Synthesis and in Vitro Cytocompatibility of Segmented Poly(Ester-Urethane)s and Poly(Ester-Urea-Urethane)s for Bone Tissue Engineering

 ,
,  , ,
, ,
Abstract
:
1. Introduction.
2. Experimental
2.1. Materials
2.2. Synthesis of Polyurethanes
2.3. Preparation of Polyurethane Films
2.4. Characterization Techniques
2.4.1. Size Exclusion Chromatography (SEC)
2.4.2. Proton Nuclear Magnetic Resonance (1H-NMR)
2.4.3. Infrared Spectroscopy (FT-IR)
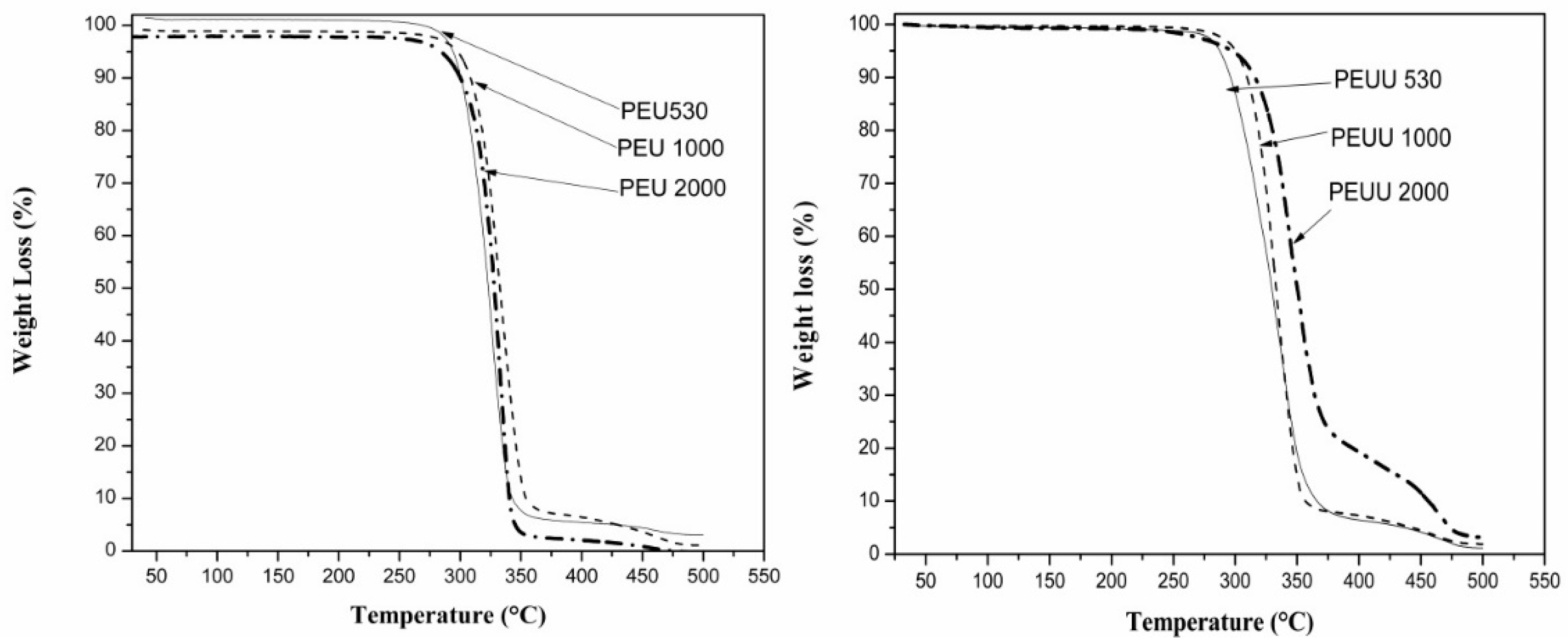
2.4.4. Thermogravimetric Analysis (TGA)
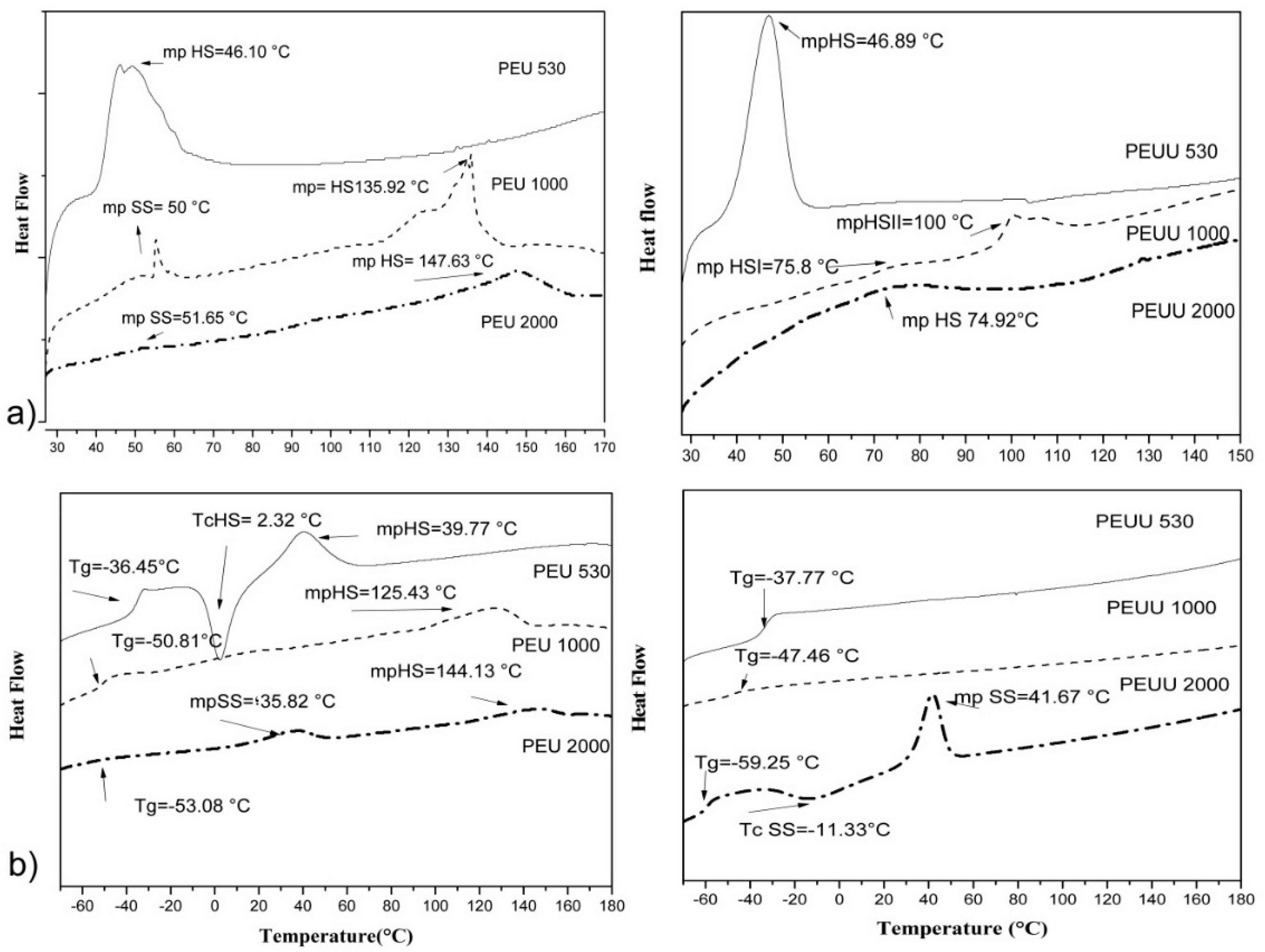
2.4.5. Differential Scanning Calorimetry (DSC)
2.4.6. X-ray Diffraction (XRD)
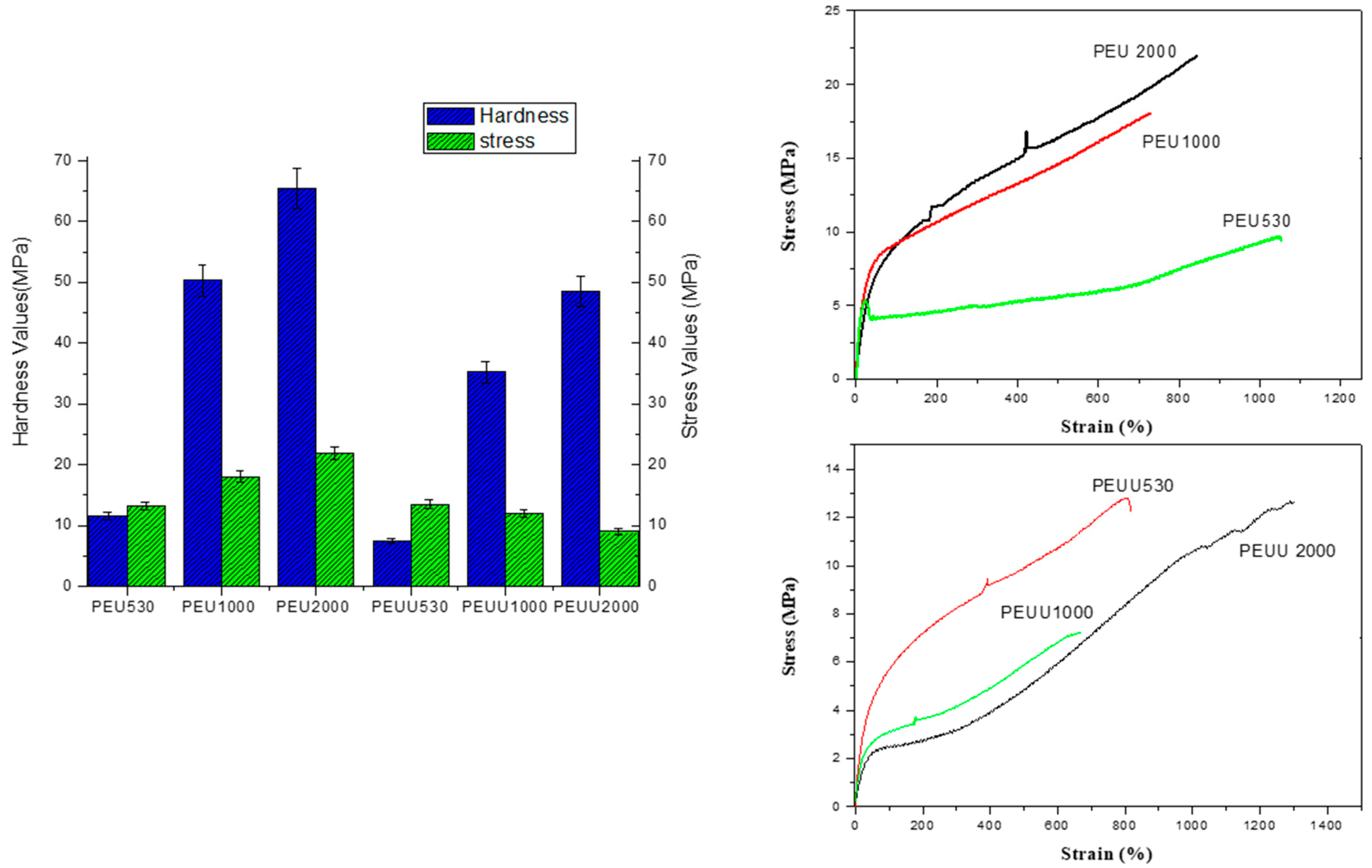
2.4.7. Mechanical Properties
2.4.8. Scanning Electron Microscopy (SEM)
2.5. In Vitro Test
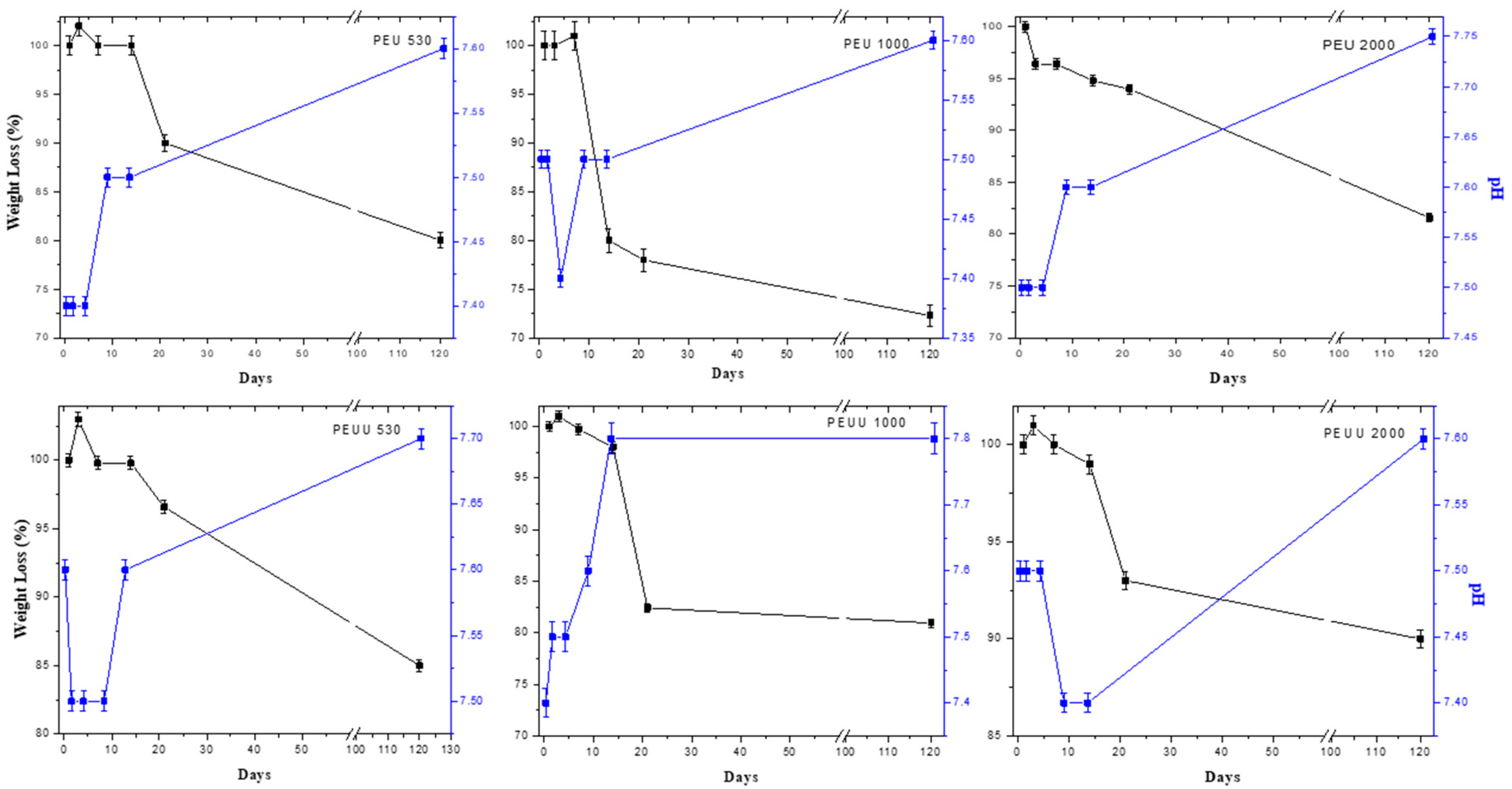
2.5.1. Hydrolitic Degradation
2.5.2. Alamar Blue Test
2.5.3. Statistical Analysis of Biocompatibility
3. Results
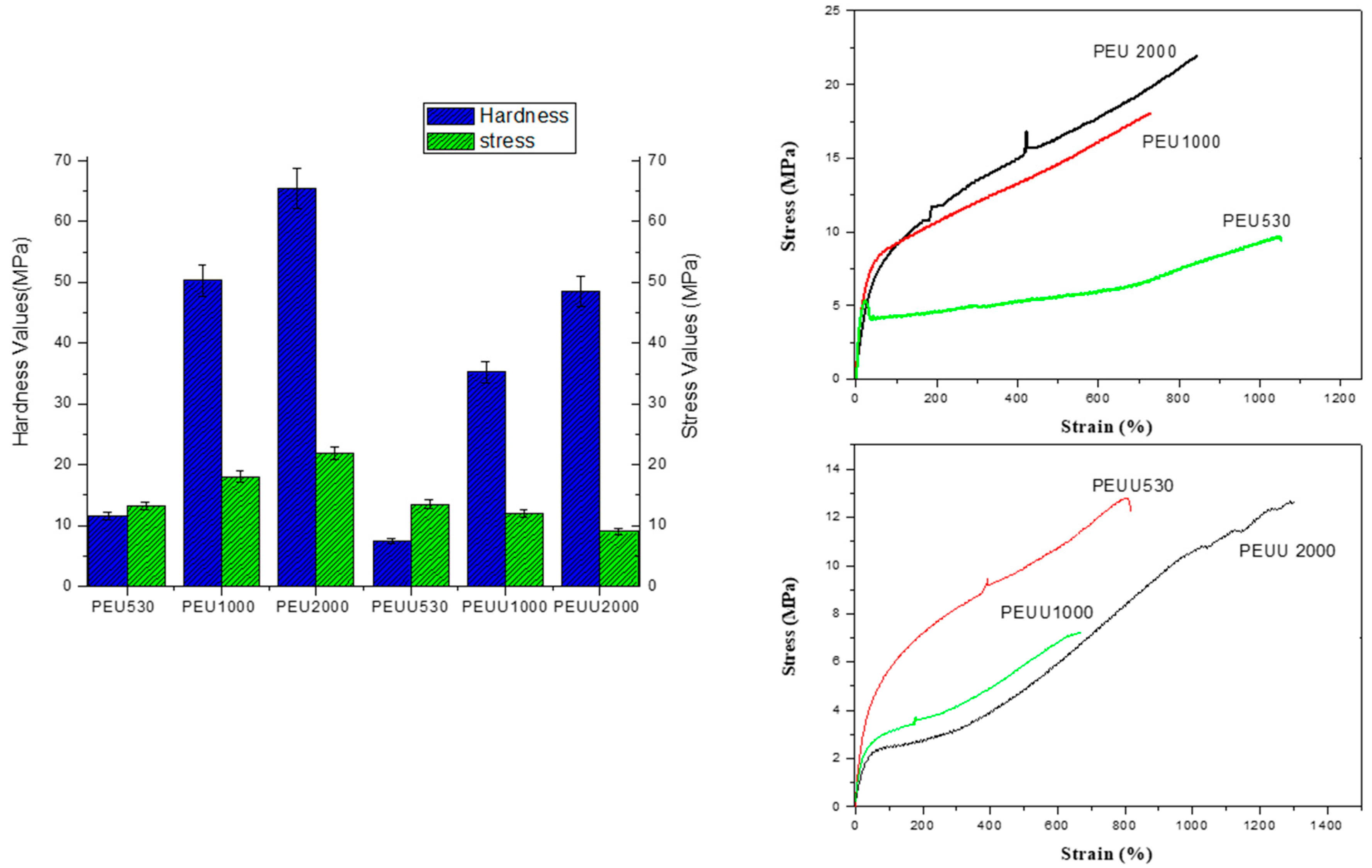
3.1. PEUs and PEUUs Synthesis
3.2. Biodegradability and Surface Wettability
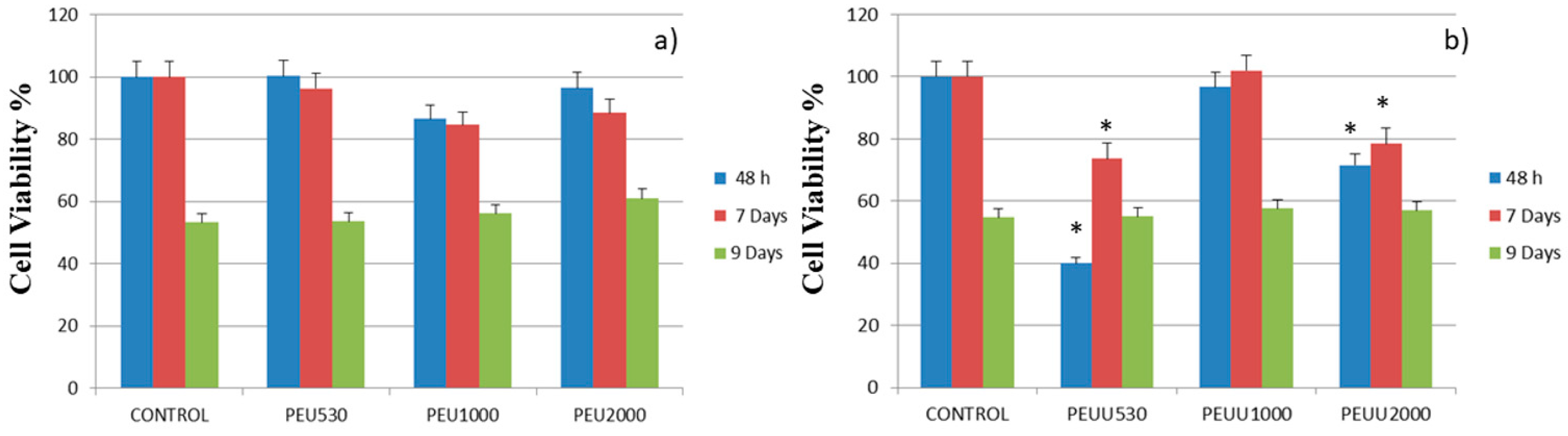
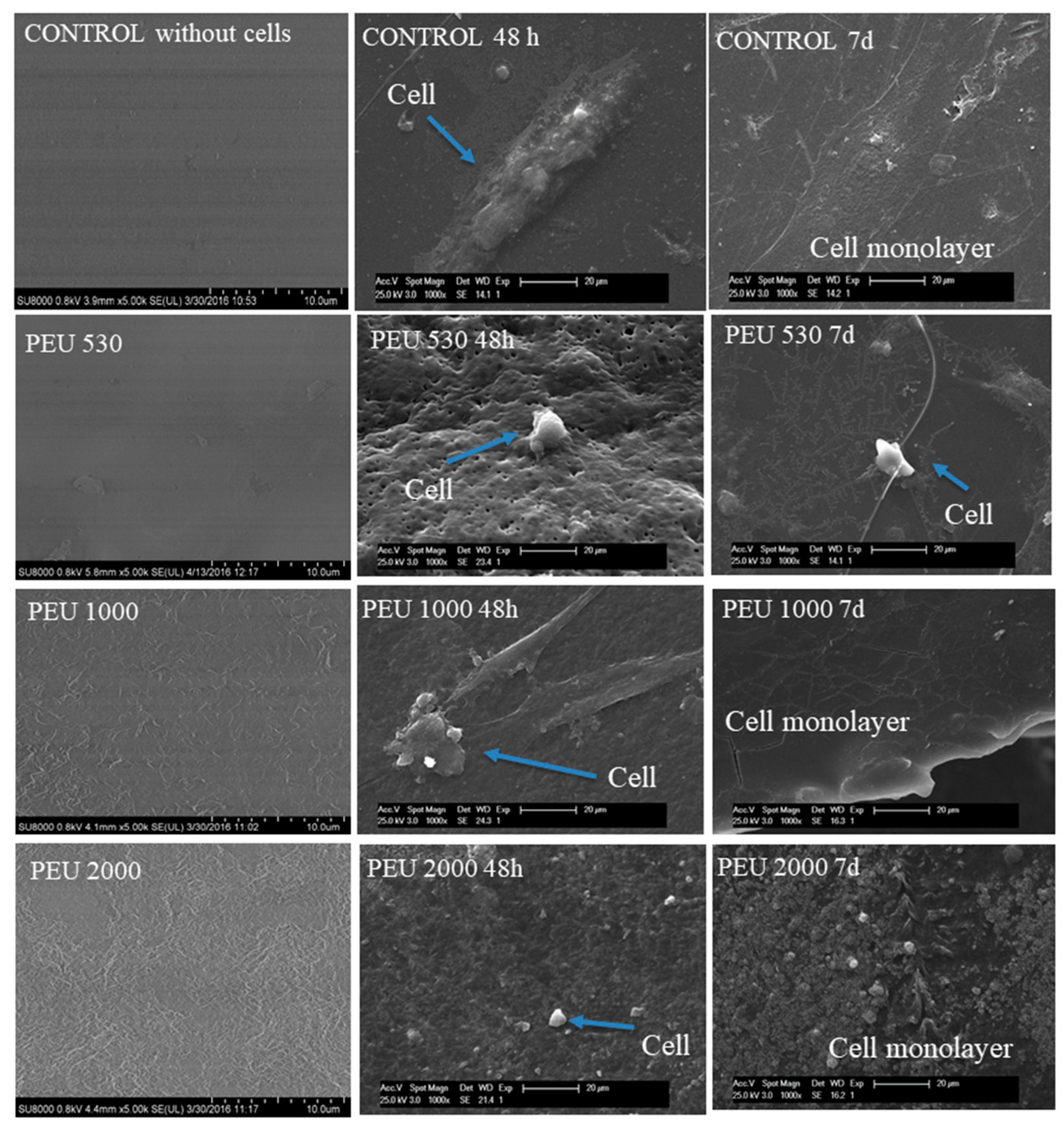
3.3. Cytocompatibility
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Roseti, L.; Parisi, V.; Petretta, M.; Cavallo, C.; Desando, G.; Bartolotti, I.; Grigolo, B. Scaffolds for bone tissue engineering: State of the art and new perspectives. Mater. Sci. Eng. C-Mater. Biol. Appl. 2017, 78, 1246–1262. [Google Scholar] [CrossRef] [PubMed]
- Guelcher, S.A. Biodegradable polyurethanes: Synthesis and applications in regenerative medicine. Tissue Eng. Part B-Rev. 2008, 14, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Loh, X.J.; Sng, K.B.C.; Li, J. Synthesis and water-swelling of thermo-responsive poly(ester urethane)s containing poly(epsilon-caprolactone), poly(ethylene glycol) and poly(propylene glycol). Biomaterials 2008, 29, 3185–3194. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Foster, C.J.; Liu, X.; Wang, S. Enhanced bone cell functions on poly(ε-caprolactone) triacrylate networks grafted with polyhedral oligomeric silsesquioxane nanocages. Polymer 2014, 55, 3836–3845. [Google Scholar] [CrossRef]
- Liu, X.; Miller, A.L.; Fundora, K.A.; Yaszemski, M.J.; Lu, L. Poly (ε-caprolactone) dendrimer cross-linked via metal-free click chemistry: Injectable hydrophobic platform for tissue engineering. ACS Macro Lett. 2016, 5, 1261–1265. [Google Scholar] [CrossRef]
- Gonzalez-Garcia, D.M.; Jurado, L.T.; Jimenez-Gallegos, R.; Rodriguez-Lorenzo, L.M. Novel non-cytotoxic, bioactive and biodegradable hybrid materials based on polyurethanes/tio2 for biomedical applications. Mater. Sci. Eng. C-Mater. Biol. Appl. 2017, 75, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Kishan, A.P.; Wilems, T.; Mohiuddin, S.; Cosgriff-Hernandez, E.M. Synthesis and characterization of plug-and-play polyurethane urea elastomers as biodegradable matrixes for tissue engineering applications. ACS Biomater. Sci. Eng. 2017, 3, 3493–3502. [Google Scholar] [CrossRef]
- Sanchez-Tellez, D.A.; Tellez-Jurado, L.; Rodriguez-Lorenzo, L.M. Hydrogels for cartilage regeneration, from polysaccharides to hybrids. Polymers 2017, 9, 671. [Google Scholar] [CrossRef]
- Filardo, G.; Kon, E.; Perdisa, F.; Sessa, A.; Di Martino, A.; Busacca, M.; Zaffagnini, S.; Marcacci, M. Polyurethane-based cell-free scaffold for the treatment of painful partial meniscus loss. Knee Sur. Sports Traumatol. Arthrosco. 2017, 25, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Reyes, R.; Delgado, A.; Solis, R.; Sanchez, E.; Hernandez, A.; San Roman, J.; Evora, C. Cartilage repair by local delivery of transforming growth factor- beta 1 or bone morphogenetic protein-2 from a novel, segmented polyurethane/polylactic-co-glycolic bilayered scaffold. J. Biomed. Mater. Res. Part A 2014, 102, 1110–1120. [Google Scholar] [CrossRef]
- Sobczak, M. Biodegradable polyurethane elastomers for biomedical applications—synthesis methods and properties. Polym.-Plast. Technol. Eng. 2015, 54, 155–172. [Google Scholar] [CrossRef]
- Gogolewski, S.; Gorna, K.; Turner, A.S. Regeneration of bicortical defects in the iliac crest of estrogen-deficient sheep, using new biodegradable polyurethane bone graft substitutes. J. Biomed. Mater. Res. Part A 2006, 77A, 802–810. [Google Scholar] [CrossRef] [PubMed]
- Fernández-d’Arlas, B.; Alonso-Varona, A.; Palomares, T.; Corcuera, M.A.; Eceiza, A. Studies on the morphology, properties and biocompatibility of aliphatic diisocyanate-polycarbonate polyurethanes. Polym. Degrad. Stabil. 2015, 122, 153–160. [Google Scholar] [CrossRef]
- Zieleniewska, M.; Auguścik, M.; Prociak, A.; Rojek, P.; Ryszkowska, J. Polyurethane-urea substrates from rapeseed oil-based polyol for bone tissue cultures intended for application in tissue engineering. Polym. Degrad. Stabil. 2014, 108, 241–249. [Google Scholar] [CrossRef]
- Castonguay, M.; Koberstein, J.T.; Zhang, Z.; Laroche, G. Synthesis, Physicochemical and Surface Characteristics of Polyurethanes in Biomed Applications of Polyurethanes; Landes Bioscience: Austin, TX, USA, 2001; pp. 1–19. [Google Scholar]
- Abraham, G.A.; Marcos-Fernández, A.; Román, J.S. Bioresorbable poly(ester-ether urethane) s from l-lysine diisocyanate and triblock copolymers with different hydrophilic character. J. Biomed. Mater. Res. Part A 2006, 76, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Sacks, M.S.; Beckman, E.J.; Wagner, W.R. Synthesis, characterization, and cytocompatibility of elastomeric, biodegradable poly(ester-urethane) ureas based on poly(caprolactone) and putrescine. J. Biomed. Mater. Res. 2002, 61, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.H.; Wei, J.; Chang, C.Y.; Laiw, R.F.; Lee, Y.D. Studies on segmented polyetherurethane for biomedical application: Effects of composition and hard-segment content on biocompatibility. J. Biomed. Mater. Res. 1998, 41, 633–648. [Google Scholar] [CrossRef]
- Kavlock, K.D.; Pechar, T.W.; Hollinger, J.O.; Guelcher, S.A.; Goldstein, A.S. Synthesis and characterization of segmented poly(esterurethane urea) elastomers for bone tissue engineering. Acta Biomater. 2007, 3, 475–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krol, P. Synthesis methods, chemical structures and phase structures of linear polyurethanes. Properties and applications of linear polyurethanes in polyurethane elastomers, copolymers and ionomers. Prog. Mater. Sci. 2007, 52, 915–1015. [Google Scholar] [CrossRef]
- Ganta, S.R.; Piesco, N.P.; Long, P.; Gassner, R.; Motta, L.F.; Papworth, G.D.; Stolz, D.B.; Watkins, S.C.; Agarwal, S. Vascularization and tissue infiltration of a biodegradable polyurethane matrix. J. Biomed. Mater. Res. Part A 2003, 64, 242–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skarja, G.; Woodhouse, K. Synthesis and characterization of degradable polyurethane elastomers containing an amino acid-based chain extender. J. Biomater. Sci. 1998, 9, 271–295. [Google Scholar] [CrossRef]
- Skarja, G.; Woodhouse, K. Structure-property relationships of degradable polyurethane elastomers containing an amino acid-based chain extender. J. Appl. Polym. Sci. 2000, 75, 1522–1534. [Google Scholar] [CrossRef]
- Bruin, P.; Veenstra, G.; Nijenhuis, A.; Pennings, A. Design and synthesis of biodegradable poly(ester-urethane) elastomer networks composed of non-toxic building blocks. Macromol. Rapid Commun. 1988, 9, 589–594. [Google Scholar] [CrossRef]
- Marcos-Fernández, A.; Abraham, G.A.; Valentín, J.; San Román, J. Synthesis and characterization of biodegradable non-toxic poly(ester-urethane-urea) s based on poly(ε-caprolactone) and amino acid derivatives. Polymer 2006, 47, 785–798. [Google Scholar] [CrossRef]
- Chen, Q.; Liang, S.; Thouas, G.A. Elastomeric biomaterials for tissue engineering. Prog. Polym. Sci. 2013, 38, 584–671. [Google Scholar] [CrossRef]
- Tatai, L.; Moore, T.G.; Adhikari, R.; Malherbe, F.; Jayasekara, R.; Griffiths, I.; Gunatillake, P.A. Thermoplastic biodegradable polyurethanes: The effect of chain extender structure on properties and in-vitro degradation. Biomaterials 2007, 28, 5407–5417. [Google Scholar] [CrossRef] [PubMed]
- Báez, J.E.; Ramírez, D.; Valentín, J.L.; Marcos-Fernández, A.N. Biodegradable poly(ester–urethane–amide) s based on poly(ε-caprolactone) and diamide–diol chain extenders with crystalline hard segments. Synthesis and characterization. Macromolecules 2012, 45, 6966–6980. [Google Scholar] [CrossRef]
- Stokes, K.; McVenes, R.; Anderson, J.M. Polyurethane elastomer biostability. J. Biomater. Appl. 1995, 9, 321–354. [Google Scholar] [CrossRef] [PubMed]
- Kuang, W.; Mather, P.T. A latent crosslinkable PCL-based polyurethane: Synthesis, shape memory, and enzymatic degradation. J. Mater. Res. 2018, 17, 1–14. [Google Scholar] [CrossRef]
- Li, H.; Chang, J.; Cao, A.; Wang, J. In vitro evaluation of biodegradable poly(butylene succinate) as a novel biomaterial. Macromol. Biosci. 2005, 5, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Xynos, I.; Hukkanen, M.; Batten, J.; Buttery, L.; Hench, L.; Polak, J. Bioglass® 45s5 stimulates osteoblast turnover and enhances bone formation in vitro: Implications and applications for bone tissue engineering. Calcif. Tissue Int. 2000, 67, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Akasaka, T.; Yokoyama, A.; Matsuoka, M.; Hashimoto, T.; Abe, S.; Uo, M.; Watari, F. Adhesion of human osteoblast-like cells (saos-2) to carbon nanotube sheets. Bio-Med. Mater. Eng. 2009, 19, 147–153. [Google Scholar]
- Tsai, K.J.; Dixon, S.; Hale, L.R.; Darbyshire, A.; Martin, D.; de Mel, A. Biomimetic heterogenous elastic tissue development. NPJ Regen. Med. 2017, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lui, Y.F.; Ip, W.Y. Biological evaluation of flexible polyurethane/poly l-lactic acid composite scaffold as a potential filler for bone regeneration. Materials 2017, 10, 1042. [Google Scholar] [CrossRef]
- Muggli, D.S.; Burkoth, A.K.; Anseth, K.S. Crosslinked polyanhydrides for use in orthopedic applications: Degradation behavior and mechanics. J. Biomed. Mater. Res. 1999, 46, 271–278. [Google Scholar] [CrossRef]
- Fernando, S.; McEnery, M.; Guelcher, S. Polyurethanes for bone tissue engineering. Adv. Poly. Biomater. 2016, 481–501. [Google Scholar] [CrossRef]
- Rodriguez-Lorenzo, L.M.; Garcia-Carrodeguas, R.; Rodriguez, M.A.; De Aza, S.; Jimenez, J.; Lopez-Bravo, A.; Fernandez, M.; Roman, J.S. Synthesis, characterization, bioactivity and biocompatibility of nanostructured materials based on the wollastonite-poly(ethylmethacrylate-co-vinylpyrrolidone) system. J. Biomed. Mater. Res. Part A 2009, 88A, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Lorenzo, L.M.; Vallet-Regi, M.; Ferreira, J.M.F.; Ginebra, M.P.; Aparicio, C.; Planell, J.A. Hydroxyapatite ceramic bodies with tailored mechanical properties for different applications. J. Biomed. Mater. Res. 2002, 60, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.F.; Liu, J.; Luo, M.; Xing, J.; Wu, J.C.; Pan, H.B.; Ruan, C.S.; Luo, Y.F. Incorporating isosorbide as the chain extender improves mechanical properties of linear biodegradable polyurethanes as potential bone regeneration materials. Rsc Adv. 2017, 7, 13886–13895. [Google Scholar] [CrossRef] [Green Version]
- Rodrigo, J.-G.; Luis, M.R.-L.; Julio San, R.; Lucía, T.-J. Preparation, bioactivity, and cytotoxicity studies of poly(ester urethane)s/SiO2 nanocomposites. J. Thermoplast. Compos. Mater. 2017, 0892705717744831. [Google Scholar] [CrossRef]
- Jimenez-Gallegos, R.; Tellez-Jurado, L.; Rodriguez-Lorenzo, L.M.; San Roman, J. Modulation of the hydrophilic character and influence on the biocompatibility of polyurethane-siloxane based hybrids. Boletín de la Sociedad Española de Cerámica Y Vidrio 2011, 50, 1–8. [Google Scholar]
- Marzec, M.; Kucinska-Lipka, J.; Kalaszczynska, I.; Janik, H. Development of polyurethanes for bone repair. Mater. Sci. Eng. C-Mater. Biol. Appl. 2017, 80, 736–747. [Google Scholar] [CrossRef] [PubMed]
- Bil, M.; Ryszkowska, J.; Woźniak, P.; Kurzydłowski, K.J.; Lewandowska-Szumieł, M. Optimization of the structure of polyurethanes for bone tissue engineering applications. Acta Biomater. 2010, 6, 2501–2510. [Google Scholar] [CrossRef] [PubMed]
- Middleton, J.C.; Tipton, A.J. Synthetic biodegradable polymers as orthopedic devices. Biomaterials 2000, 21, 2335–2346. [Google Scholar] [CrossRef]
- Gorna, K.; Gogolewski, S. Molecular stability, mechanical properties, surface characteristics and sterility of biodegradable polyurethanes treated with low-temperature plasma. Polym. Degrad. Stab. 2003, 79, 475–485. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PCL Mn | PEU’s Code | Molar Ratio PCL:HDI:BD | PEUs Mw | PEUs Mw/Mn | PEUs Contact Angle | PEUs Code | Molar Ratio PCL:HDI:LYS | PEUUs Mw | PEUUs Mw/Mn | PEUUs Contact Angle |
|---|---|---|---|---|---|---|---|---|---|---|
| 530 | PEU 530 | 1:1.21:0.21 | 120,800 | 1.65 | 67 | PEUU 530 | 1:1.17:0.17 | 27,600 | 1.2 | 70 |
| 1000 | PEU 1090 | 1:2.16:1.16 | 125,300 | 1.7 | 70 | PEUU 1000 | 1:1.87:0.87 | 136,700 | 1.8 | 73 |
| 2000 | PEU 2000 | 1:3.67:2.67 | 139,200 | 1.7 | 70 | PEUU 2000 | 1:3.01:2.01 | 196,300 | 2.0 | 77 |
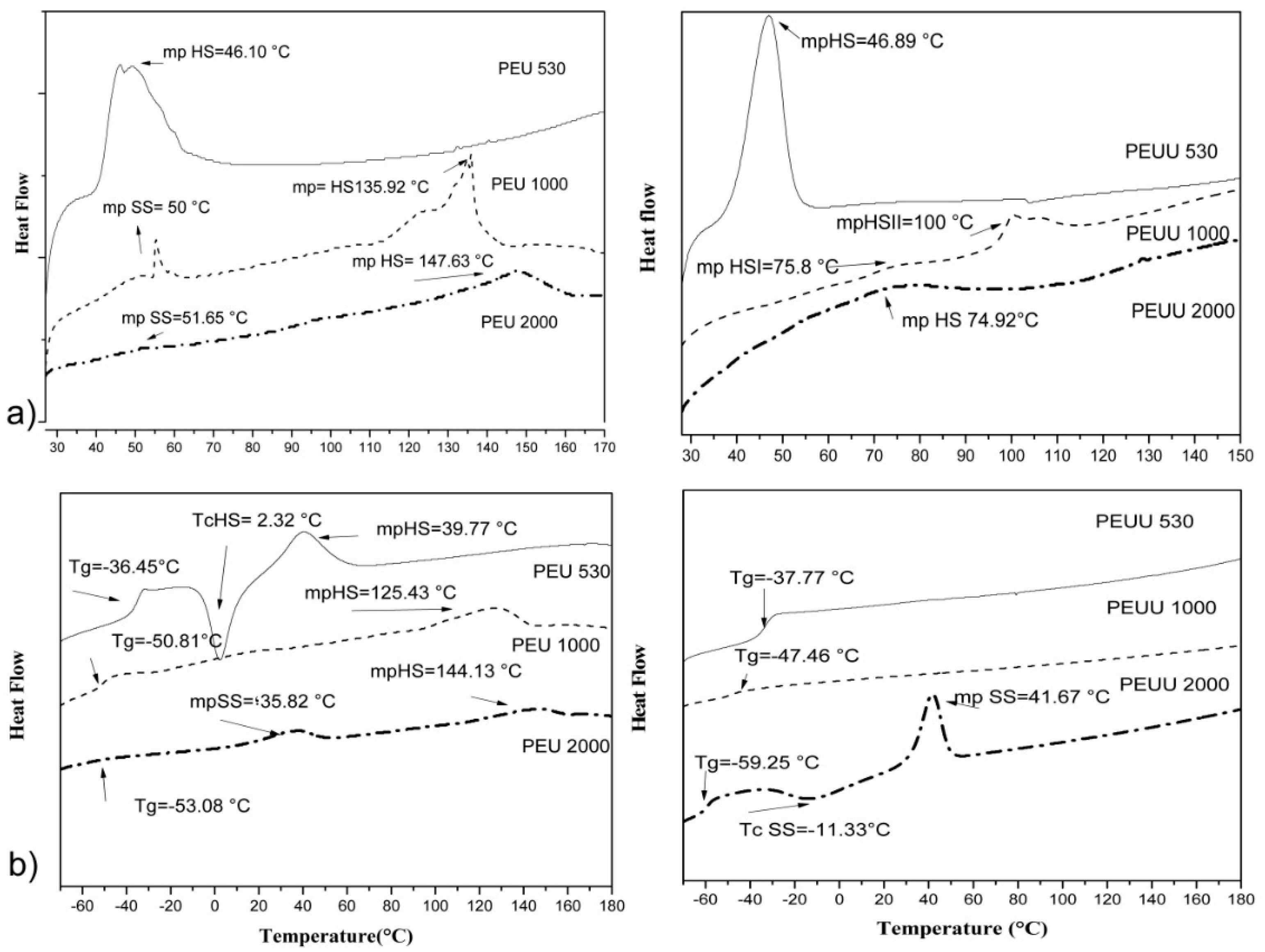
| Polymer Codes | First Scan (20–90 °C) | Second SCAN (−80–180 °C) | ||||
|---|---|---|---|---|---|---|
| Mp SS | Mp HS | Tc | Mp SS | Mp HS | Tg | |
| PEU 530 | - | 46.1 | 2.3 (HS) | - | 39.8 | −36.45 |
| PEU 1000 | 55.3 | 135.9 | - | - | 125.4 | −50.8 |
| PEU 2000 | 51.6 | 147.6 | - | 35.8 | 144.1 | −53.1 |
| PEUU 530 | - | 46.9 | - | - | - | −33.77 |
| PEUU 1000 | - | 75.8, 100.1 | - | - | - | −47.5 |
| PEUU 2000 | - | 74.9 | 11.3 (SS) | 41.7 | - | −59.2 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-García, D.M.; Marcos-Fernández, Á.; Rodríguez-Lorenzo, L.M.; Jiménez-Gallegos, R.; Vargas-Becerril, N.; Téllez-Jurado, L. Synthesis and in Vitro Cytocompatibility of Segmented Poly(Ester-Urethane)s and Poly(Ester-Urea-Urethane)s for Bone Tissue Engineering. Polymers 2018, 10, 991. https://doi.org/10.3390/polym10090991
González-García DM, Marcos-Fernández Á, Rodríguez-Lorenzo LM, Jiménez-Gallegos R, Vargas-Becerril N, Téllez-Jurado L. Synthesis and in Vitro Cytocompatibility of Segmented Poly(Ester-Urethane)s and Poly(Ester-Urea-Urethane)s for Bone Tissue Engineering. Polymers. 2018; 10(9):991. https://doi.org/10.3390/polym10090991
Chicago/Turabian StyleGonzález-García, Dulce María, Ángel Marcos-Fernández, Luis M. Rodríguez-Lorenzo, Rodrigo Jiménez-Gallegos, Nancy Vargas-Becerril, and Lucía Téllez-Jurado. 2018. "Synthesis and in Vitro Cytocompatibility of Segmented Poly(Ester-Urethane)s and Poly(Ester-Urea-Urethane)s for Bone Tissue Engineering" Polymers 10, no. 9: 991. https://doi.org/10.3390/polym10090991
APA StyleGonzález-García, D. M., Marcos-Fernández, Á., Rodríguez-Lorenzo, L. M., Jiménez-Gallegos, R., Vargas-Becerril, N., & Téllez-Jurado, L. (2018). Synthesis and in Vitro Cytocompatibility of Segmented Poly(Ester-Urethane)s and Poly(Ester-Urea-Urethane)s for Bone Tissue Engineering. Polymers, 10(9), 991. https://doi.org/10.3390/polym10090991