109, 5528–5578. [Google Scholar] [CrossRef]
- Tian, H.Y.; Tang, Z.H.; Zhuang, X.L.; Chen, X.S.; Jing, X.B. Biodegradable Synthetic Polymers: Preparation, Functionalization and Biomedical Application. Prog. Polym. Sci. 2012, 37, 237–280. [Google Scholar] [CrossRef]
- Becker, G.; Wurm, F.R. Functional Biodegradable Polymers via Ring-Opening Polymerization of Monomers without Protective Groups. Chem. Soc. Rev. 2018, 47, 7739–7782. [Google Scholar] [CrossRef]
- Irimia-Vladu, M. “Green” Electronics: Biodegradable and Biocompatible Materials and Devices for Sustainable Future. Chem. Soc. Rev. 2014, 43, 588–610. [Google Scholar] [CrossRef]
- Mülhaupt, R. Green Polymer Chemistry and Bio-based Plastics: Dreams and Reality. Macromol. Chem. Phys. 2013, 214, 159–174. [Google Scholar] [CrossRef]
- Nair, L.S.; Laurencin, C.T. Biodegradable Polymers as Biomaterials. Prog. Polym. Sci. 2007, 32, 762–798. [Google Scholar] [CrossRef]
- Kiesewetter, M.K.; Shin, E.J.; Hedrick, J.L.; Waymouth, R.M. Organocatalysis: Opportunities and Challenges for Polymer Synthesis. Macromolecules 2010, 43, 2093–2107. [Google Scholar] [CrossRef]
- Boileau, S.; Illy, N. Activation in Anionic Polymerization: Why Phosphazene Bases are Very Exciting Promoters. Prog. Polym. Sci. 2011, 36, 1132–1151. [Google Scholar] [CrossRef]
- Wei, J.; Meng, H.; Guo, B.; Zhong, Z.; Meng, F. Organocatalytic Ring-opening Copolymerization of Trimethylene Carbonate and Dithiolane Trimethylene Carbonate: Impact of Organocatalysts on Copolymerization Kinetics and Copolymer Microstructures. Biomacromolecules 2018, 19, 2294–2301. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Ren, C.; Li, H.; Li, Y.; Liu, S.; Li, Z. Selective Ring-Opening Polymerization of Non-strained γ-Butyrolactone Catalyzed by A Cyclic Trimeric Phosphazene Base. Angew. Chem. Int. Ed. Engl. 2017, 56, 12987–12990. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.N.; Shen, J.Z.; Alamri, H.; Hadjichristidis, N.; Zhao, J.P.; Wang, Y.C.; Zhang, G.Z. Revealing the Cytotoxicity of Residues of Phosphazene Catalysts Used for the Synthesis of Poly(ethylene oxide). Biomacromolecules 2017, 18, 3233–3237. [Google Scholar] [CrossRef] [PubMed]
- Clément, B.; Grignard, B.; Koole, L.; Jérôme, C.; Lecomte, P. Metal-Free Strategies for the Synthesis of Functional and Well-defined Polyphosphoesters. Macromolecules 2012, 45, 4476–4486. [Google Scholar] [CrossRef]
- Teng, C.J.; Weber, W.P.; Cai, G. Anionic and Cationic Ring-Opening Polymerization of 2, 2, 4, 4, 6, 6-Hexamethyl-8, 8-divinylcyclotetrasiloxane. Macromolecules 2003, 36, 5126–5130. [Google Scholar] [CrossRef]
- Shen, Y.; Zhang, J.; Zhao, N.; Liu, F.; Li, Z. Preparation of Biorenewable Poly(γ-butyrolactone)-b-Poly(l-lactide) Diblock Copolyesters via One-Pot Sequential Metal-Free Ring-Opening Polymerization. Polym. Chem. 2018, 9, 2936–2941. [Google Scholar] [CrossRef]
- Ladelta, V.; Kim, J.D.; Bilalis, P.; Gnanou, Y.; Hadjichristidis, N. Block Copolymers of Macrolactones/Small Lactones by a “Catalyst-Switch” Organocatalytic Strategy. Thermal Properties and Phase Behavior. Macromolecules 2018, 51, 2428–2436. [Google Scholar] [CrossRef]
- Nederberg, F.; Lohmeijer, B.G.G.; Leibfarth, F.; Pratt, R.C.; Choi, J.; Dove, A.P.; Waymouth, R.M.; Hedrick, J.L. Organocatalytic Ring Opening Polymerization of Trimethylene Carbonate. Biomacromolecules 2006, 8, 153–160. [Google Scholar] [CrossRef]
- Yuen, A.Y.; Lopez-Martinez, E.; Gomez-Bengoa, E.; Cortajarena, A.L.; Aguirresarobe, R.H.; Bossion, A.; Mecerreyes, D.; Hedrick, J.L.; Yang, Y.Y.; Sardon, H. Preparation of Biodegradable Cationic Polycarbonates and Hydrogels through the Direct Polymerization of Quaternized Cyclic Carbonates. ACS Biomater. Sci. Eng. 2017, 3, 1567–1575. [Google Scholar] [CrossRef]
- Feng, J.; Zhuo, R.X.; Zhang, X.Z. Construction of Functional Aliphatic Polycarbonates for Biomedical Applications. Prog. Polym. Sci. 2012, 37, 211–236. [Google Scholar] [CrossRef]
- Lohmeijer, B.G.G.; Pratt, R.C.; Leibfarth, F.; Logan, J.W.; Long, D.A.; Dove, A.P.; Nederberg, F.; Choi, J.; Wade, C.; Waymouth, R.M. Guanidine and Amidine Organocatalysts for Ring-Opening Polymerization of Cyclic Esters. Macromolecules 2006, 39, 8574–8583. [Google Scholar] [CrossRef]
- Pratt, R.C.; Lohmeijer, B.G.G.; Long, D.A.; Waymouth, R.M.; Hedrick, J.L. Triazabicyclodecene: A Simple Bifunctional Organocatalyst for Acyl Transfer and Ring-Opening Polymerization of Cyclic Esters. J. Am. Chem. Soc. 2006, 128, 4556–4557. [Google Scholar] [CrossRef] [PubMed]
- Brown, H.A.; de Crisci, A.G.; Hedrick, J.L.; Waymouth, R.M. Amidine-Mediated Zwitterionic Polymerization of Lactide. ACS Macro Lett. 2012, 1, 1113–1115. [Google Scholar] [CrossRef]
- Yang, H.; Xu, J.; Pispas, S.; Zhang, G. Hybrid Copolymerization of ε-Caprolactone and Methyl Methacrylate. Macromolecules 2012, 45, 3312–3317. [Google Scholar] [CrossRef]
- Zhao, J.; Schlaad, H.; Weidner, S.; Antonietti, M. Synthesis of Terpene–Poly(ethylene oxide)s by t-BuP4-Promoted Anionic Ring-Opening Polymerization. Polym. Chem. 2012, 3, 1763–1768. [Google Scholar] [CrossRef]
- Xia, Y.N.; Chen, Y.; Song, Q.L.; Hu, S.Y.; Zhao, J.P.; Zhang, G.Z. Base-to-Base Organocatalytic Approach for One-Pot Construction of Poly(ethylene oxide)-based Macromolecular Structures. Macromolecules 2016, 49, 6817–6825. [Google Scholar] [CrossRef]
- Li, H.; Luo, H.; Zhao, J.; Zhang, G. One-Step Approach to Polyester–Polyether Block Copolymers Using Highly Tunable Bicomponent Catalyst. ACS Macro Lett. 2018, 7, 1420–1425. [Google Scholar] [CrossRef]
- Loefgren, A.; Albertsson, A.C.; Dubois, P.; Jerome, R.; Teyssie, P. Synthesis and Characterization of Biodegradable Homopolymers and Block Copolymers based on 1, 5-Dioxepan-2-one. Macromolecules 1994, 27, 5556–5562. [Google Scholar] [CrossRef]
- Andronova, N.; Srivastava, R.K.; Albertsson, A.C. Potential Tissue Implants from the Networks based on 1, 5-Dioxepan-2-one and ε-Caprolactone. Polymer 2005, 46, 6746–6755. [Google Scholar] [CrossRef]
- Van der Meulen, I.; Li, Y.; Deumens, R.; Joosten, E.A.; Koning, C.E.; Heise, A. Copolymers from Unsaturated Macrolactones: Toward the Design of Cross-linked Biodegradable Polyesters. Biomacromolecules 2011, 12, 837–843. [Google Scholar] [CrossRef]
- Stjerndahl, A.; Wistrand, A.F.; Albertsson, A.C. Industrial Utilization of Tin-initiated Resorbable Polymers: Synthesis on A Large Scale with A Low Amount of Initiator Residue. Biomacromolecules 2007, 8, 937–940. [Google Scholar] [CrossRef]
- Odelius, K.; Plikk, P.; Albertsson, A.C. Elastomeric Hydrolyzable Porous Scaffolds: Copolymers of Aliphatic Polyesters and A Polyether−ester. Biomacromolecules 2005, 6, 2718–2725. [Google Scholar] [CrossRef] [PubMed]
- Mathisen, T.; Albertsson, A.C. Polymerization of 1,5-Dioxepan-2-one. I. Synthesis and Characterization of the Monomer 1,5-Dioxepan-2-one and Its Cyclic Dimer 1,5,8,12-Tetraoxacyclotetradecane-2,9-dione. Macromolecules 1989, 22, 3838–3842. [Google Scholar] [CrossRef]
- Mathisen, T.; Masus, K.; Albertsson, A.C. Polymerization of 1,5-Dioxepan-2-one. II. Polymerization of 1,5-Dioxepan-2-one and Its Cyclic Dimer, Including a New Procedure for the Synthesis of 1,5-Dioxepan-2-one. Macromolecules 1989, 22, 3842–3846. [Google Scholar] [CrossRef]
- Löfgren, A.; Albertsson, A.C. Synthesis and Characterization of High Molecular Weight Poly(1,5-dioxepan-2-one) with Narrow Molecular Weight Distribution. Polymer 1995, 36, 3753–3759. [Google Scholar] [CrossRef]
- Macdonald, J.P.; Sidera, M.; Fletcher, S.P.; Shaver, M.P. Living and Immortal Polymerization of Seven and Six Membered Lactones to High Molecular Weights with Aluminum Salen and Salan Catalysts. Eur. Polym. J. 2016, 74, 287–295. [Google Scholar] [CrossRef]
- Höglund, A.; Albertsson, A.C. Spontaneous Crosslinking of Poly(1,5-dioxepan-2-one) Originating from Ether Bond Fragmentation. J. Polym. Sci. Part. A Polym. Chem. 2008, 46, 7258–7267. [Google Scholar] [CrossRef]
- Kiesewetter, M.K.; Scholten, M.D.; Kirn, N.; Weber, R.L.; Hedrick, J.L.; Waymouth, R.M. Cyclic Guanidine Organic Catalysts: What is Magic about Triazabicyclodecene. J. Org. Chem. 2009, 74, 9490–9496. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Cat. | fb | T (°C) | Time (min) | Conv. (%) c | Mn,NMR’ × 10−4 (g/mol) c | Mn,theo’ × 10−4 (g/mol) d | Mn,SEC × 10−4 (g/mol) e | PDI e |
|---|---|---|---|---|---|---|---|---|---|
| H1 | t-BuP4 | 50 | 25 | 5 | 95 | 0.55 | 0.56 | 0.60 | 1.24 |
| H2 | t-BuP4 | 100 | 25 | 15 | 96 | 1.10 | 1.17 | 1.08 | 1.28 |
| H3 | t-BuP4 | 200 | 25 | 120 | >99 | 1.84 | 2.32 | 2.04 | 1.40 |
| H4 | t-BuP4 | 300 | 25 | 180 | >99 | 2.45 | 3.49 | 3.25 | 1.35 |
| H5 | t-BuP4 | 500 | 25 | 600 | >99 | 3.41 | 5.81 | 3.92 | 1.42 |
| H6 | t-BuP4 | 100 | 60 | 480 | -- | -- | -- | gel | -- |
| H7 | t-BuP4 | 50 | 60 | 800 | -- | -- | -- | gel | -- |
| H9 | t-BuP4 | 100 | 40 | 600 | >99 | 1.13 | 1.17 | 1.22 | 1.28 |
| H10 | t-BuP4 | 50 | −20 | 15 | >99 | 0.58 | 0.59 | 0.61 | 1.31 |
| H11 | TBD | 50 | 25 | 30 | >99 | 0.56 | 0.59 | 0.78 | 1.15 |
| H12 | TBD | 100 | 25 | 50 | >99 | 1.10 | 1.17 | 1.04 | 1.12 |
| H13 | TBD | 500 | 25 | 600 | >99 | 3.25 | 5.81 | 3.34 | 1.15 |
| H14 | TBD | 50 | 60 | 1080 | -- | -- | -- | gel | -- |
| H15 | DBU | 50 | 25 | 180 | >99 | 0.56 | 0.59 | 0.59 | 1.15 |
| H16 | DBU | 100 | 25 | 600 | >99 | 1.15 | 1.17 | 0.98 | 1.10 |
| H17 | DBU | 500 | 25 | 1200 | 98 | 4.01 | 5.81 | 2.98 | 1.13 |
| H18 | DBU | 50 | 60 | 1440 | -- | -- | -- | gel | -- |
| Samples | Initiators | Catalysts | fb | Mn × 10−4 (g/mol) c | PDI c |
|---|---|---|---|---|---|
| S1 | mPEG2000-OH | t-BuP4 | DXO | 0.72 | 1.24 |
| S2 | mPEG2000-OH | TBD | DXO | 0.70 | 1.14 |
| S3 | mPEG2000-OH | DBU | DXO | 0.73 | 1.10 |
| S4 c | BnOH | t-BuP4 | CL/DXO | 1.03 | 1.37 |
| S5 | BnOH | TBD | CL/DXO | 1.03 | 1.27 |
| S6 | BnOH | DBU | CL/DXO | 1.12 | 1.09 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, J.; Chen, Y.; Xiao, W.; Zhang, J.; Bu, M.; Zhang, X.; Lei, C. Studying the Ring-Opening Polymerization of 1,5-Dioxepan-2-one with Organocatalysts. Polymers 2019, 11, 1642. https://doi.org/10.3390/polym11101642
Xu J, Chen Y, Xiao W, Zhang J, Bu M, Zhang X, Lei C. Studying the Ring-Opening Polymerization of 1,5-Dioxepan-2-one with Organocatalysts. Polymers. 2019; 11(10):1642. https://doi.org/10.3390/polym11101642
Chicago/Turabian StyleXu, Jinbao, Yang Chen, Wenhao Xiao, Jie Zhang, Minglu Bu, Xiaoqing Zhang, and Caihong Lei. 2019. "Studying the Ring-Opening Polymerization of 1,5-Dioxepan-2-one with Organocatalysts" Polymers 11, no. 10: 1642. https://doi.org/10.3390/polym11101642
APA StyleXu, J., Chen, Y., Xiao, W., Zhang, J., Bu, M., Zhang, X., & Lei, C. (2019). Studying the Ring-Opening Polymerization of 1,5-Dioxepan-2-one with Organocatalysts. Polymers, 11(10), 1642. https://doi.org/10.3390/polym11101642