Methoxy-Group Control of Helical Pitch in Stereoregular Poly(2-ethynylmethoxynaphthalene) Prepared by Rhodium Complex Catalyst


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Measurements
2.2. Materials
2.2.1. General
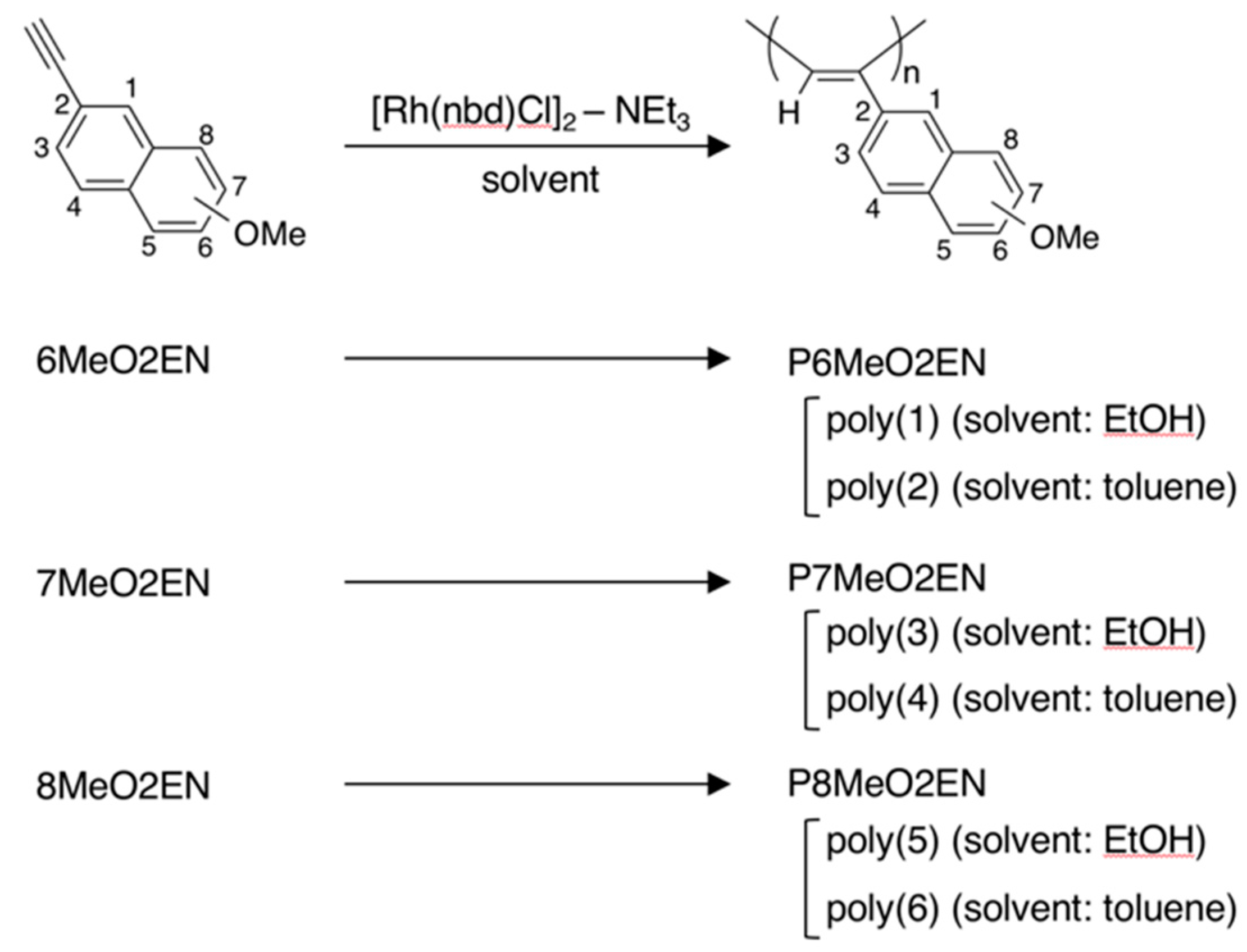
2.2.2. Synthesis of Monomers
- 6MeO2EN: pale yellow solid. 1H NMR (500 MHz, CDCl3): δ 7.94 (s, 1H, Ar), 7.69 (d, J = 9.0 Hz, 1H, Ar), 7.66 (d, J = 8.5 Hz, 1H, Ar), 7.49 (dd, J = 8.3 Hz, 1.8 Hz, 1H, Ar), 7.15 (dd, J = 9.0 Hz, 2.5 Hz, 1H, Ar), 7.10 (d, J = 2.5 Hz, 1H, Ar), 3.92 (s, 3H, O–CH3), 3.10 (s, 1H, –C≡C–H); 13C NMR (125 MHz, CDCl3): δ 158.44, 134.37, 132.07, 129.31, 129.14, 128.28, 126.80, 119.48, 116.91, 105.75, 84.18, 76.68, 55.32.
- 7MeO2EN: pale yellow solid. 1H NMR (500 MHz, CDCl3): δ 7.91 (s, 1H, Ar), 7.71 (d, J = 2.0 Hz, 1H, Ar), 7.69 (d, J = 1.5 Hz, 1H, Ar), 7.39 (dd, J = 8.3 Hz, 1.8 Hz, 1H, Ar), 7.16 (dd, J = 8.8 Hz, 2.8 Hz, 1H, Ar), 7.07 (d, J = 2.5 Hz, 1H, Ar), 3.91 (s, 3H, O–CH3), 3.14 (s, 1H, –C≡C–H); 13C NMR (125 MHz, CDCl3): δ 158.10, 134.02, 131.05, 129.22, 128.56, 127.73, 126.40, 119.82, 119.75, 105.52, 84.14, 77.29, 55.32.
- 8MeO2EN: pale yellow solid. 1H NMR (500 MHz, CDCl3): δ 8.45 (s, 1H, Ar), 7.72 (dd, J = 8.5 Hz, 0.5 Hz, 1H, Ar), 7.52 (dd, J = 8.5 Hz, 1.5 Hz, 1H, Ar), 7.42–7.35 (m, 2H, Ar), 6.81 (dd, J = 6.5 Hz, 2.0 Hz, 1H, Ar), 3.98 (s, 3H, O–CH3), 3.12 (s, 1H, –C≡C–H); 13C NMR (125 MHz, CDCl3): δ 155.13, 140.00, 129.05, 127.54, 127.10, 126.78, 125.05, 119.94, 118.55, 104.46, 84.37, 76.95, 55.53.
2.3. Polymerization Procedure
2.4. Computation
3. Results and Discussion
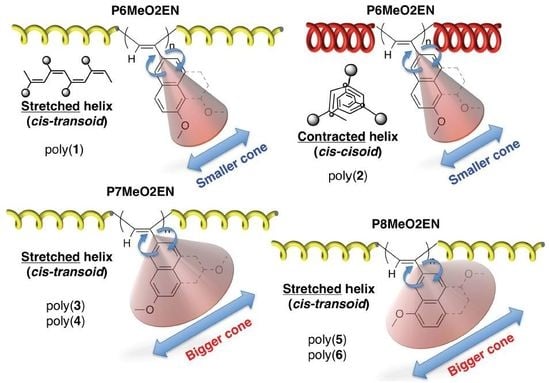
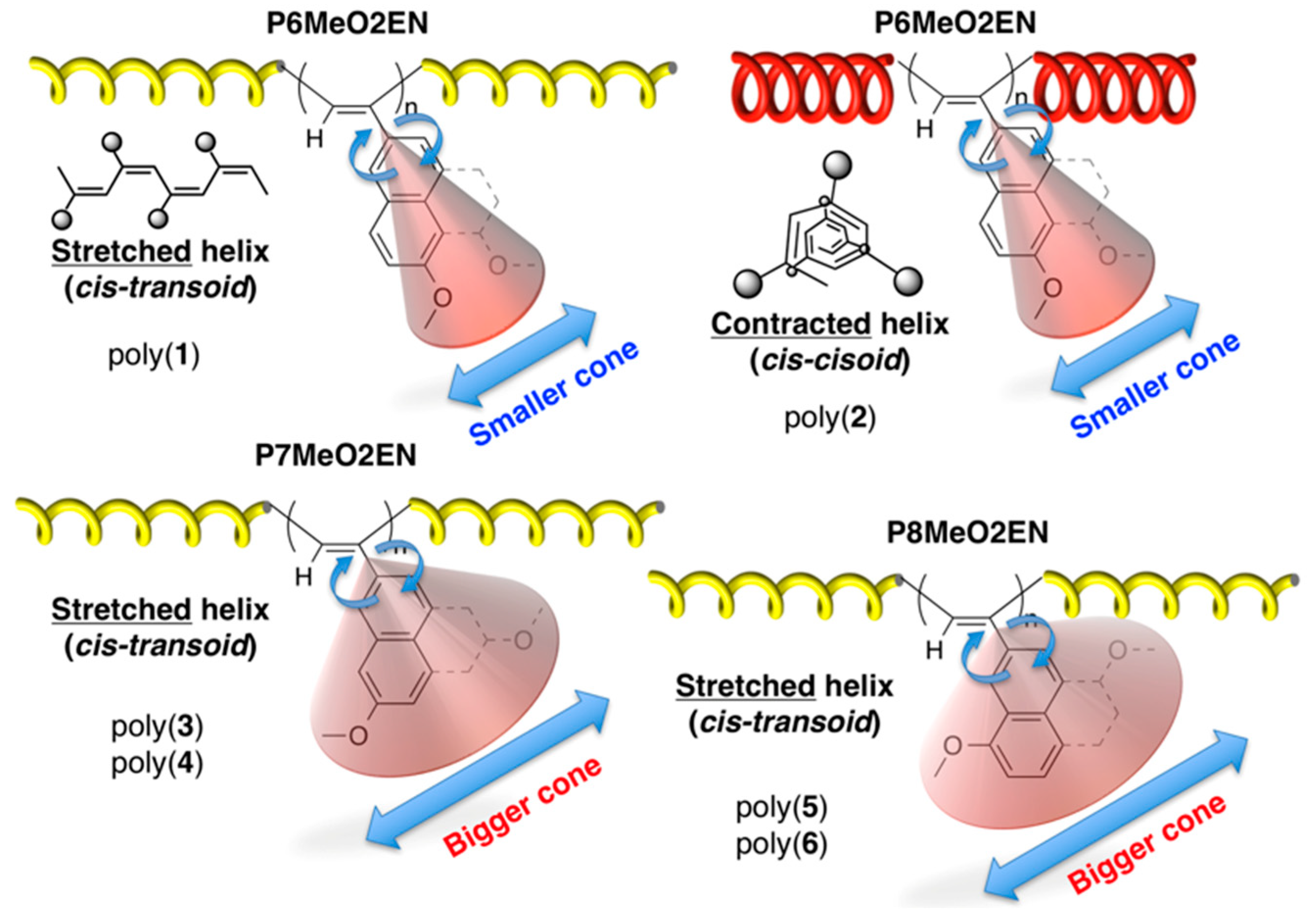
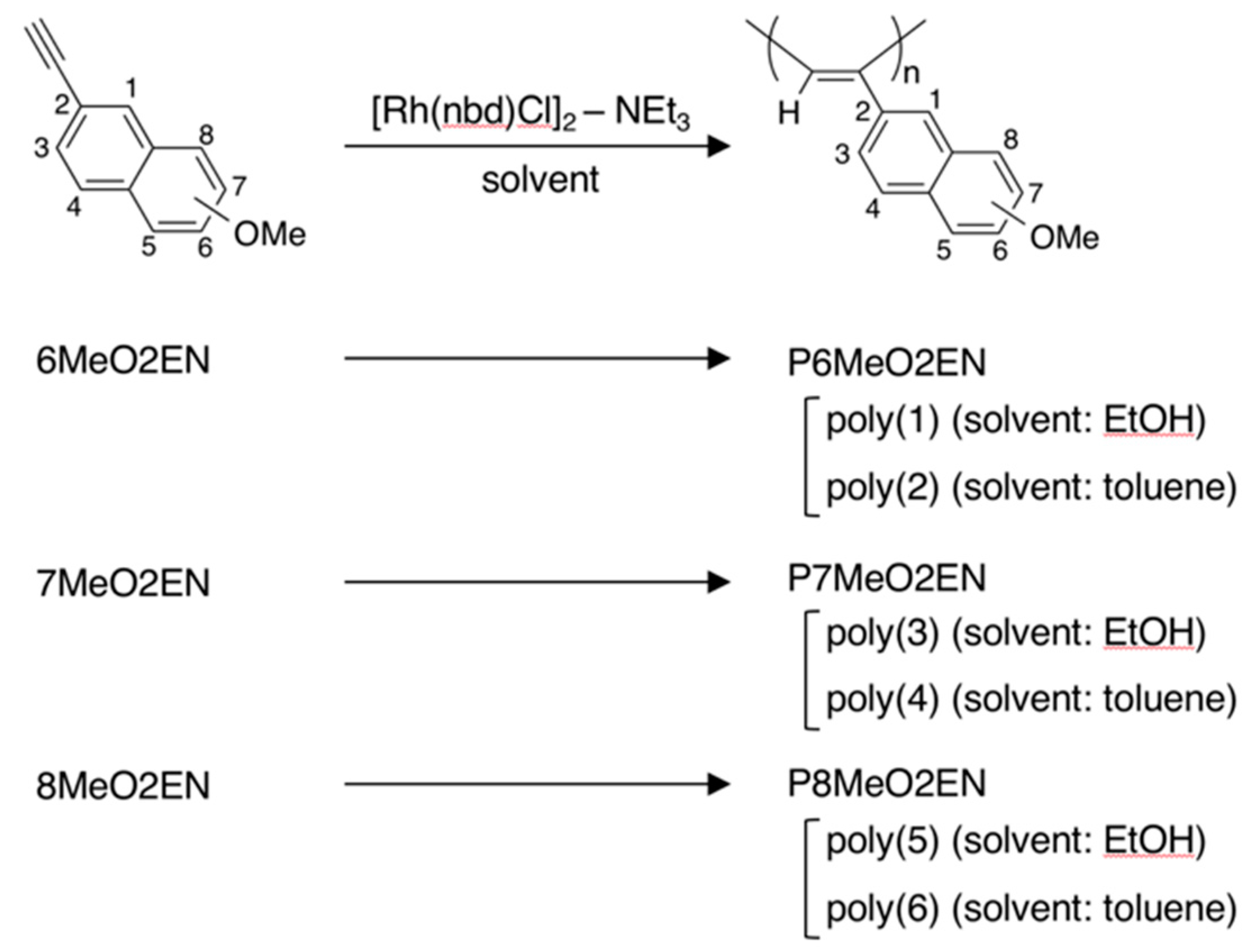
3.1. Synthesis of MeO-Substituted P2ENs (PnMeO2ENs)
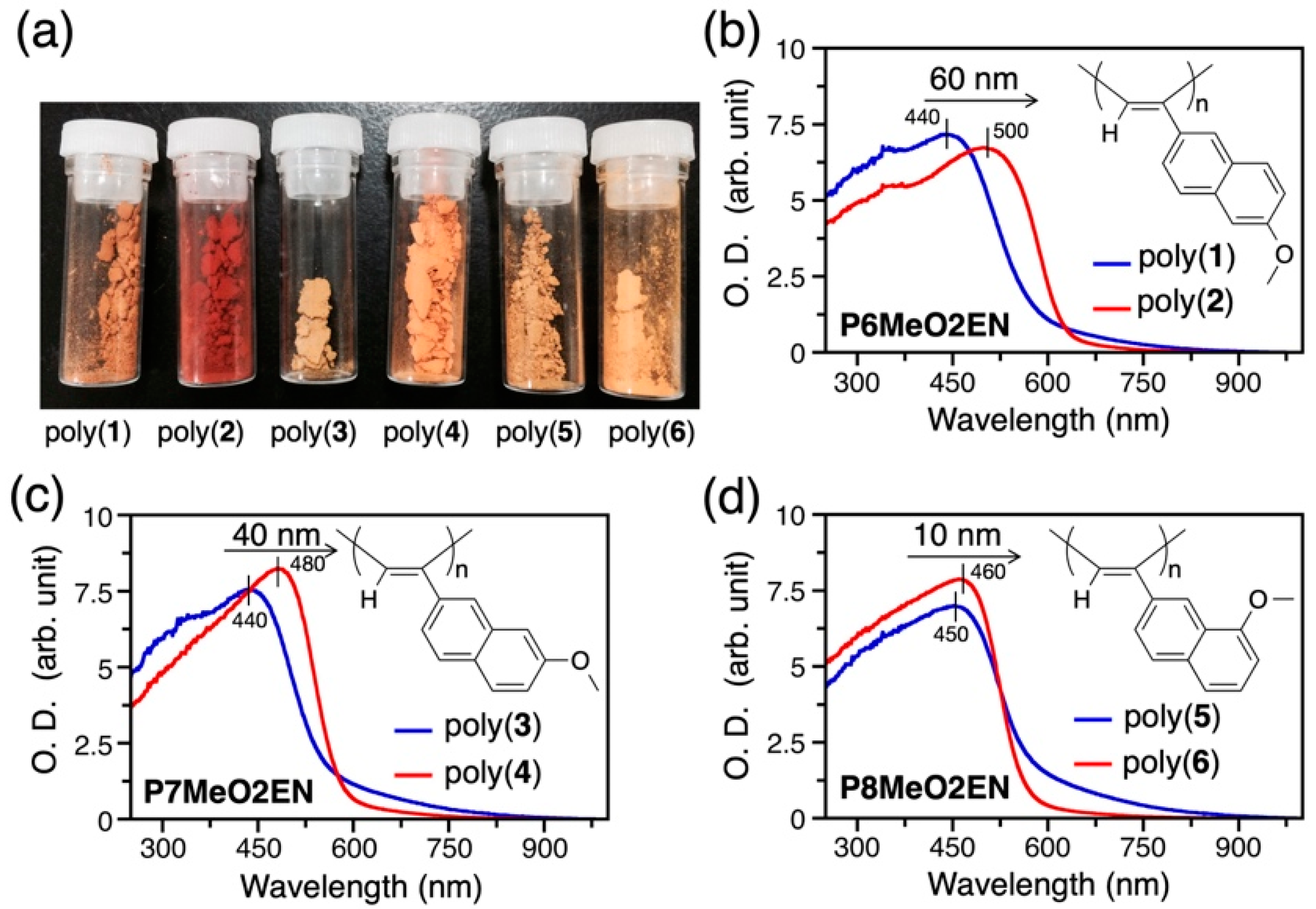
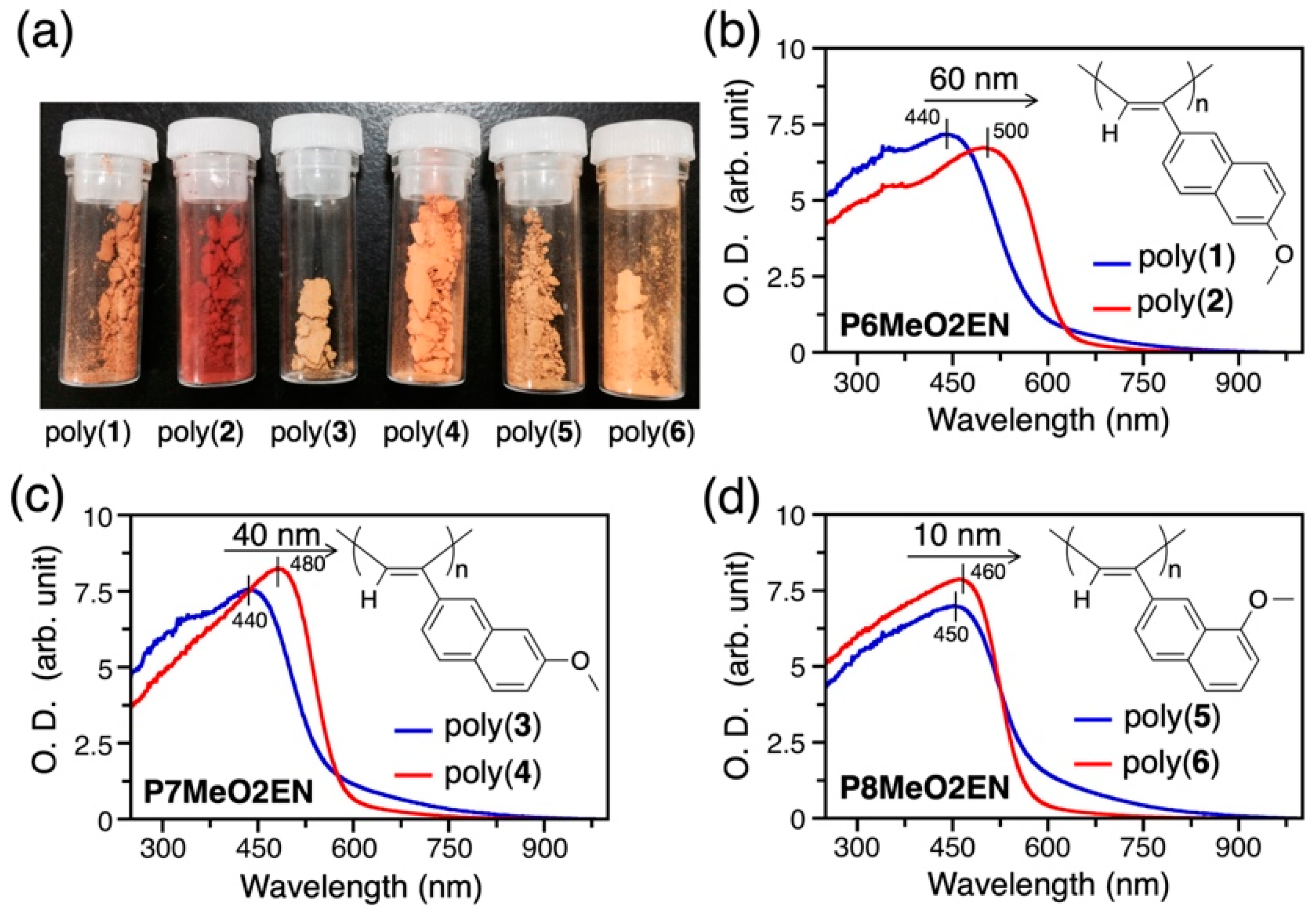
3.2. Difference in Color of Polymer Powders
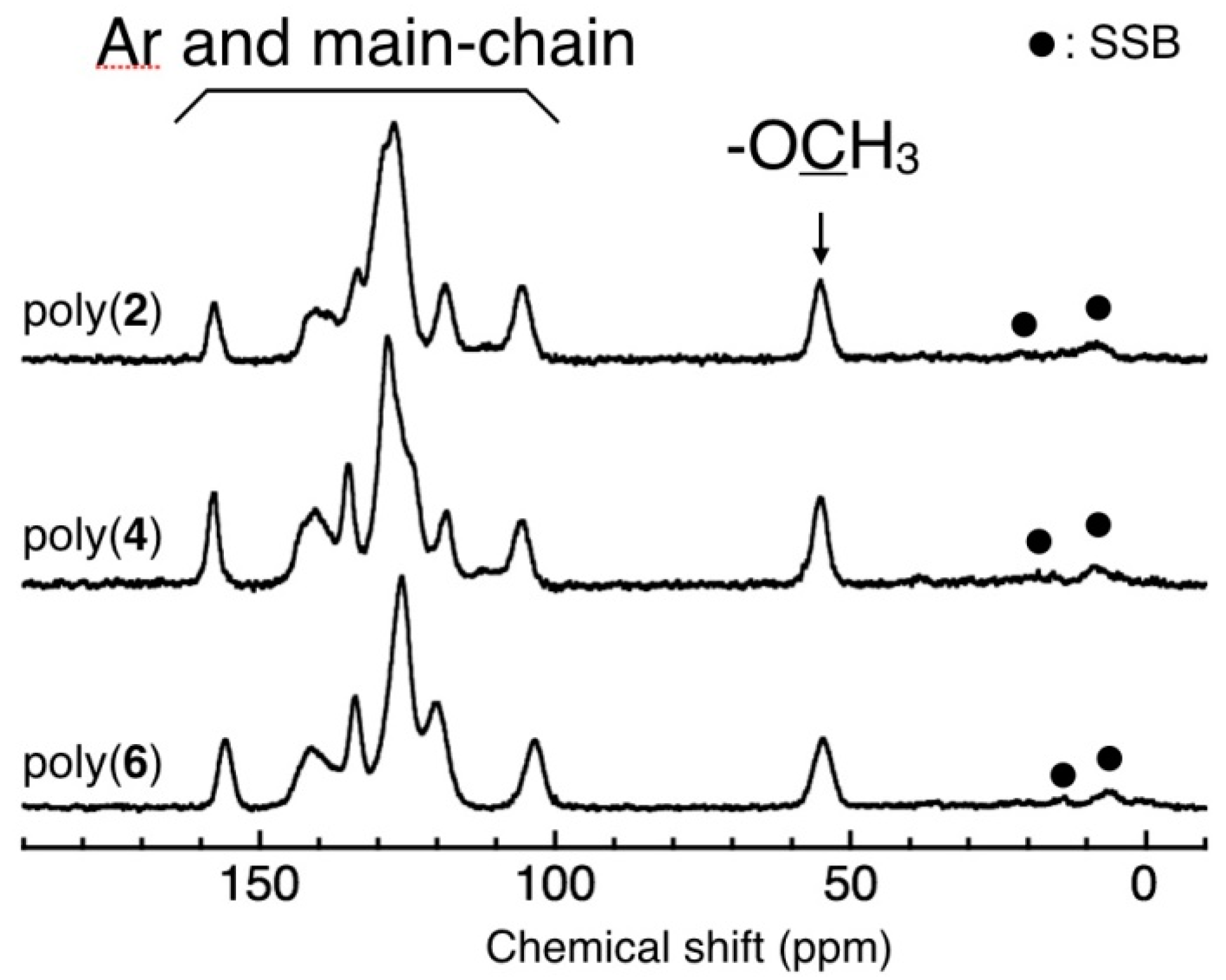
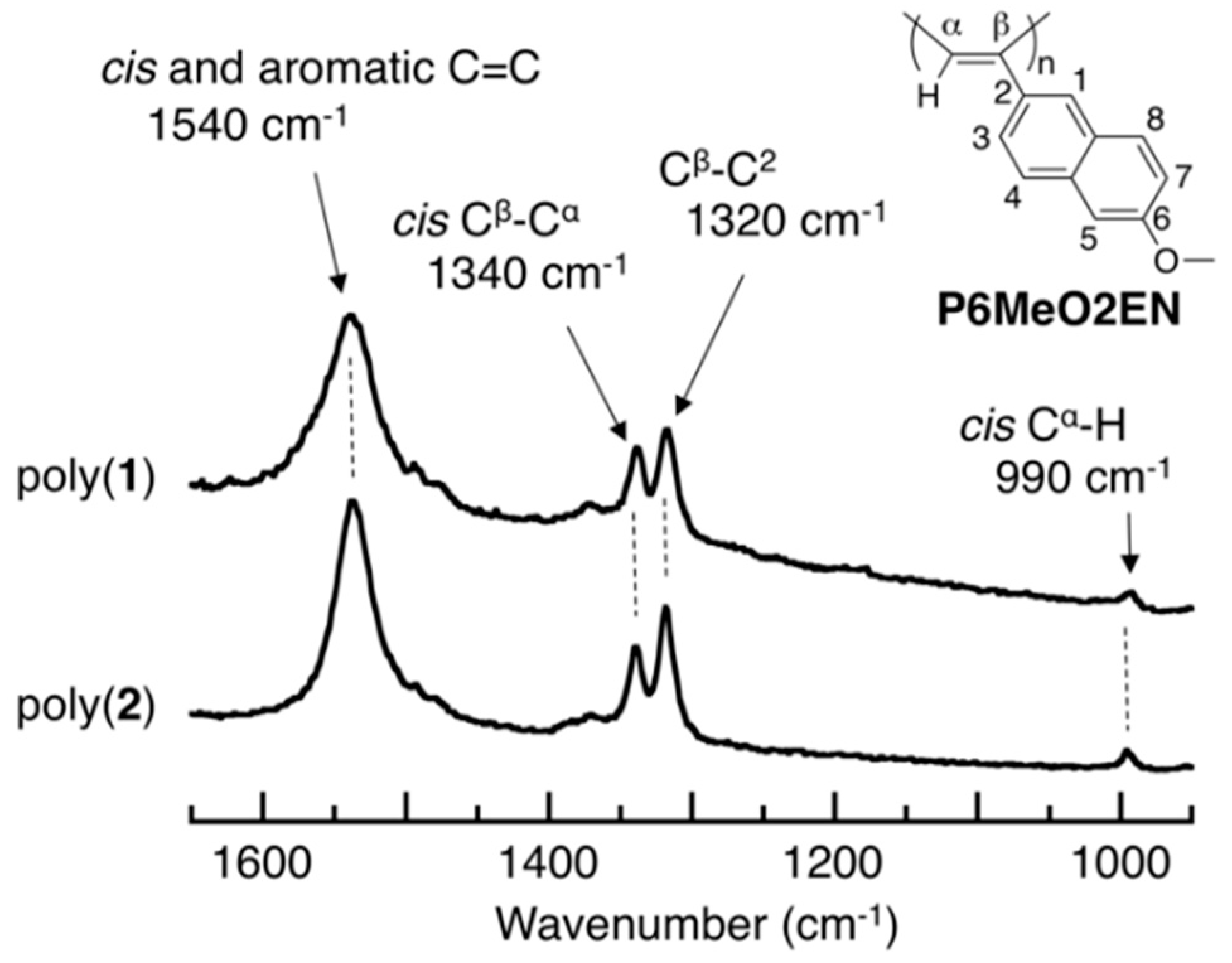
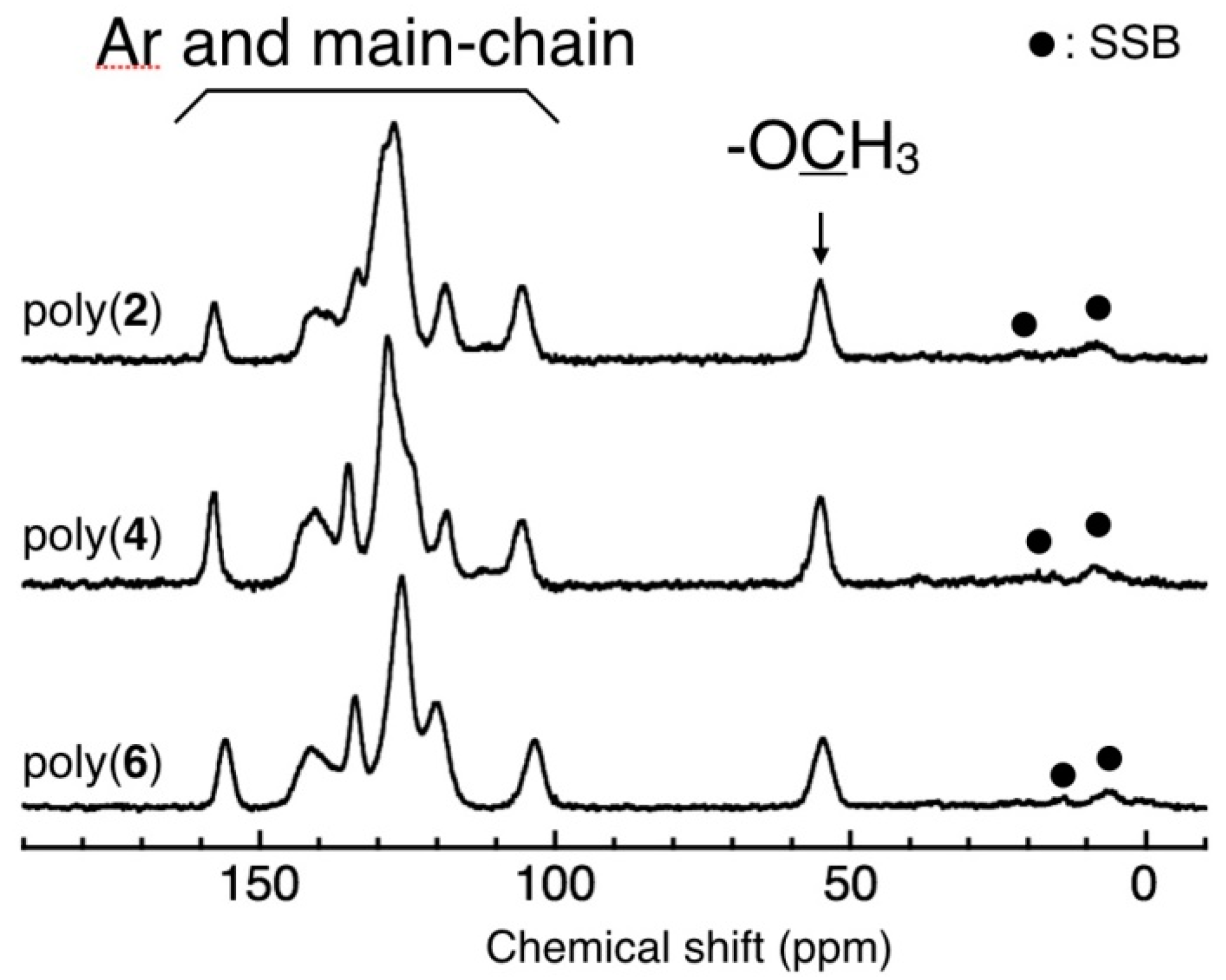
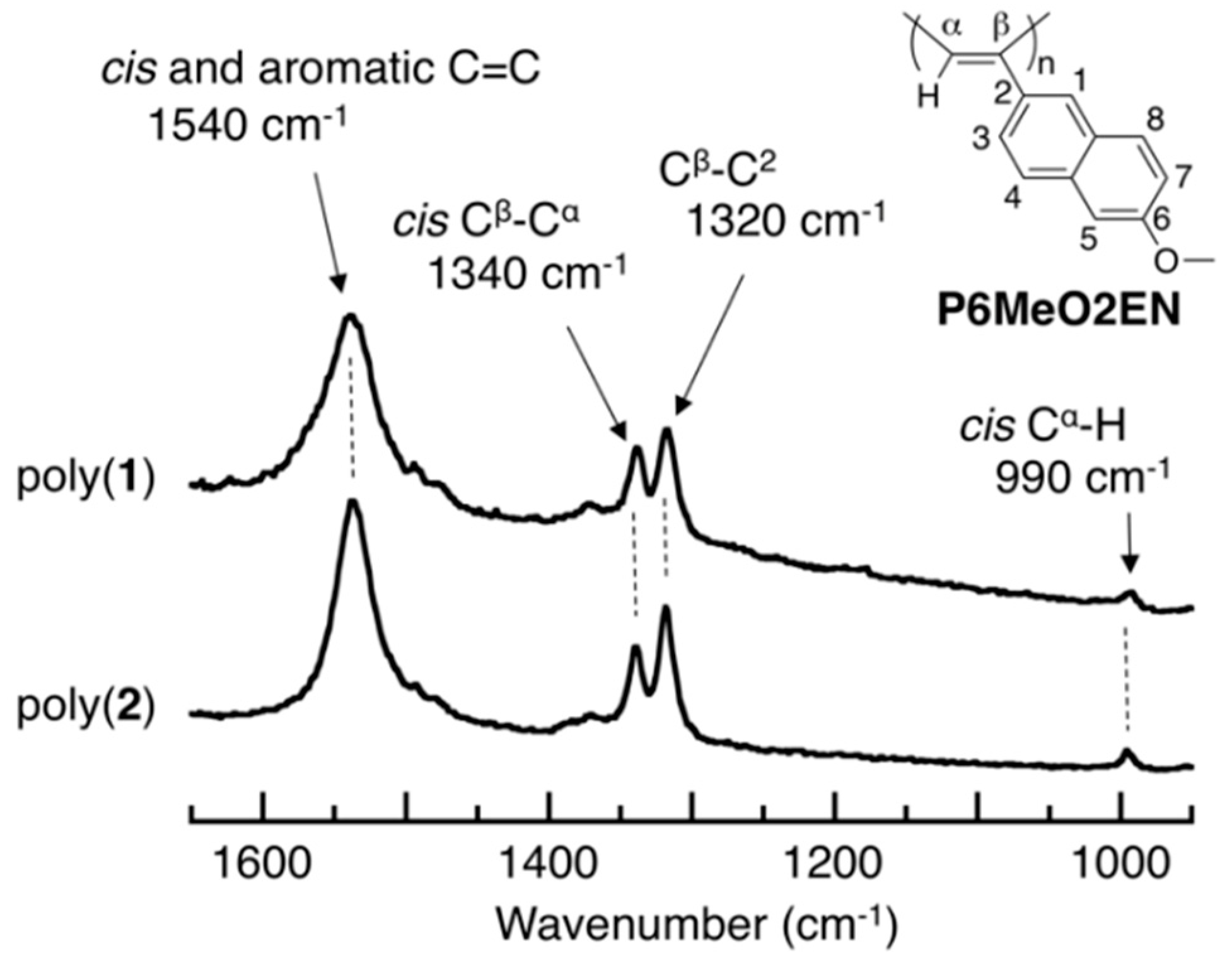
3.3. Main-Chain Structure
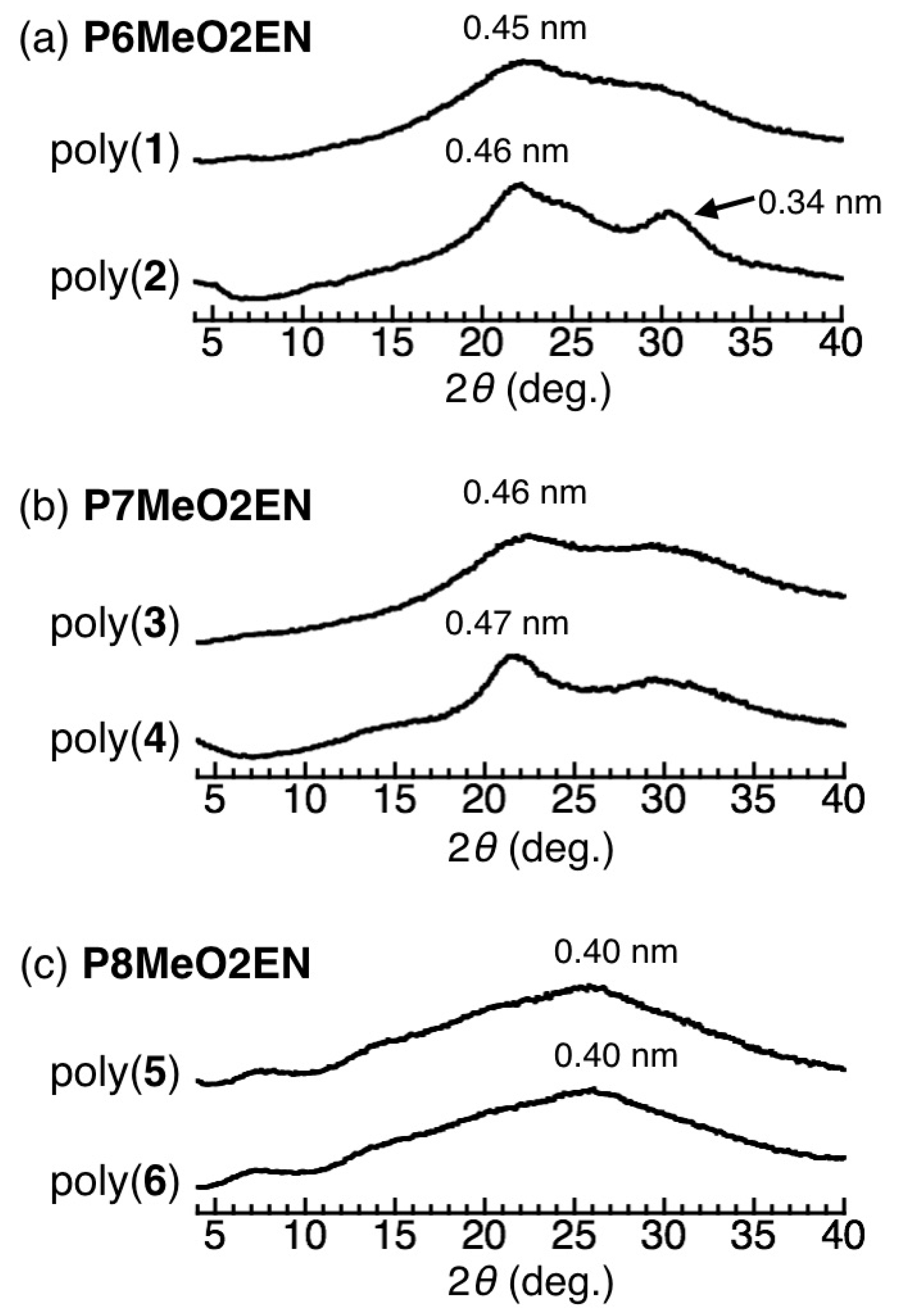
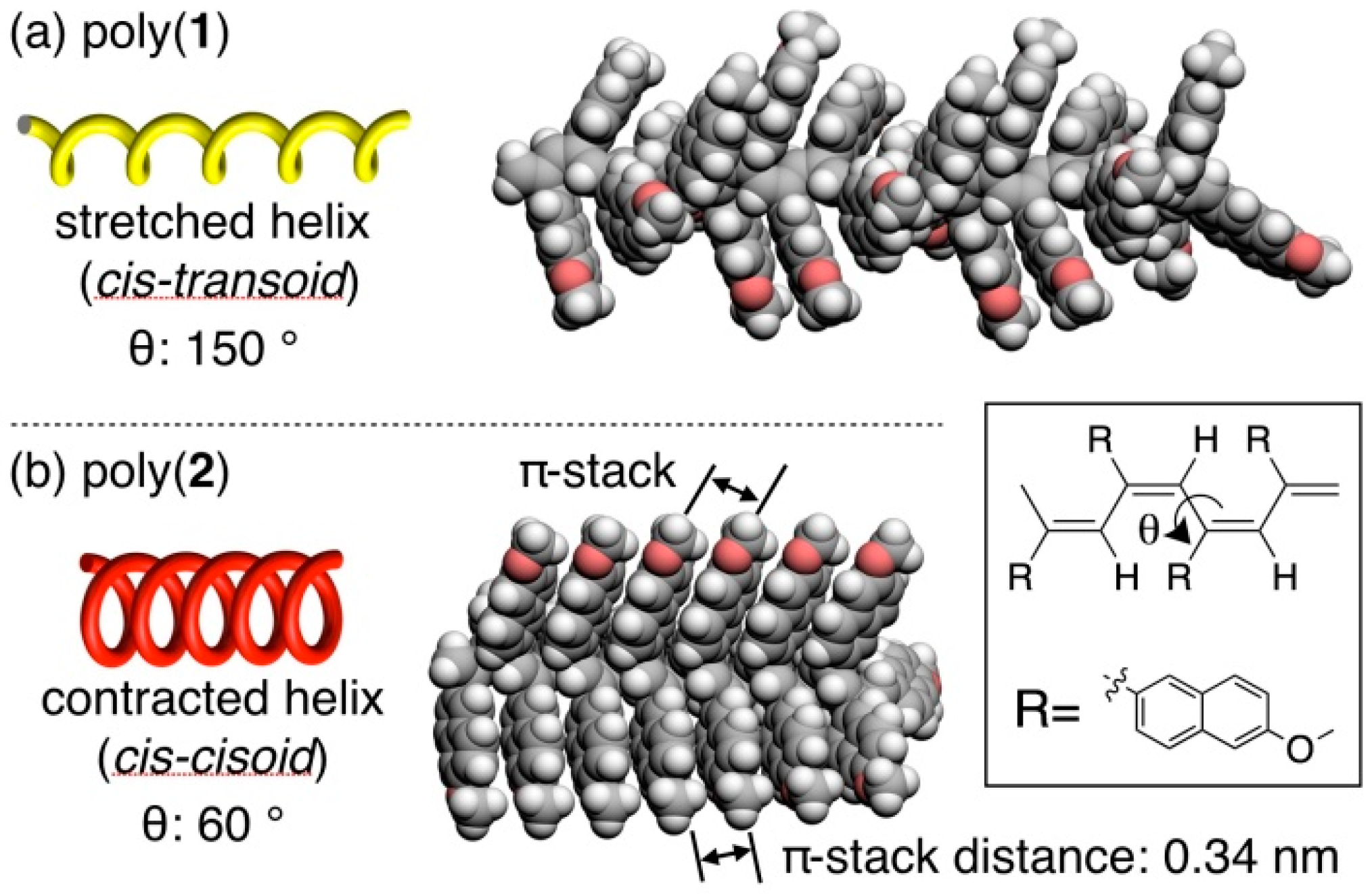
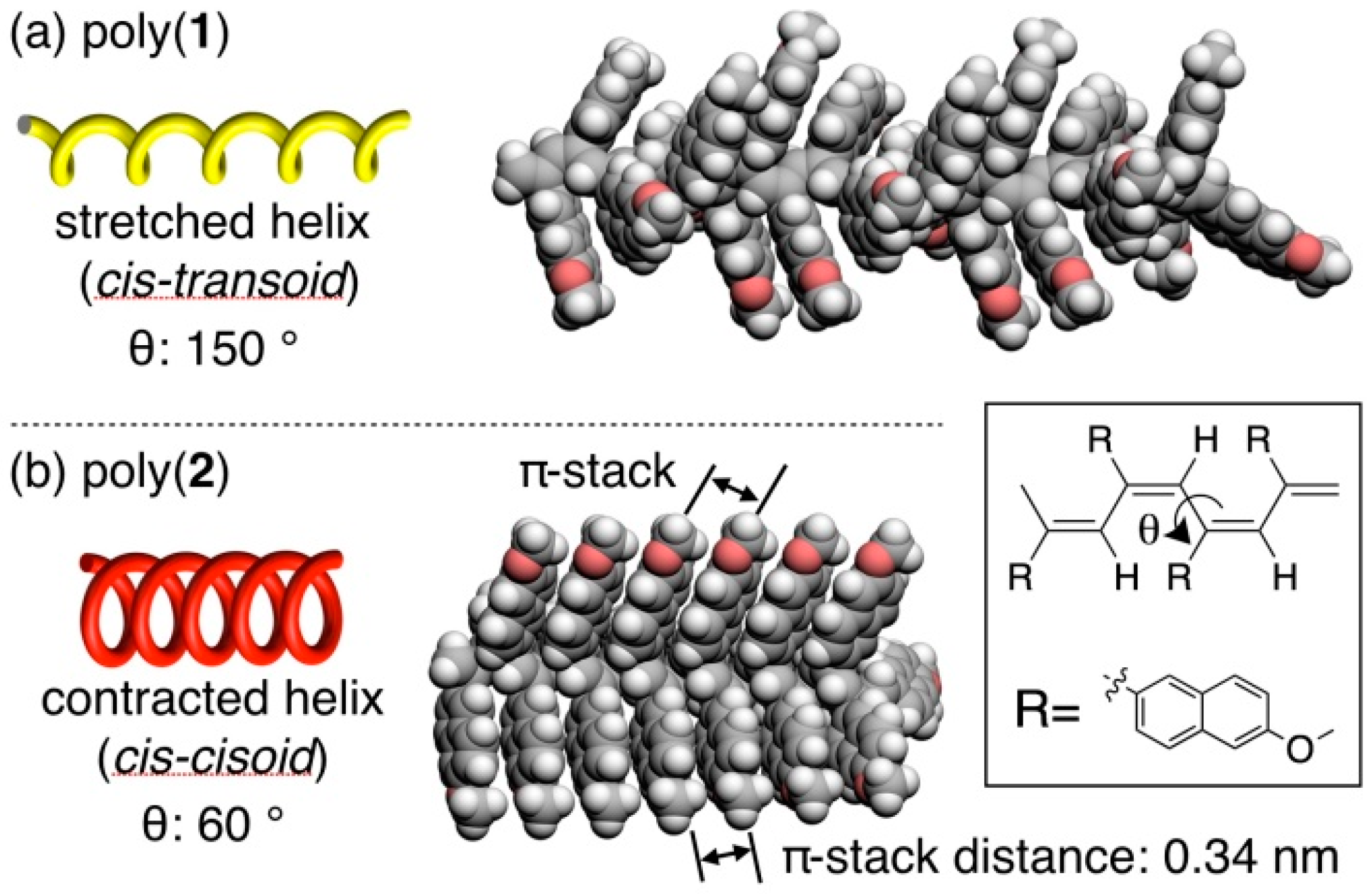
3.4. Secondary Structure
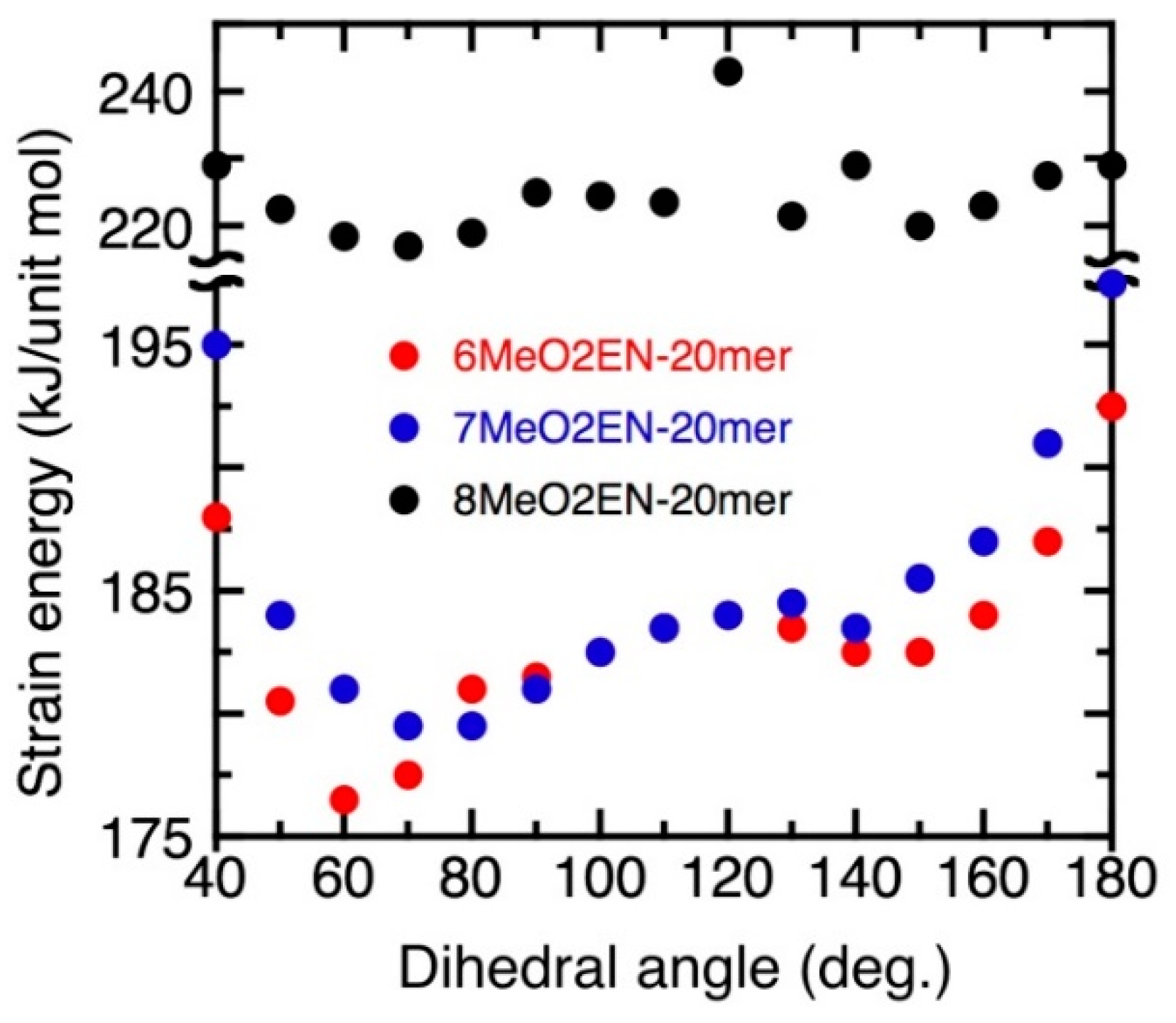
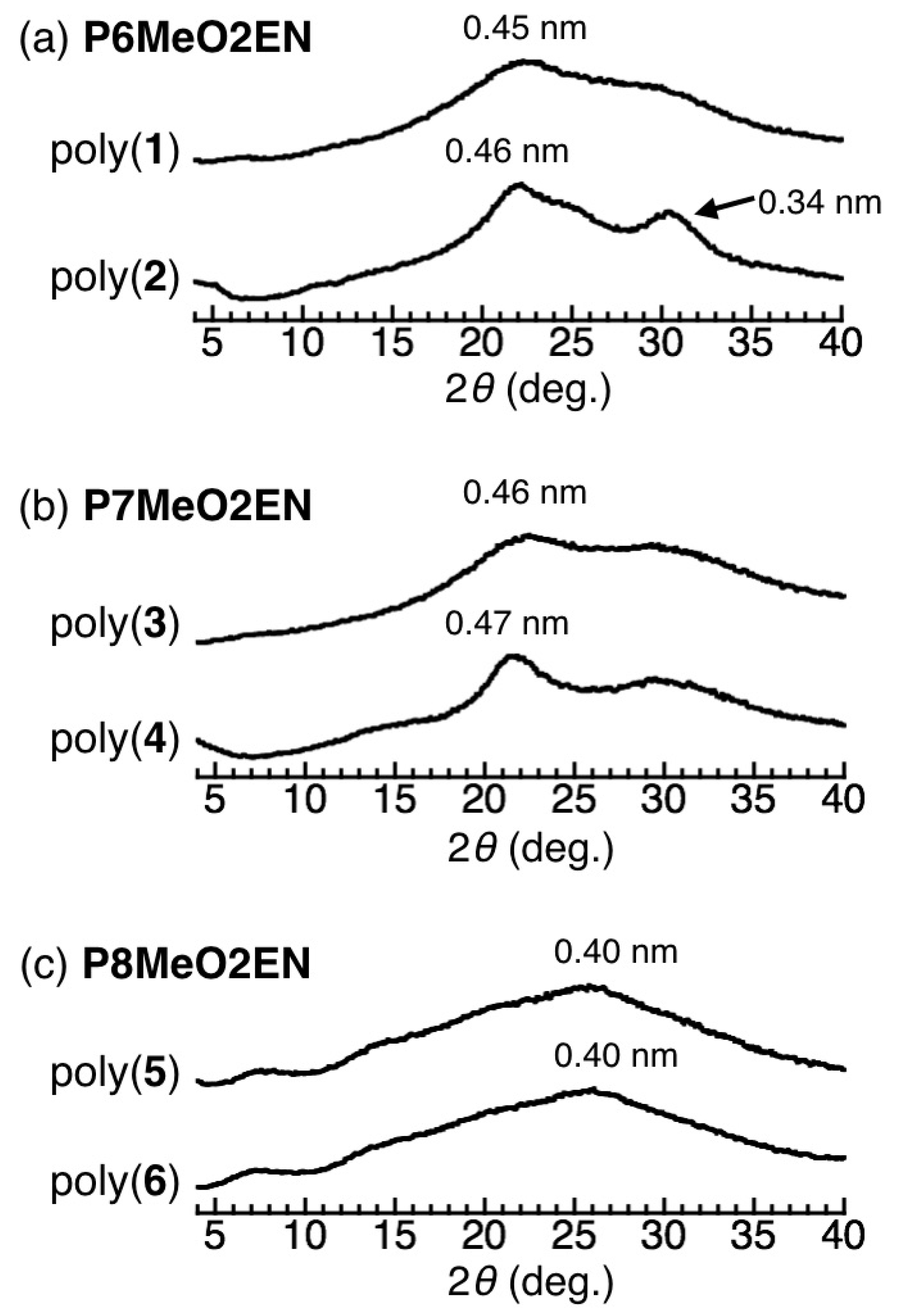
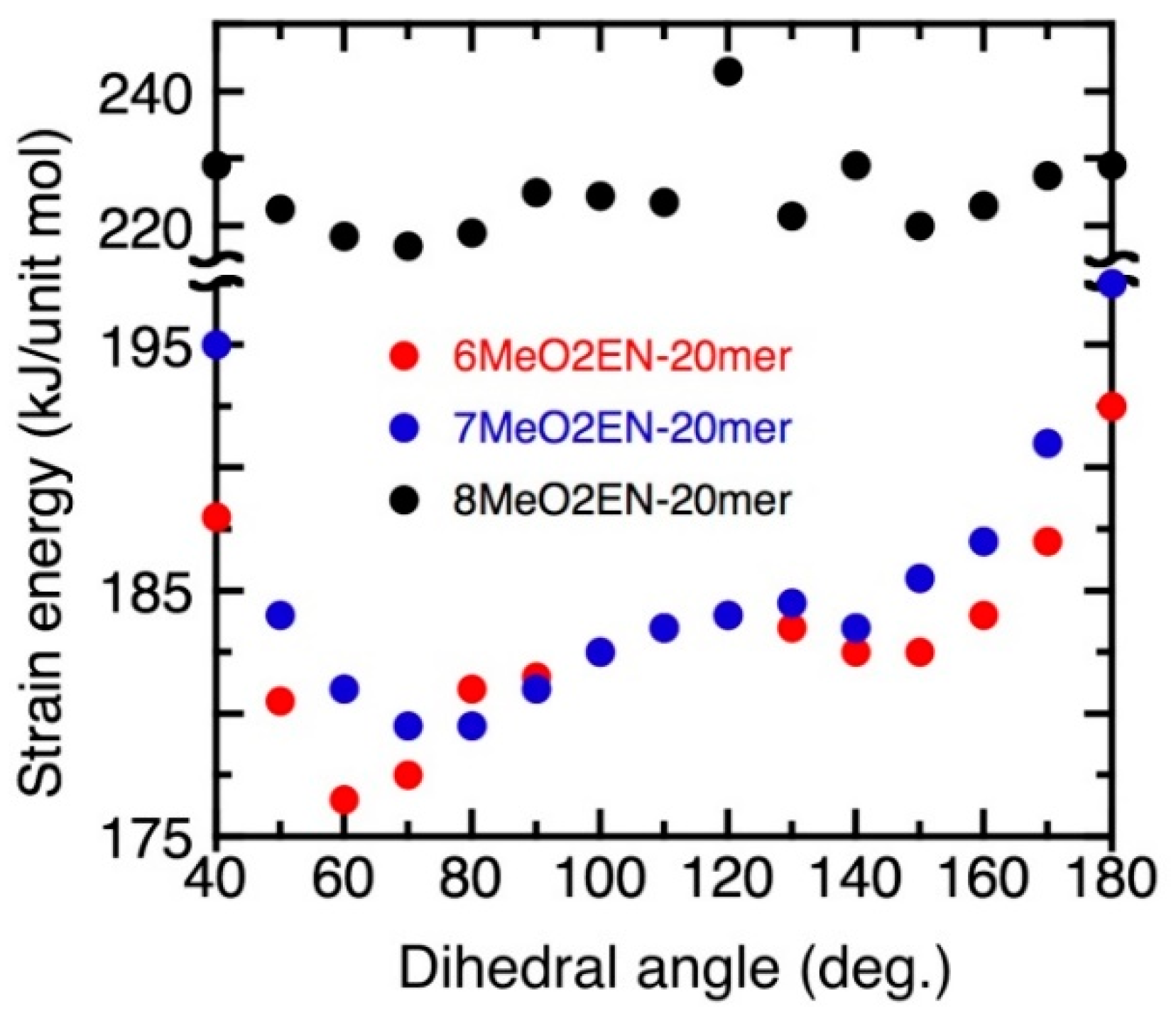
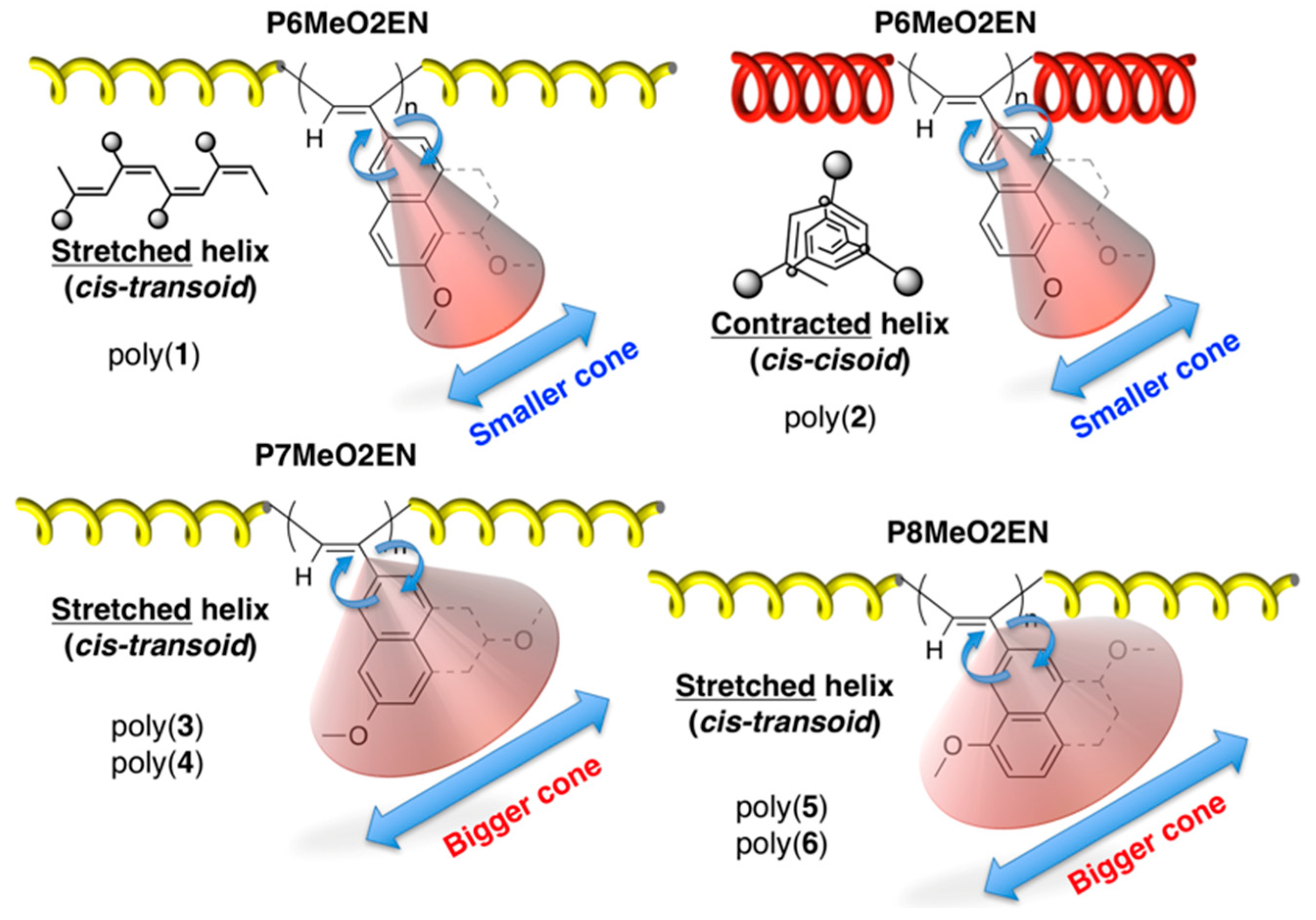
3.5. Helical Pitch Analysis
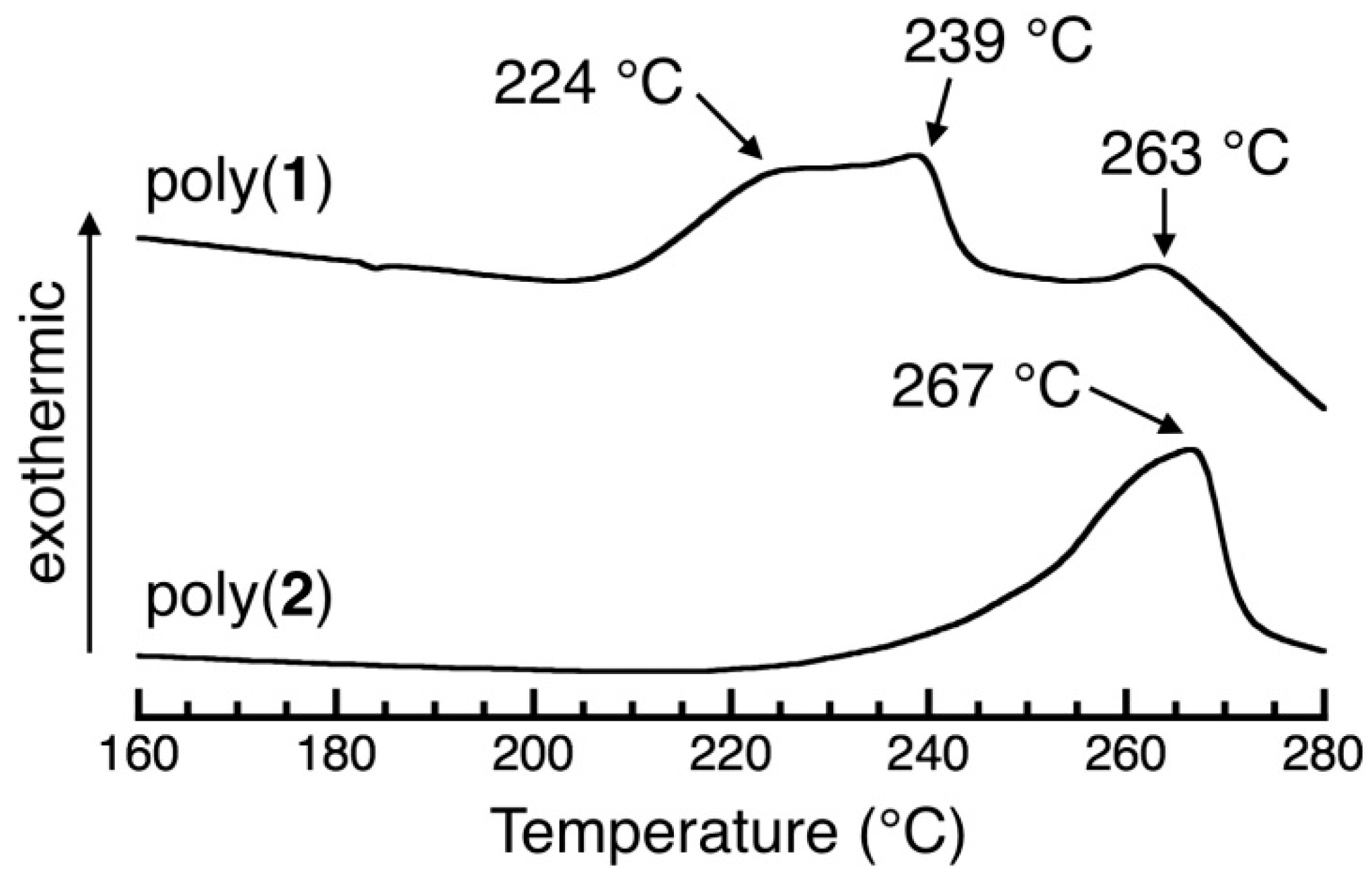
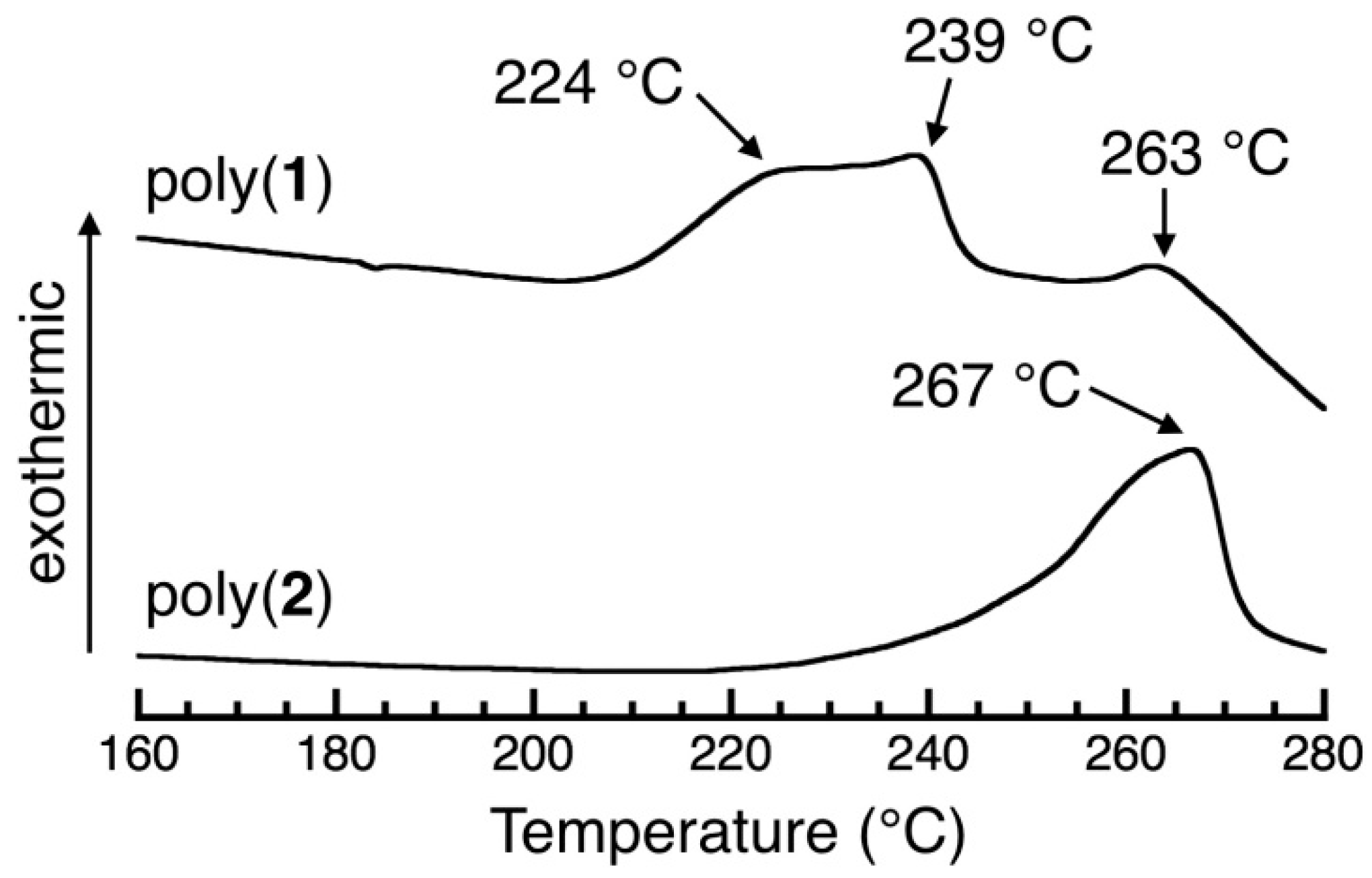
3.6. DSC Study
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Yashima, E.; Ousaka, N.; Taura, D.; Shimomura, K.; Ikai, T.; Maeda, K. Supramolecular Helical Systems: Helical Assemblies of Small Molecules, Foldamers, and Polymers with Chiral Amplification and Their Functions. Chem. Rev. 2016, 116, 13752–13990. [Google Scholar] [CrossRef]
- Yashima, E.; Maeda, K.; Iida, H.; Furusho, Y.; Nagai, K. Helical Polymers: Synthesis, Structures, and Functions. Chem. Rev. 2009, 109, 6102–6211. [Google Scholar] [CrossRef]
- Yashima, E.; Maeda, K. Chirality-Responsive Helical Polymers. Macromolecules 2008, 41, 3–12. [Google Scholar] [CrossRef]
- Yamada, T.; Nomura, K.; Fujiki, M. Noticeable Chiral Center Dependence of Signs and Magnitudes in Circular Dichroism (CD) and Circularly Polarized Luminescence (CPL) Spectra of all-trans -Poly(9,9-dialkylfluorene-2,7-vinylene)s Bearing Chiral Alkyl Side Chains in Solution, Aggregates, and Thin Films. Macromolecules 2018, 51, 2377–2387. [Google Scholar] [CrossRef]
- Nagata, Y.; Nishikawa, T.; Suginome, M.; Sato, S.; Sugiyama, M.; Porcar, L.; Martel, A.; Inoue, R.; Sato, N. Elucidating the Solvent Effect on the Switch of the Helicity of Poly(quinoxaline-2,3-diyl)s: A Conformational Analysis by Small-Angle Neutron Scattering. J. Am. Chem. Soc. 2018, 140, 2722–2726. [Google Scholar] [CrossRef]
- Jin, Y.-J.; Park, H.; Lee, C.-L.; Teraguchi, M.; Kaneko, T.; Aoki, T.; Kwak, G. Fluorescence emission and image patterning from selective photocyclic aromatization of cis–cisoid helical poly(phenylacetylene)s in situ in a film via top-down photodegradation. Dyes Pigment. 2018, 149, 444–448. [Google Scholar] [CrossRef]
- Xu, C.; Aoki, T.; Ma, L.; Jia, H.; Teraguchi, M.; Kaneko, T. Synthesis and Ultrahigh Oxygen Permeability of Silicon-containing cis-cisoidal Poly(substituted phenylacetylene)s. Chem. Lett. 2018, 47, 1314–1317. [Google Scholar] [CrossRef]
- Freire, F.; Quiñoá, E.; Riguera, R. Supramolecular Assemblies from Poly(phenylacetylene)s. Chem. Rev. 2016, 116, 1242–1271. [Google Scholar] [CrossRef]
- Mawatari, Y.; Tabata, M. Synthetic Molecular Springs: Stretched and Contracted Helices with Their Interconversions of Monosubstituted Polyacetylenes Prepared with a Rhodium Complex Catalyst. In Carbon-Related Materials in Recognition of Nobel Lectures by Prof. Akira Suzuki in ICCE; Springer International Publishing: Cham, Switzerland, 2017; pp. 305–326. [Google Scholar]
- Zhao, B.; Deng, J. Emulsion Polymerization of Acetylenics for Constructing Optically Active Helical Polymer Nanoparticles. Polym. Rev. 2017, 57, 119–137. [Google Scholar] [CrossRef]
- Xu, A.; Masuda, T.; Zhang, A. Stimuli-Responsive Polyacetylenes and Dendronized Poly(phenylacetylene)s. Polym. Rev. 2017, 57, 138–158. [Google Scholar] [CrossRef]
- Sedláček, J.; Balcar, H. Substituted Polyacetylenes Prepared with Rh Catalysts: From Linear to Network-Type Conjugated Polymers. Polym. Rev. 2017, 57, 31–51. [Google Scholar] [CrossRef]
- Liu, L.; Zang, Y.; Jia, H.; Aoki, T.; Kaneko, T.; Hadano, S.; Teraguchi, M.; Miyata, M.; Zhang, G.; Namikoshi, T. Helix-Sense-Selective Polymerization of Achiral Phenylacetylenes and Unique Properties of the Resulting Cis-cisoidal Polymers. Polym. Rev. 2017, 57, 89–118. [Google Scholar] [CrossRef]
- Sakai, R.; Satoh, T.; Kakuchi, T. Polyacetylenes as Colorimetric and Fluorescent Chemosensor for Anions. Polym. Rev. 2017, 57, 159–174. [Google Scholar] [CrossRef]
- Wang, S.; Shi, G.; Guan, X.; Zhang, J.; Wan, X. Cis-Cisoid Helical Structures of Poly(3,5-disubstituted phenylacetylene)s Stabilized by Intramolecular n → π* Interactions. Macromolecules 2018, 51, 1251–1259. [Google Scholar] [CrossRef]
- Tabata, M.; Yang, W.; Yokota, K. 1H-NMR and UV studies of Rh complexes as a stereoregular polymerization catalysts for phenylacetylenes: Effects of ligands and solvents on its catalyst activity. J. Polym. Sci. Part A Polym. Chem. 1994, 32, 1113–1120. [Google Scholar] [CrossRef]
- Tabata, M.; Sone, T.; Sadahiro, Y. Precise synthesis of monosubstituted polyacetylenes using Rh complex catalysts. Control of solid structure and π-conjugation length. Macromol. Chem. Phys. 1999, 200, 265–282. [Google Scholar] [CrossRef]
- Maeda, K.; Mochizuki, H.; Watanabe, M.; Yashima, E. Switching of Macromolecular Helicity of Optically Active Poly(phenylacetylene)s Bearing Cyclodextrin Pendants Induced by Various External Stimuli. J. Am. Chem. Soc. 2006, 128, 7639–7650. [Google Scholar] [CrossRef]
- Maeda, K.; Mochizuki, H.; Osato, K.; Yashima, E. Stimuli-Responsive Helical Poly(phenylacetylene)s Bearing Cyclodextrin Pendants that Exhibit Enantioselective Gelation in Response to Chirality of a Chiral Amine and Hierarchical Super-Structured Helix Formation. Macromolecules 2011, 44, 3217–3226. [Google Scholar] [CrossRef]
- Shimomura, K.; Ikai, T.; Kanoh, S.; Yashima, E.; Maeda, K. Switchable enantioseparation based on macromolecular memory of a helical polyacetylene in the solid state. Nat. Chem. 2014, 6, 429–434. [Google Scholar] [CrossRef]
- Wang, N.; Yano, K.; Durkan, C.; Plank, N.; Welland, M.E.; Zhang, Y.; Unalan, H.E.; Mann, M.; Amaratunga, G.A.J.; Milne, W.I. Direct measurement of charge transport through helical poly(ethyl propiolate) nanorods wired into gaps in single walled carbon nanotubes. Nanotechnology 2009, 20, 105201. [Google Scholar] [CrossRef]
- Motoshige, A.; Mawatari, Y.; Yoshida, Y.; Seki, C.; Matsuyama, H.; Tabata, M. Irreversible helix rearrangement from Cis-transoid to Cis-cisoid in poly(p-n-hexyloxyphenylacetylene) induced by heat-treatment in solid phase. J. Polym. Sci. Part A Polym. Chem. 2012, 50, 3008–3015. [Google Scholar] [CrossRef]
- Motoshige, A.; Mawatari, Y.; Yoshida, Y.; Motoshige, R.; Tabata, M. Synthesis and solid state helix to helix rearrangement of poly(phenylacetylene) bearing n-octyl alkyl side chains. Polym. Chem. 2014, 5, 971–978. [Google Scholar] [CrossRef]
- Motoshige, R.; Mawatari, Y.; Motoshige, A.; Yoshida, Y.; Sasaki, T.; Yoshimizu, H.; Suzuki, T.; Tsujita, Y.; Tabata, M. Mutual conversion between stretched and contracted helices accompanied by a drastic change in color and spatial structure of poly(phenylacetylene) prepared with a [Rh(nbd)Cl]2-amine catalyst. J. Polym. Sci. Part A Polym. Chem. 2014, 52, 752–759. [Google Scholar] [CrossRef]
- Mawatari, Y.; Motoshige, A.; Yoshida, Y.; Motoshige, R.; Sasaki, T.; Tabata, M. Structural determination of stretched helix and contracted helix having yellow and red colors of poly(2-ethynylnaphthalene) prepared with a [Rh(norbornadiene)Cl]2-triethylamine catalyst. Polymer 2014, 55, 2356–2361. [Google Scholar] [CrossRef]
- Yoshida, Y.; Mawatari, Y.; Motoshige, A.; Motoshige, R.; Hiraoki, T.; Wagner, M.; Müllen, K.; Tabata, M. Accordion-like oscillation of contracted and stretched helices of polyacetylenes synchronized with the restricted rotation of side chains. J. Am. Chem. Soc. 2013, 135, 4110–4116. [Google Scholar] [CrossRef]
- Yoshida, Y.; Mawatari, Y.; Motoshige, A.; Motodshige, R.; Hiraoki, T.; Tabata, M. Helix oscillation of polyacetylene esters detected by dynamic 1H NMR, IR, and UV-vis methods in solution. Polym. Chem. 2013, 4. [Google Scholar] [CrossRef]
- Tabata, M.; Sadahiro, Y.; Sone, T.; Yokota, K.; Ishikawa, Y. Unusually facilcis totrans isomerization of a polypropiolate bearing a long alkyl chain polymerized using a [Rh(norbornadiene)Cl]2 catalyst: An ESR study. J. Polym. Sci. Part A Polym. Chem. 1998, 36, 2457–2461. [Google Scholar] [CrossRef]
- Huang, K.; Mawatari, Y.; Miyasaka, A.; Sadahiro, Y.; Tabata, M.; Kashiwaya, Y. Generation of polyacetylene sulfoxide radicals through spin migration from the main-chain to the sulfoxide moiety in the side chain of poly[p-(n-butylsulfoxide)phenylacetylene] prepared with a [Rh(norbornadiene)Cl]2 catalyst. Polymer 2007, 48, 6366–6373. [Google Scholar] [CrossRef]
- Miyasaka, A.; Sone, T.; Mawatari, Y.; Setayesh, S.; Müllen, K.; Tabata, M. Poly[(para-halogenated)phenylacetylene]s prepared with a [Rh(norbornadiene)Cl]2 catalyst: Syntheses and structure elucidation. Macromol. Chem. Phys. 2006, 207, 1938–1944. [Google Scholar] [CrossRef]
- Mawatari, Y.; Tabata, M.; Sone, T.; Ito, K.; Sadahiro, Y. Origin of color of π-conjugated columnar polymers. 1. Poly(p-3-methylbutoxy)phenylacetylene prepared using a [Rh(norbornadiene)Cl]2 catalyst. Macromolecules 2001, 34. [Google Scholar] [CrossRef]
- Kozuka, M.; Sone, T.; Tabata, M.; Sadahiro, Y.; Enoto, T. Radiation-induced Cis to Trans isomerization of poly(n-butylpropiolate) prepared with a [Rh(norbornadiene)Cl]2 complex as a stereospecific catalyst. Radiat. Phys. Chem. 2002, 63, 59–61. [Google Scholar] [CrossRef]
- Nakamura, M.; Tabata, M.; Sone, T.; Mawatari, Y.; Miyasaka, A. Photoinduced cis-to-trans isomerization of poly(2-ethynylthiophene) prepared with a [Rh(norbornadiene)Cl]2 catalyst. 1H NMR, UV, and ESR studies. Macromolecules 2002, 35, 2000–2004. [Google Scholar] [CrossRef]
- Percec, V.; Rudick, J.G.; Peterca, M.; Aqad, E.; Imam, M.R.; Heiney, P.A. Synthesis, structural, and retrostructural analysis of helical dendronized poly(1-naphthylacetylene)s. J. Polym. Sci. Part A Polym. Chem. 2007, 45, 4974–4987. [Google Scholar] [CrossRef]
- Rodríguez, R.; Quiñoá, E.; Riguera, R.; Freire, F. Architecture of Chiral Poly(phenylacetylene)s: From Compressed/Highly Dynamic to Stretched/Quasi-Static Helices. J. Am. Chem. Soc. 2016, 138, 9620–9628. [Google Scholar] [CrossRef]
- Peebles, C.; Alvey, P.M.; Lynch, V.; Iverson, B.L. Time-Dependent Solid-State Polymorphism of a Series of Donor–Acceptor Dyads. Cryst. Growth Des. 2014, 14, 290–299. [Google Scholar] [CrossRef]
- Tabata, M.; Inaba, Y.; Yokota, K.; Nozaki, Y. Stereoregular Polymerization of Alkyl Propiolate Catalyzed by Rh Complex. J. Macromol. Sci. Part A 1994, 31, 465–475. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Sone, T.; Asako, R.; Masuda, T.; Tabata, M.; Wada, T.; Sasabe, H. Polymerization of o-Trifluoromethyl(phenylacetylene) Initiated by [Rh(norbornadiene)Cl]2 and MoOCl4−n-Bu4Sn−EtOH Catalysts. Formation of Order and Disorder Trans Sequences. Macromolecules 2001, 34, 1586–1592. [Google Scholar] [CrossRef]
- Tabata, M.; Yokota, K.; Namioka, M. An electron spin resonance study of poly(α-ethynylnaphthalene) polymerized with [Rh(norbornadiene)Cl]2 and WCl6 as catalysts. Macromol. Chem. Phys. 1995, 196, 2969–2977. [Google Scholar] [CrossRef]
- Tabata, M.; Namioka, M.; Yokota, K.; Minakawa, H. Structural differences of poly(α-ethynylnaphthalene)s obtained with [Rh(norbornadiene)Cl]2 and WCl6 catalysts: An electron spin resonance and Raman study. Polymer 1996, 37, 1959–1963. [Google Scholar] [CrossRef]
- Minakawa, H.; Tabata, M.; Yokota, K. Structural Differences between Polypentynoates Bearing Mesogenic Moieties Polymerized with Rh Complex and WCl6 Catalysts. A 13C-NMR and Raman Study. J. Macromol. Sci. Part A 1996, 33, 291–303. [Google Scholar] [CrossRef]
- Sone, T.; D’Amato, R.; Mawatari, Y.; Tabata, M.; Furlani, A.; Russo, M.V. Copolymers of phenylacetylene and para-nitrophenylacetylene with nonlinear optical properties: Further insight on the conformational structure. J. Polym. Sci. Part A Polym. Chem. 2004, 42, 2365–2376. [Google Scholar] [CrossRef]
- Tabata, M.; Sone, T.; Sadahiro, Y.; Yokota, K. Pressure-induced cis to trans isomerization of poly(ortho- and para-methoxyphenylacetylene)s prepared by [Rh(norbornadiene)Cl]2 catalyst. A Raman, UV, and ESR study. Macromol. Chem. Phys. 1998, 199, 1161–1166. [Google Scholar] [CrossRef]
- Tabata, M.; Mawatari, Y. Emerging π-Conjugated Stretched and Contracted Helices and their Mutual Conversions of Substituted Polyacetylenes Prepared using an Organo-rhodium Catalyst. Polym. Rev. 2017, 57, 65–88. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Monomer | Polymer | Polymerization Solvent | Yield (%) b | λmax(nm) c |
|---|---|---|---|---|
| 6MeO2EN | Poly(1) | Ethanol (EtOH) | 60 | 440 |
| 6MeO2EN | Poly(2) | Toluene | 74 | 500 |
| 7MeO2EN | Poly(3) | EtOH | 54 | 440 |
| 7MeO2EN | Poly(4) | Toluene | 91 | 480 |
| 8MeO2EN | Poly(5) | EtOH | 70 | 450 |
| 8MeO2EN | Poly(6) | Toluene | 98 | 460 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mawatari, Y.; Yoshida, Y.; Huang, K.; Tabata, M. Methoxy-Group Control of Helical Pitch in Stereoregular Poly(2-ethynylmethoxynaphthalene) Prepared by Rhodium Complex Catalyst. Polymers 2019, 11, 94. https://doi.org/10.3390/polym11010094
Mawatari Y, Yoshida Y, Huang K, Tabata M. Methoxy-Group Control of Helical Pitch in Stereoregular Poly(2-ethynylmethoxynaphthalene) Prepared by Rhodium Complex Catalyst. Polymers. 2019; 11(1):94. https://doi.org/10.3390/polym11010094
Chicago/Turabian StyleMawatari, Yasuteru, Yoshiaki Yoshida, Kai Huang, and Masayoshi Tabata. 2019. "Methoxy-Group Control of Helical Pitch in Stereoregular Poly(2-ethynylmethoxynaphthalene) Prepared by Rhodium Complex Catalyst" Polymers 11, no. 1: 94. https://doi.org/10.3390/polym11010094
APA StyleMawatari, Y., Yoshida, Y., Huang, K., & Tabata, M. (2019). Methoxy-Group Control of Helical Pitch in Stereoregular Poly(2-ethynylmethoxynaphthalene) Prepared by Rhodium Complex Catalyst. Polymers, 11(1), 94. https://doi.org/10.3390/polym11010094