Modification of Pea Starch and Dextrin Polymers with Isocyanate Functional Groups

 ,
, 
 and
and 
Abstract
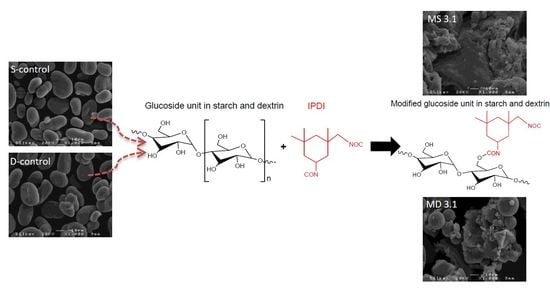
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Functionalization of Starch and Dextrin
2.3. Characterization
2.3.1. Elemental Analysis
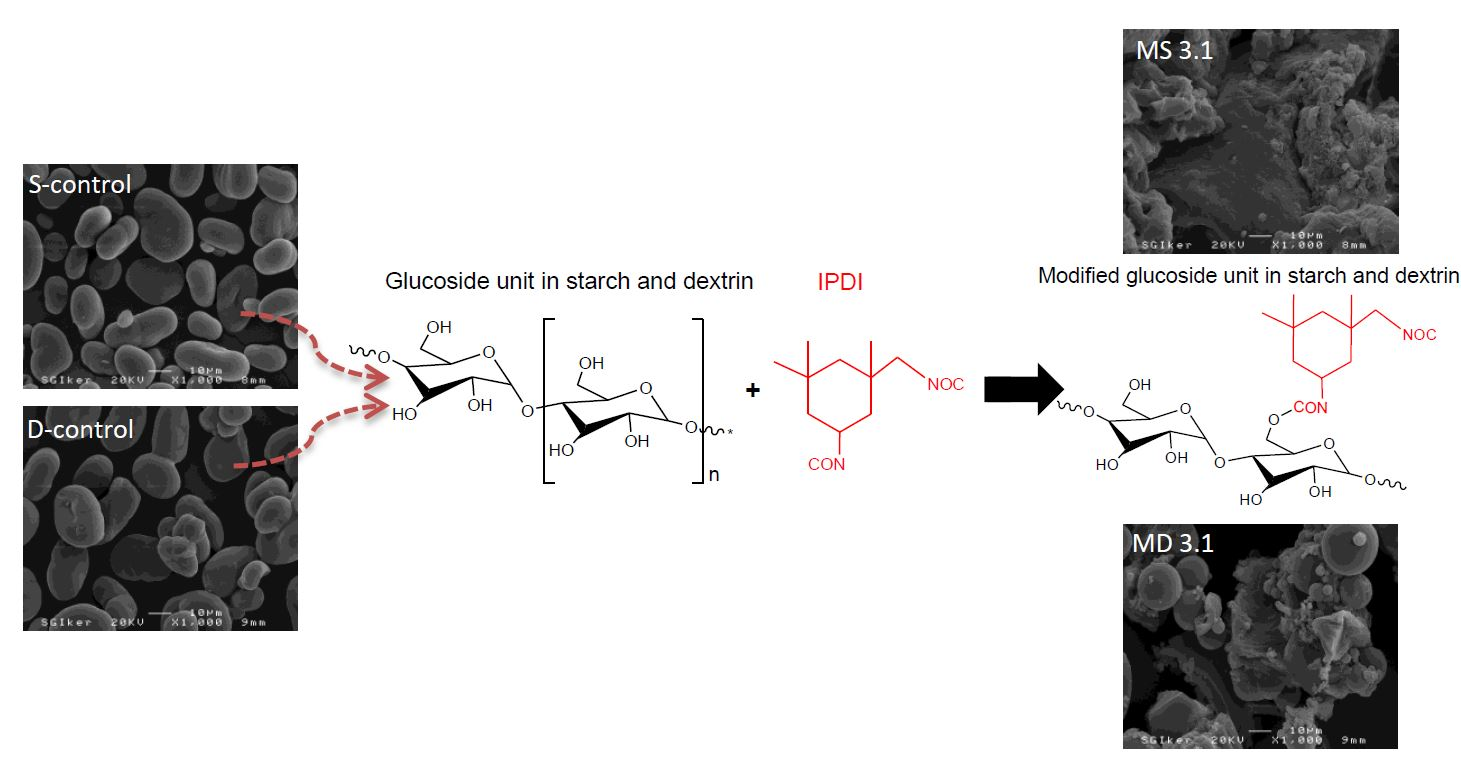
2.3.2. Fourier Transform Infrared Spectroscopy
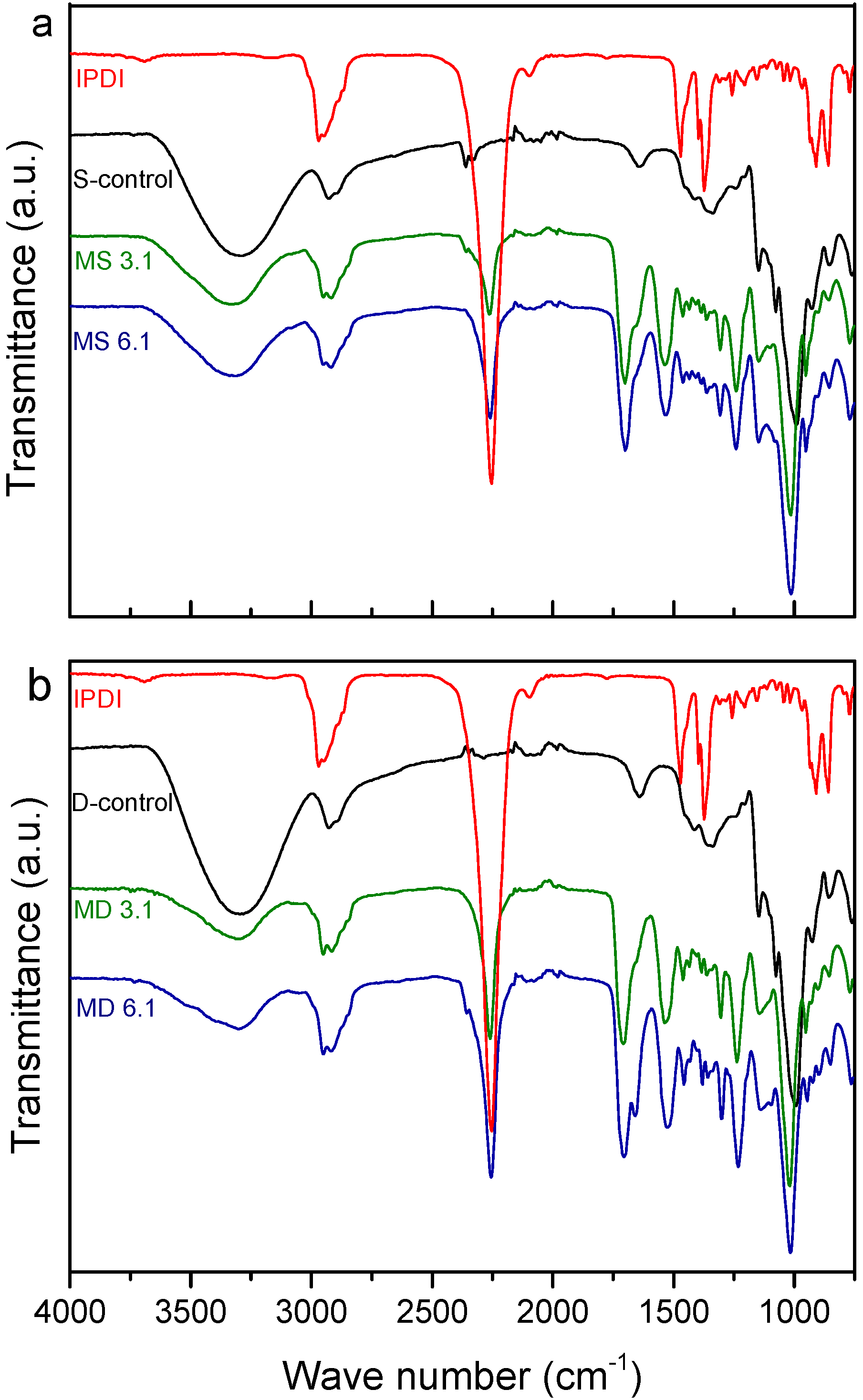
2.3.3. Nuclear Magnetic Resonance Spectroscopy
2.3.4. Thermogravimetric Analysis
2.3.5. Differential Scanning Calorimetry
2.3.6. Dynamic Vapor Sorption
2.3.7. Scanning Electron Microscopy
3. Results and Discussion
3.1. Elemental Analysis and Degree of Substitution (DS)
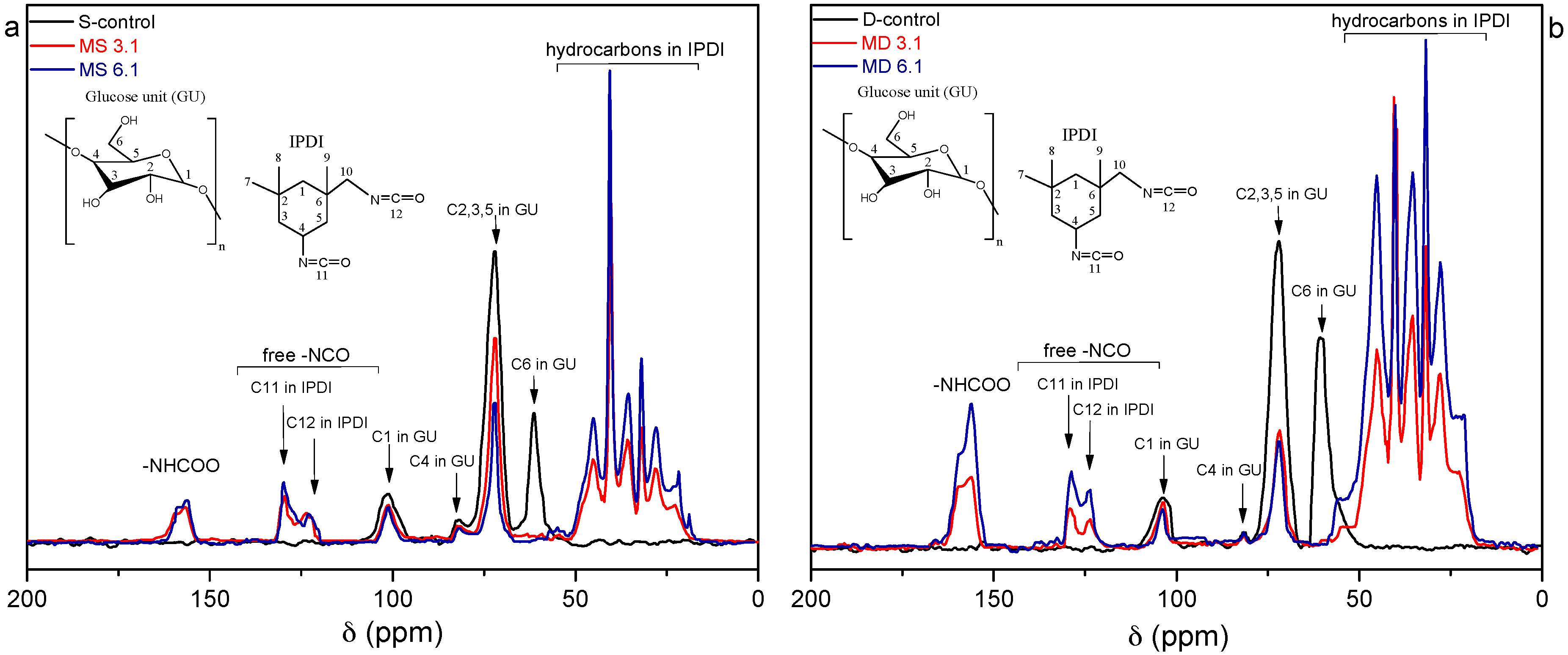
3.2. Structural Characterization
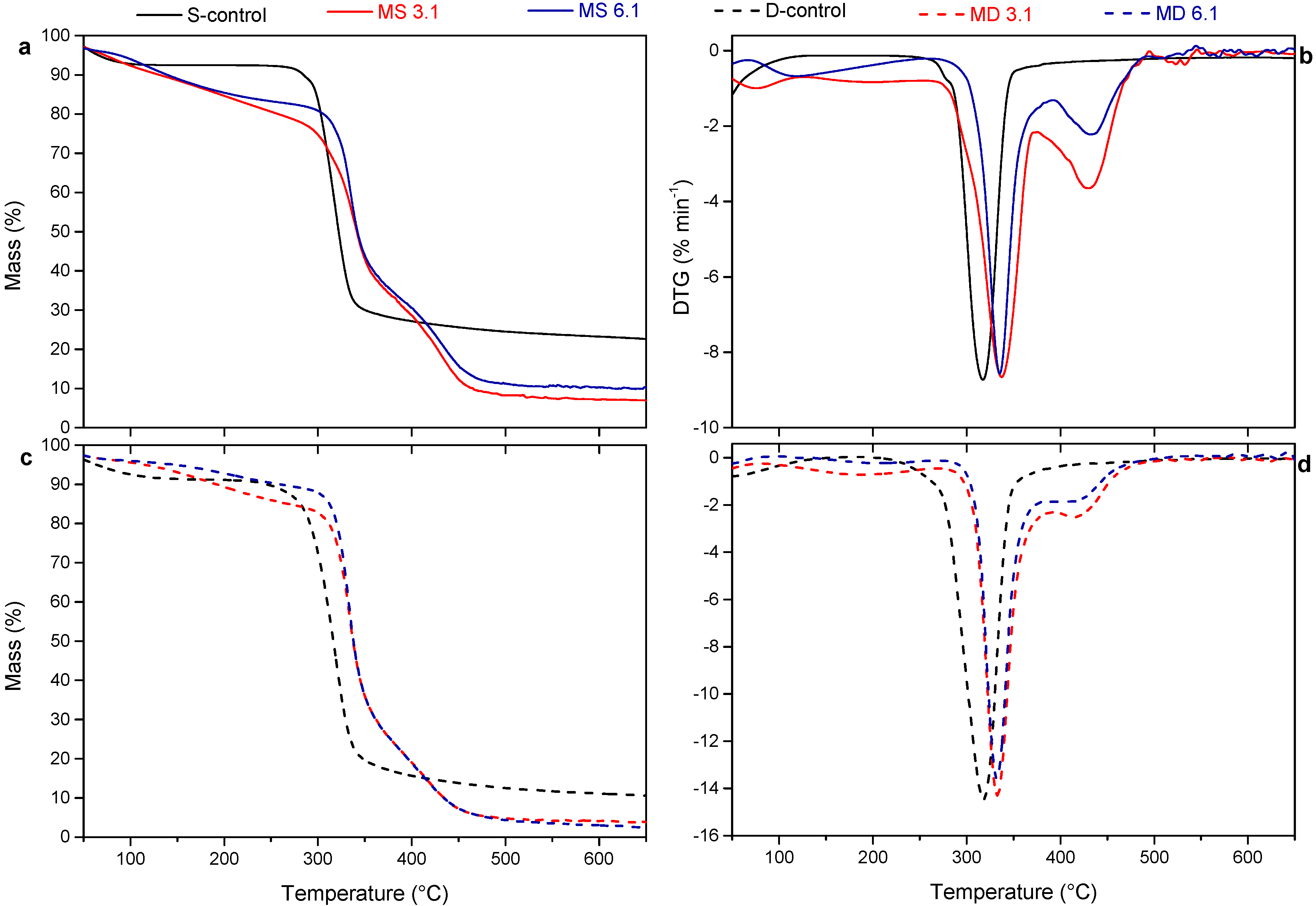
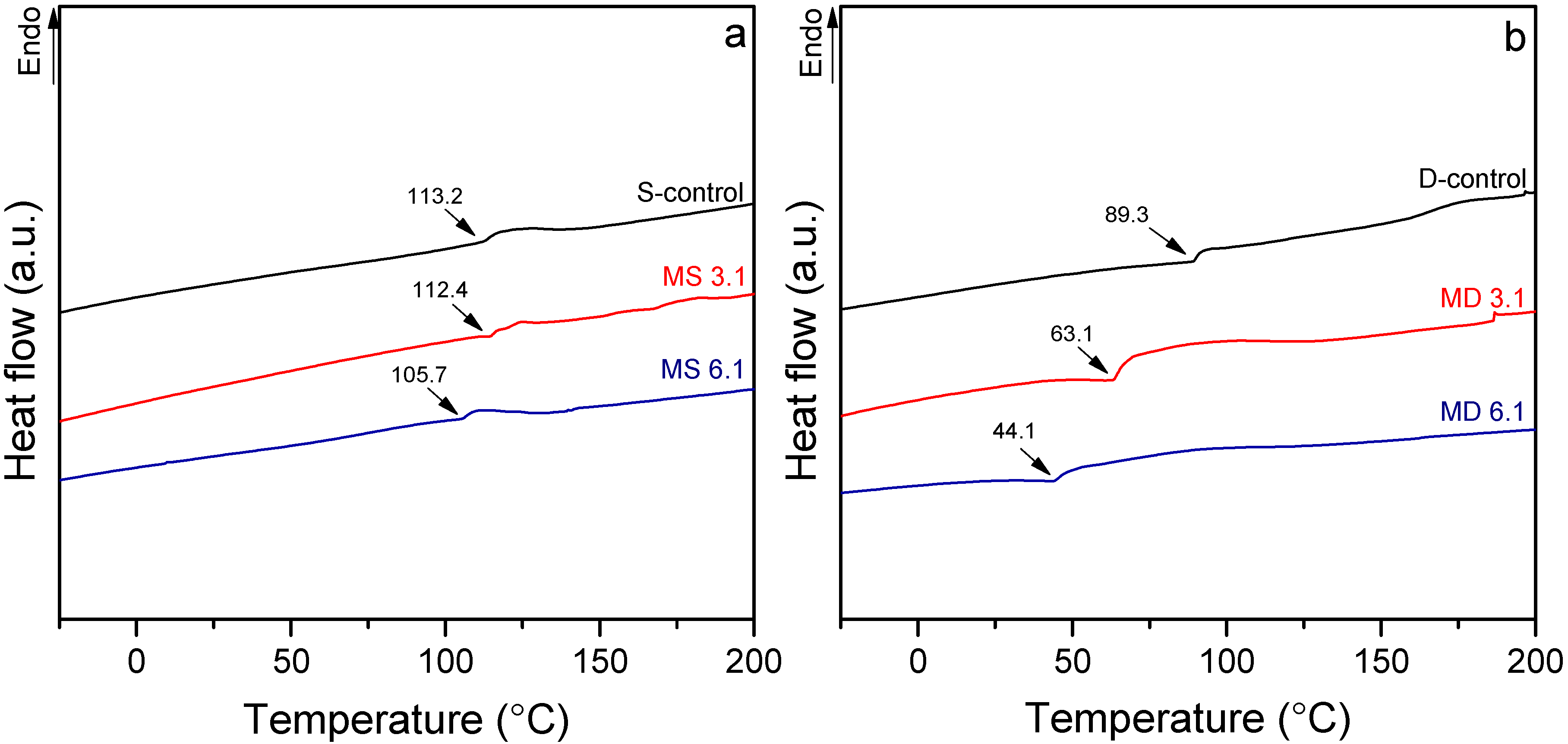
3.3. Thermal Behavior
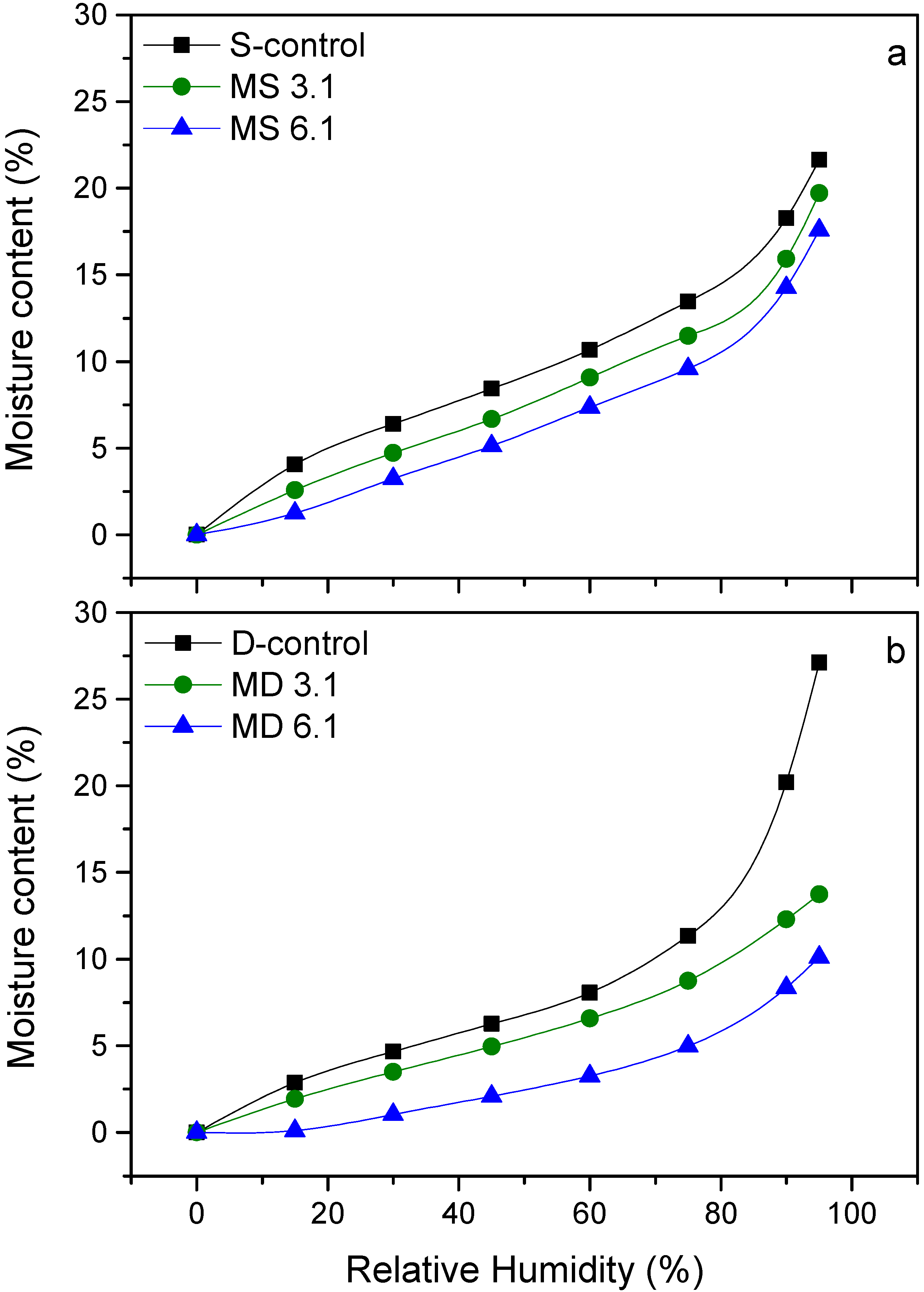
3.4. Water Vapor Sorption Behavior
3.5. Scanning Electron Microscopy (SEM)
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Biliaderis, C.G. Structures and phase transitions of starch polymers. In Polysaccharide Association Structures in Food; Walter, R.H., Ed.; Marcel-Dekker Inc.: New York, NY, USA, 1998; pp. 57–168. [Google Scholar]
- Wang, S.J.; Copeland, L. Effect of alkali treatment on structure and function of pea starch granules. Food Chem. 2012, 135, 1635–1642. [Google Scholar] [CrossRef] [PubMed]
- Bertolini, A.C. (Ed.) Trends in starch applications. In Starches: Characterization, Properties, and Applications; Taylor and Francis Group LLC: New York, NY, USA, 2010; pp. 1–20. [Google Scholar]
- De Graaf, R.A.; Karman, A.P.; Janssen, L.P.B.M. Material properties and glass transition temperatures of different thermoplastic starches after extrusion processing. Starch-Starke 2003, 55, 80–86. [Google Scholar] [CrossRef]
- Barikani, M.; Mohammadi, M. Synthesis and characterization of starch-modified polyurethane. Carbohydr. Polym. 2007, 68, 773–780. [Google Scholar] [CrossRef]
- Fang, J.M.; Fowler, P.A.; Tomkinson, J.; Hill, C.A.S. The preparation and characterisation of a series of chemically modified potato starches. Carbohydr. Polym. 2002, 47, 245–252. [Google Scholar] [CrossRef]
- Kurakake, M.; Akiyama, Y.; Hagiwara, H.; Komaki, T. Effects of cross-linking and low molecular amylose on pasting characteristics of waxy corn starch. Food Chem. 2009, 116, 66–70. [Google Scholar] [CrossRef]
- Thielemans, W.; Belgacem, M.N.; Dufresne, A. Starch nanocrystals with large chain surface modifications. Langmuir 2006, 22, 4804–4810. [Google Scholar] [CrossRef] [PubMed]
- Valodkar, M.; Thakore, S. Isocyanate crosslinked reactive starch nanoparticles for thermo-responsive conducting applications. Carbohydr. Res. 2010, 345, 2354–2360. [Google Scholar] [CrossRef] [PubMed]
- Veelaert, S.; de Wit, D.; Gotlieb, K.F.; Verhe, R. Chemical and physical transitions of periodate oxidized potato starch in water. Carbohydr. Polym. 1997, 33, 153–162. [Google Scholar] [CrossRef]
- Zdanowicz, M.; Schmidt, B.; Spychaj, T. Starch graft copolymers as superabsorbents obtained via reactive extrusion processing. Pol. J. Chem. Technol. 2010, 12, 14–17. [Google Scholar] [CrossRef]
- Zhang, S.D.; Zhang, Y.R.; Zhu, J.; Wang, X.L.; Yang, K.K.; Wang, Y.Z. Modified corn starches with improved comprehensive properties for preparing thermoplastics. Starch-Starke 2007, 59, 258–268. [Google Scholar] [CrossRef]
- Da Roz, A.L.; Curvelo, A.A.S.; Gandini, A. Preparation and characterization of cross-linked starch polyurethanes. Carbohydr. Polym. 2009, 77, 526–529. [Google Scholar] [CrossRef]
- Girouard, N.M.; Xu, S.H.; Schueneman, G.T.; Shofner, M.L.; Meredith, J.C. Site-Selective Modification of Cellulose Nanocrystals with Isophorone Diisocyanate and Formation of Polyurethane-CNC Composites. ACS Appl. Mater. Interfaces 2016, 8, 1458–1467. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Fernández, S.; Ugarte, L.; Calvo-Correas, T.; Pena-Rodriguez, C.; Corcuera, M.A.; Eceiza, A. Properties of flexible polyurethane foams containing isocyanate functionalized kraft lignin. Ind. Crops Prod. 2017, 100, 51–64. [Google Scholar] [CrossRef]
- Bialas, N.; Höcker, H.; Marschner, M.; Ritter, W. 13C NMR studies on the relative reactivity of isocyanate groups of isophorone diisocyanate isomers. Macromol. Chem. Phys. 1990, 191, 1843–1852. [Google Scholar] [CrossRef]
- Gotz, H.; Beginn, U.; Bartelink, C.F.; Grunbauer, H.J.M.; Moller, M. Preparation of isophorone diisocyanate terminated star polyethers. Macromol. Mater. Eng. 2002, 287, 223–230. [Google Scholar] [CrossRef]
- Lomolder, R.; Plogmann, F.; Speier, P. Selectivity of isophorone diisocyanate in the urethane reaction influence of temperature, catalysis, and reaction partners. J. Coat. Technol. 1997, 69, 51–57. [Google Scholar] [CrossRef]
- Ono, H.K.; Jones, F.N.; Pappas, S.P. Relative Reactivity of Isocyanate Groups of Isophorone Diisocyanate. Unexpected High Reactivity of the Secondary Isocyanate Group. J. Polym. Sci. Part C Polym. Lett. 1985, 23, 509–515. [Google Scholar] [CrossRef]
- Hosseinpourpia, R.; Adamopoulos, S.; Mai, C. Dynamic vapour sorption of wood and holocellulose modified with thermosetting resins. Wood Sci. Technol. 2016, 50, 165–178. [Google Scholar] [CrossRef]
- Hosseinpourpia, R.; Adamopoulos, S.; Holstein, N.; Mai, C. Dynamic vapour sorption and water-related properties of thermally modified Scots pine (Pinus sylvestris L.) wood pre-treated with proton acid. Polym. Degrad. Stab. 2017, 138, 161–168. [Google Scholar] [CrossRef]
- Hosseinpourpia, R.; Adamopoulos, S.; Mai, C. Effects of Acid Pre-Treatments on the Swelling and Vapor Sorption of Thermally Modified Scots Pine (Pinus sylvestris L.) Wood. Bioresources 2018, 13, 331–345. [Google Scholar] [CrossRef]
- Garg, S.; Jana, A.K. Characterization and evaluation of acylated starch with different acyl groups and degrees of substitution. Carbohydr. Polym. 2011, 83, 1623–1630. [Google Scholar] [CrossRef]
- Colussi, R.; Pinto, V.Z.; El Halal, S.L.M.; Vanier, N.L.; Villanova, F.A.; Silva, M.R.; da Rosa Zavareze, E.; Dias, A.R.G. Structural, morphological, and physicochemical properties of acetylated high-, medium-, and low-amylose rice. Carbohydr. Polym. 2014, 103, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Diop, C.I.K.; Li, H.L.; Xie, B.J.; Shi, J. Effects of acetic acid/acetic anhydride ratios on the properties of corn starch acetates. Food Chem. 2011, 126, 1662–1669. [Google Scholar] [CrossRef] [PubMed]
- Rueda, L.; d’Arlas, B.F.; Zhou, Q.; Berglund, L.A.; Corcuera, M.A.; Mondragon, I.; Eceiza, A. Isocyanate-rich cellulose nanocrystals and their selective insertion in elastomeric polyurethane. Compos. Sci. Technol. 2011, 71, 1953–1960. [Google Scholar] [CrossRef]
- Tang, H.; Hills, B.P. Use of 13C MAS NMR to Study Domain Structure and Dynamics of Polysaccharides in the Native Starch Granules. Biomacromolecules 2003, 4, 1269–1276. [Google Scholar] [CrossRef] [PubMed]
- Veregin, R.P.; Fyfe, C.A.; Marchessault, R.H.; Taylor, M.G. Characterization of the crystalline A and B starch polymorphs and investigation of starch crystallization by high-resolution 13CP/MAS NMR. Macromolecules 1986, 19, 1030–1034. [Google Scholar] [CrossRef]
- Carvalho, J.; Goncalves, C.; Gil, A.M.; Gama, F.M. Production and characterization of a new dextrin based hydrogel. Eur. Polym. J. 2007, 43, 3050–3059. [Google Scholar] [CrossRef]
- Garcia, H.; Barros, A.S.; Goncalves, C.; Gama, F.M.; Gil, A.M. Characterization of dextrin hydrogels by FTIR spectroscopy and solid state NMR spectroscopy. Eur. Polym. J. 2008, 44, 2318–2329. [Google Scholar] [CrossRef]
- Shefer, A.; Shefer, S.; Kost, J.; Langer, R. Structural characterization of starch networks in the solid state by cross-polarization magic-angle-spinning 13C NMR spectroscopy and wide angle X-ray diffraction. Macromolecules 1992, 25, 6756–6760. [Google Scholar] [CrossRef]
- Cho, A.; Choi, S.H.; Choi, H.W.; Kim, H.S.; Kim, W.; Kim, D.O.; Kim, B.Y.; Baik, M.Y. Characterization of cationic dextrin prepared by ultra high pressure (UHP)-assisted cationization reaction. Carbohydr. Polym. 2013, 97, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, Y.; Yu, L.; Tong, Z.; Chen, L.; Liu, H.; Li, X. Thermal degradation and stability of starch under different processing conditions. Starch-Starke 2013, 65, 48–60. [Google Scholar] [CrossRef]
- Rao, Y.; Munro, J.; Ge, S.; Garcia-Meitin, E. PU elastomers comprising spherical nanosilicas: Balancing rheology and properties. Polymer 2014, 55, 6076–6084. [Google Scholar] [CrossRef]
- Avaltroni, F.; Bouquerand, P.E.; Normand, V. Maltodextrin molecular weight distribution influence on the glass transition temperature and viscosity in aqueous solutions. Carbohydr. Polym. 2004, 58, 323–334. [Google Scholar] [CrossRef]
- Chaudhary, V.; Panyoyai, N.; Small, D.M.; Shanks, R.A.; Kasapis, S. Effect of the glass transition temperature on alpha-amylase activity in a starch matrix. Carbohydr. Polym. 2017, 157, 1531–1537. [Google Scholar] [CrossRef] [PubMed]
- Zeleznak, K.J.; Hoseney, R.C. The Glass-Transition in Starch. Cereal Chem. 1987, 64, 121–124. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C | H | O | N | DS | |
|---|---|---|---|---|---|
| S-control | 38.60 | 6.53 | 52.94 | 0.08 | - |
| MS 3.1 | 45.58 | 7.45 | 26.96 | 5.28 | 1.11 |
| MS 6.1 | 46.97 | 7.58 | 27.92 | 6.24 | 1.54 |
| D-control | 39.33 | 6.42 | 52.45 | 0.03 | - |
| MD 3.1 | 48.40 | 7.98 | 23.68 | 7.38 | 2.29 |
| MD 6.1 | 53.13 | 8.02 | 24.08 | 8.12 | 3.04 |
| Tonset | Tmax1 | Tmax2 | |
|---|---|---|---|
| S-control | 297.2 | 316.6 | - |
| MS 3.1 | 313.2 | 334.7 | 430.4 |
| MS 6.1 | 324.4 | 337.6 | 433.6 |
| D-control | 292.1 | 319.7 | - |
| MD 3.1 | 318.9 | 330.1 | 415.1 |
| MD 6.1 | 319.8 | 334.8 | 417.5 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hosseinpourpia, R.; Echart, A.S.; Adamopoulos, S.; Gabilondo, N.; Eceiza, A. Modification of Pea Starch and Dextrin Polymers with Isocyanate Functional Groups. Polymers 2018, 10, 939. https://doi.org/10.3390/polym10090939
Hosseinpourpia R, Echart AS, Adamopoulos S, Gabilondo N, Eceiza A. Modification of Pea Starch and Dextrin Polymers with Isocyanate Functional Groups. Polymers. 2018; 10(9):939. https://doi.org/10.3390/polym10090939
Chicago/Turabian StyleHosseinpourpia, Reza, Arantzazu Santamaria Echart, Stergios Adamopoulos, Nagore Gabilondo, and Arantxa Eceiza. 2018. "Modification of Pea Starch and Dextrin Polymers with Isocyanate Functional Groups" Polymers 10, no. 9: 939. https://doi.org/10.3390/polym10090939
APA StyleHosseinpourpia, R., Echart, A. S., Adamopoulos, S., Gabilondo, N., & Eceiza, A. (2018). Modification of Pea Starch and Dextrin Polymers with Isocyanate Functional Groups. Polymers, 10(9), 939. https://doi.org/10.3390/polym10090939