Effect of Core-Shell Morphology on the Mechanical Properties and Crystallization Behavior of HDPE/HDPE-g-MA/PA6 Ternary Blends

Abstract
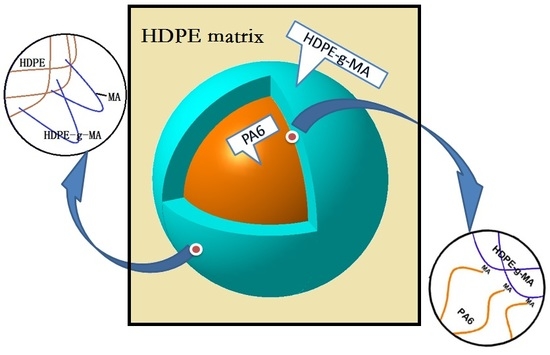
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Sample Preparation
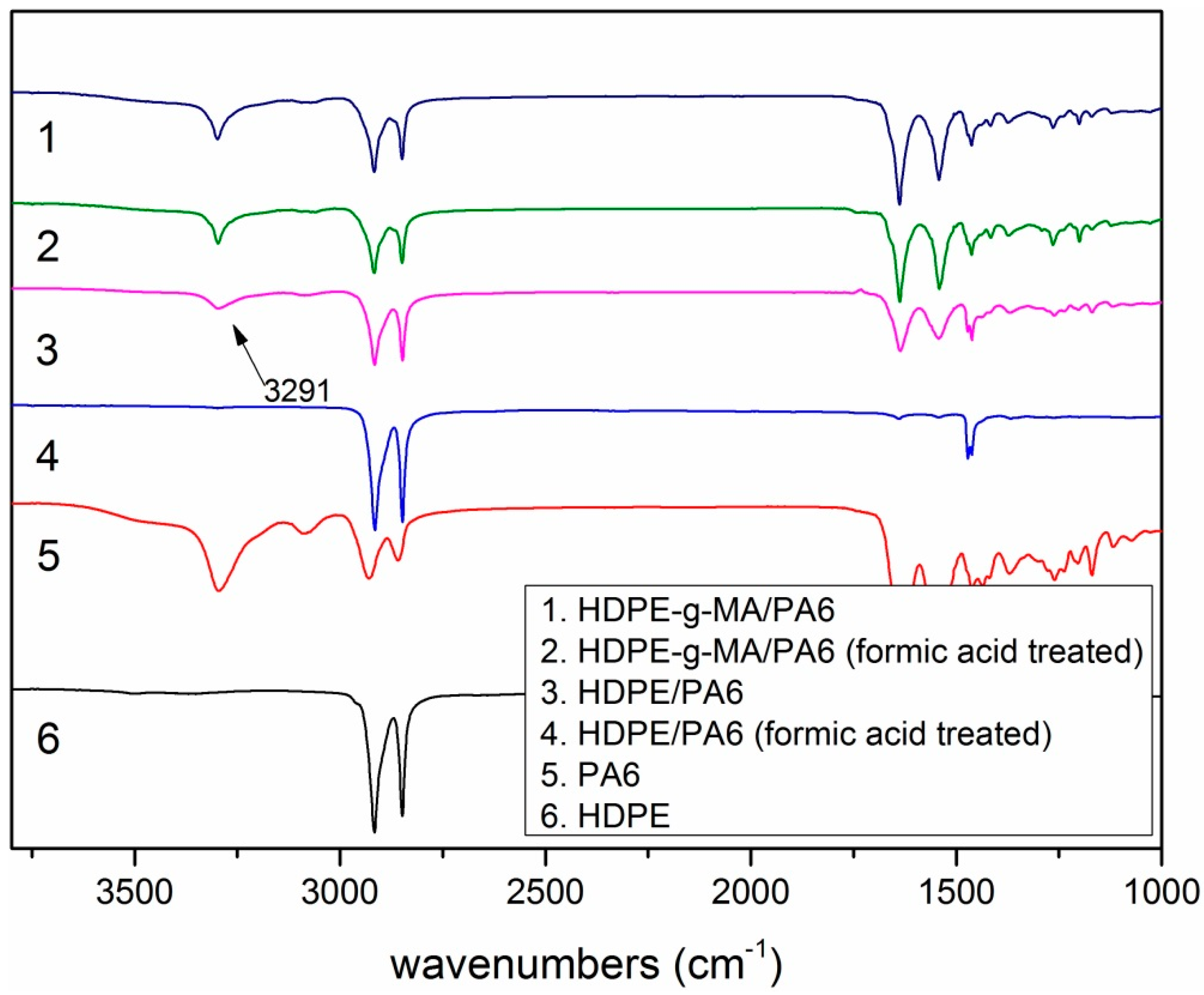
2.3. Morphology Observation
2.4. Polarized Optical Microscopy Observation
2.5. Contact Angle Measurements
2.6. Mechanical Tests
2.7. Dynamic Mechanical Analysis
2.8. Differential Scanning Calorimetry Measurement
3. Results and Discussion
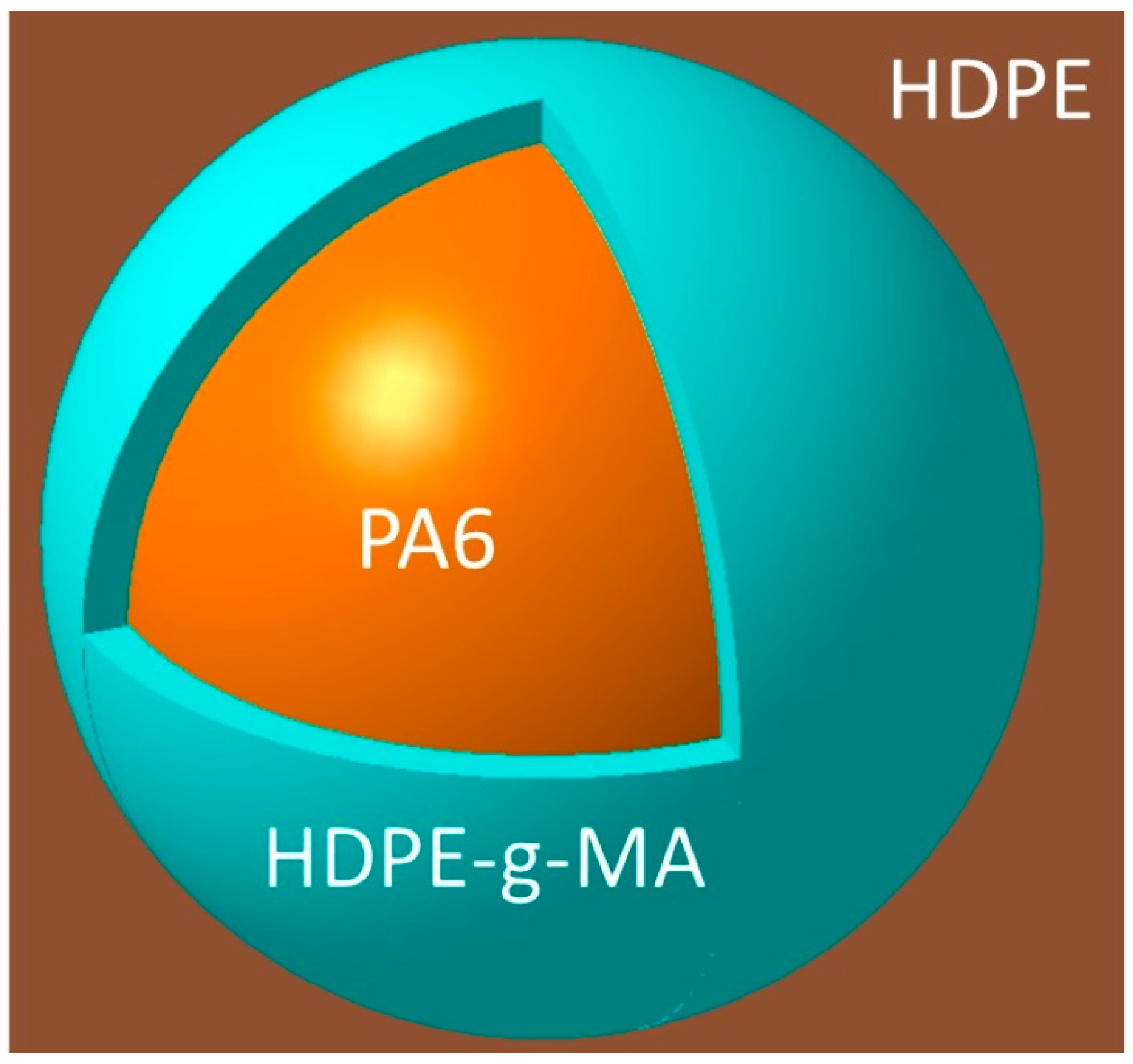
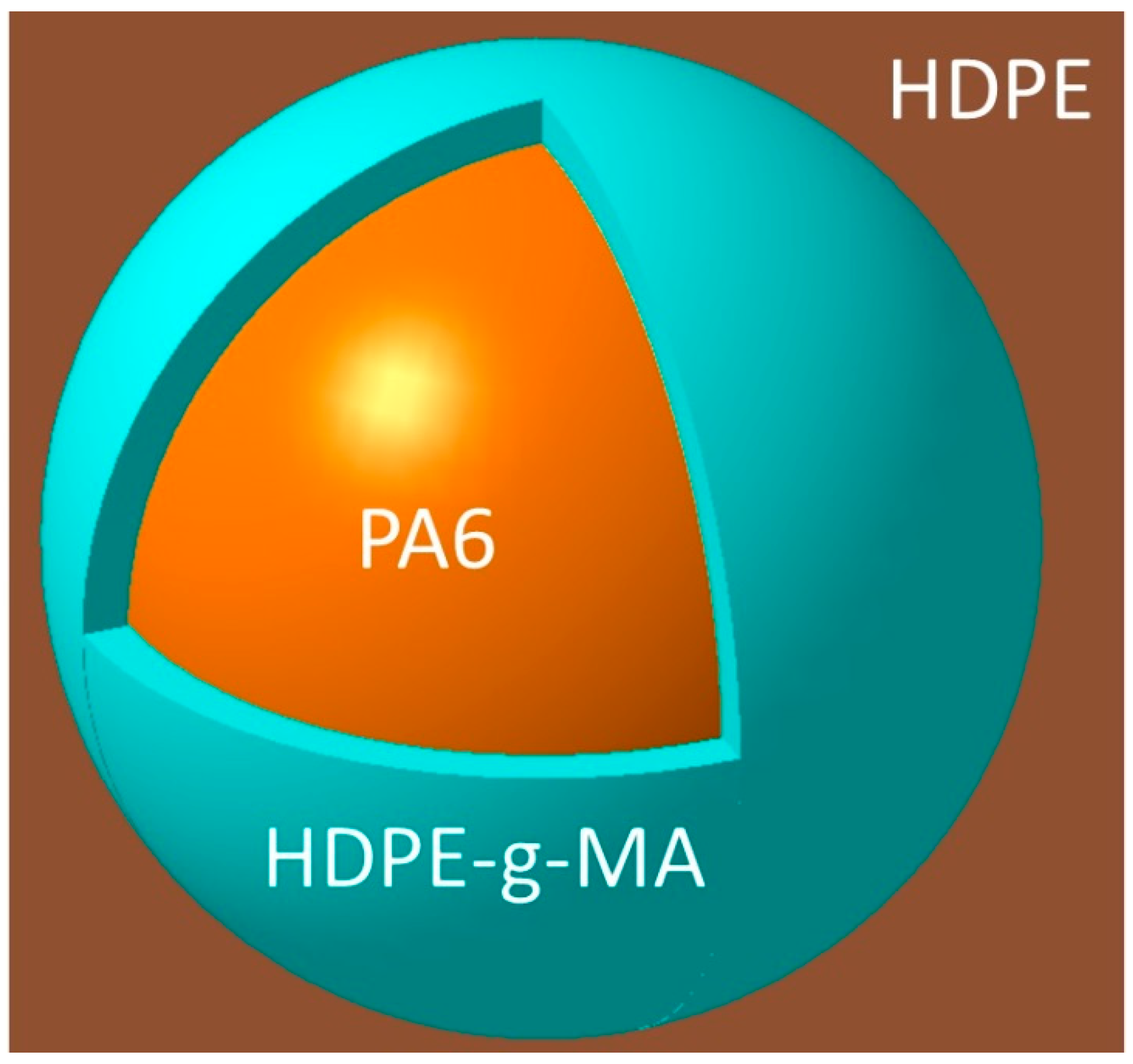
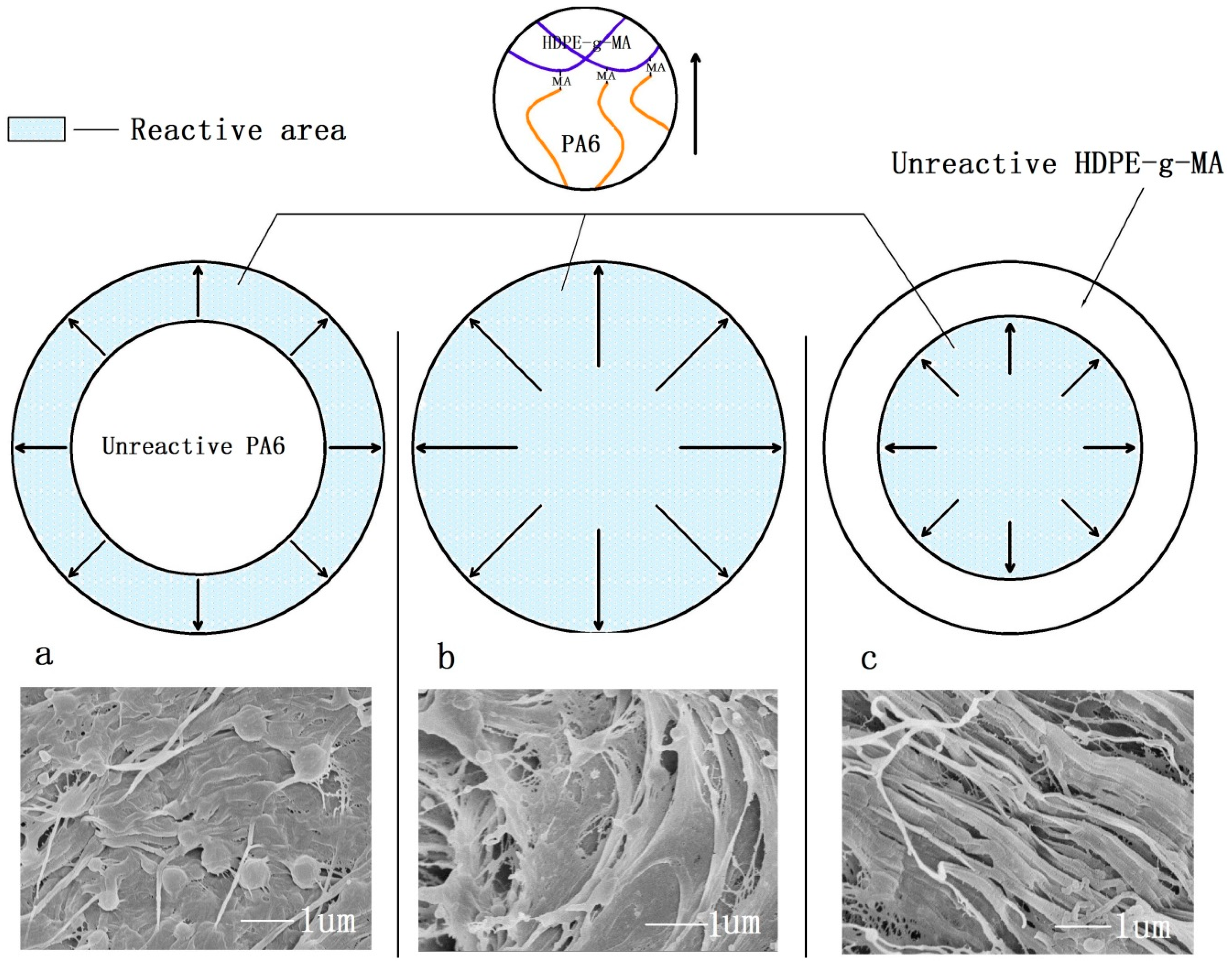
3.1. The Phase Morphology
3.1.1. The Phase Morphology Prediction
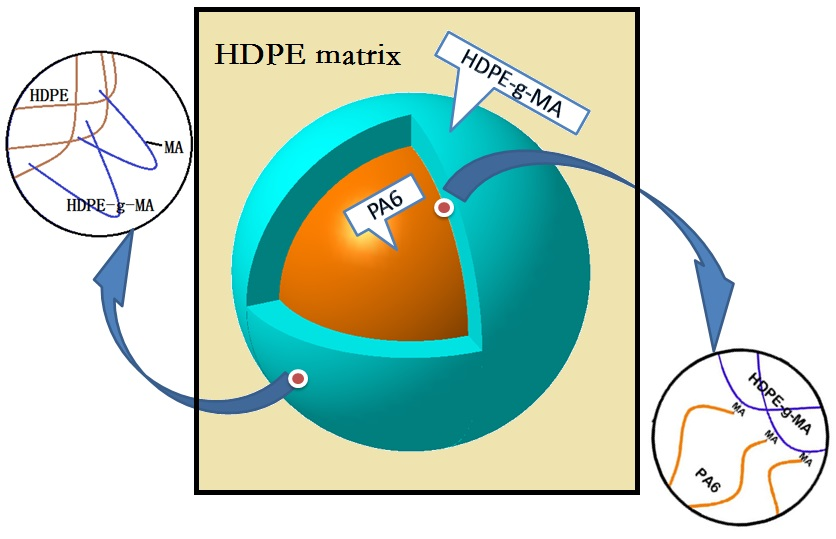
3.1.2. The Phase Morphology Analysis
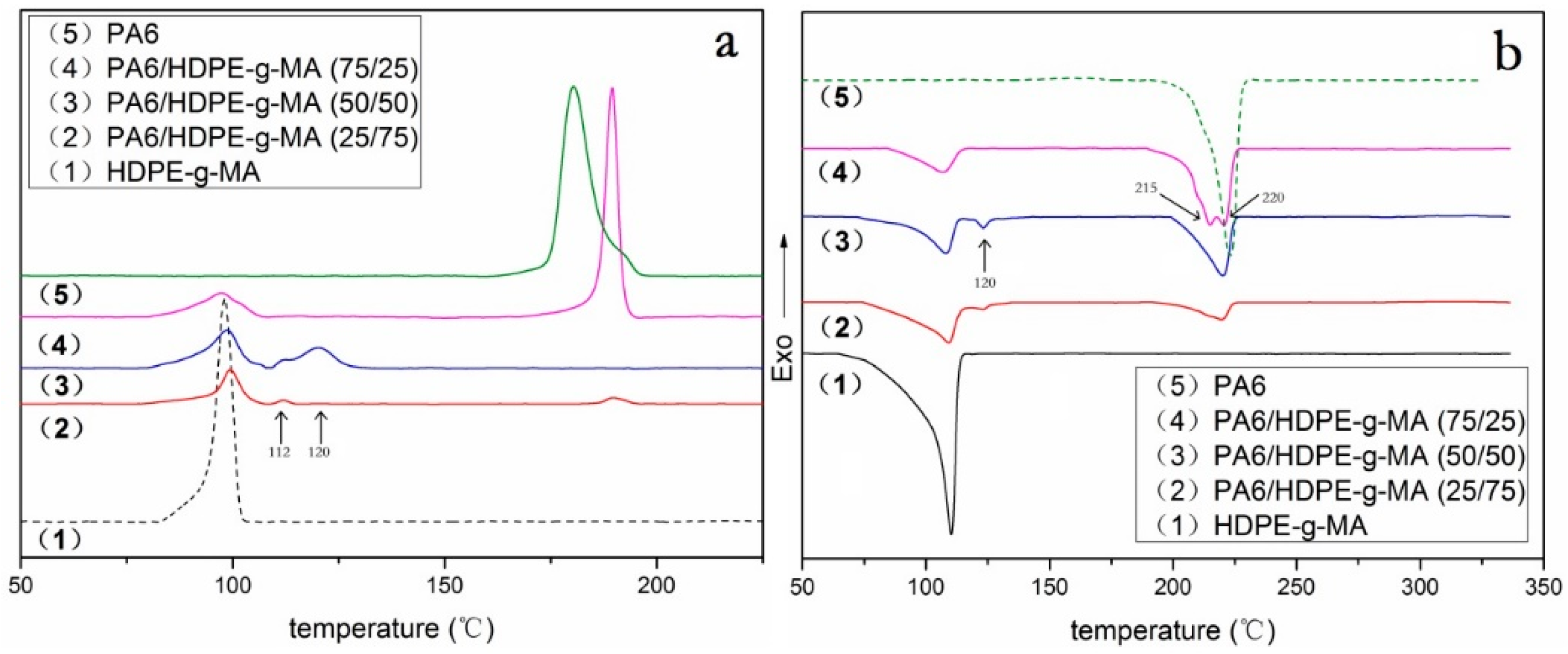
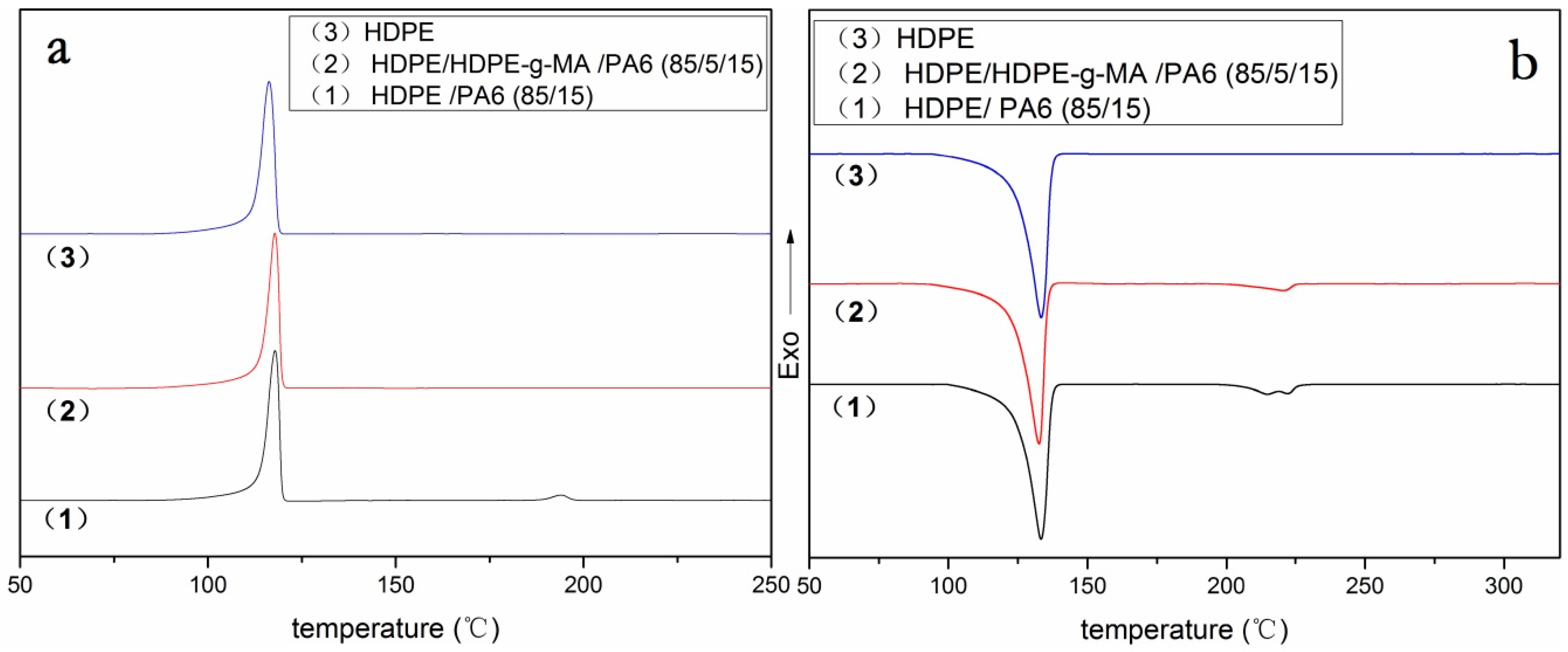
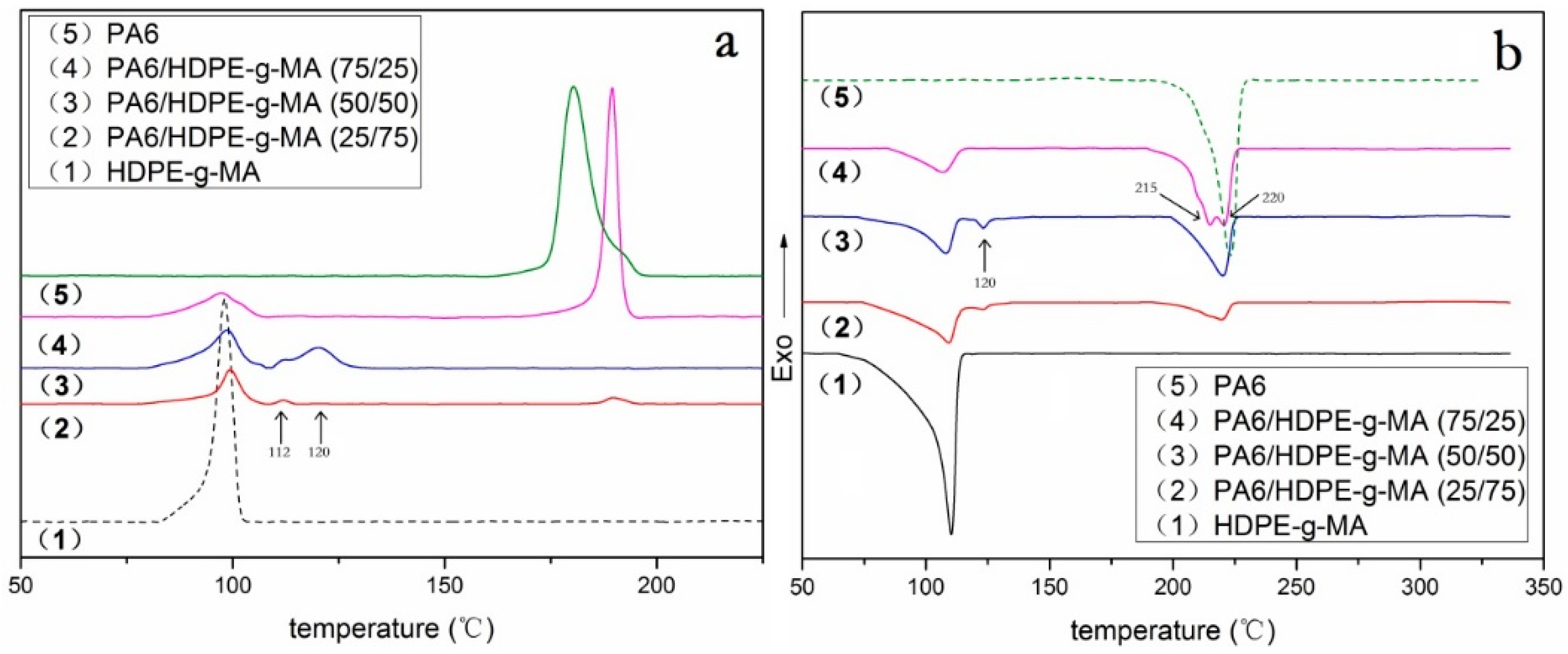
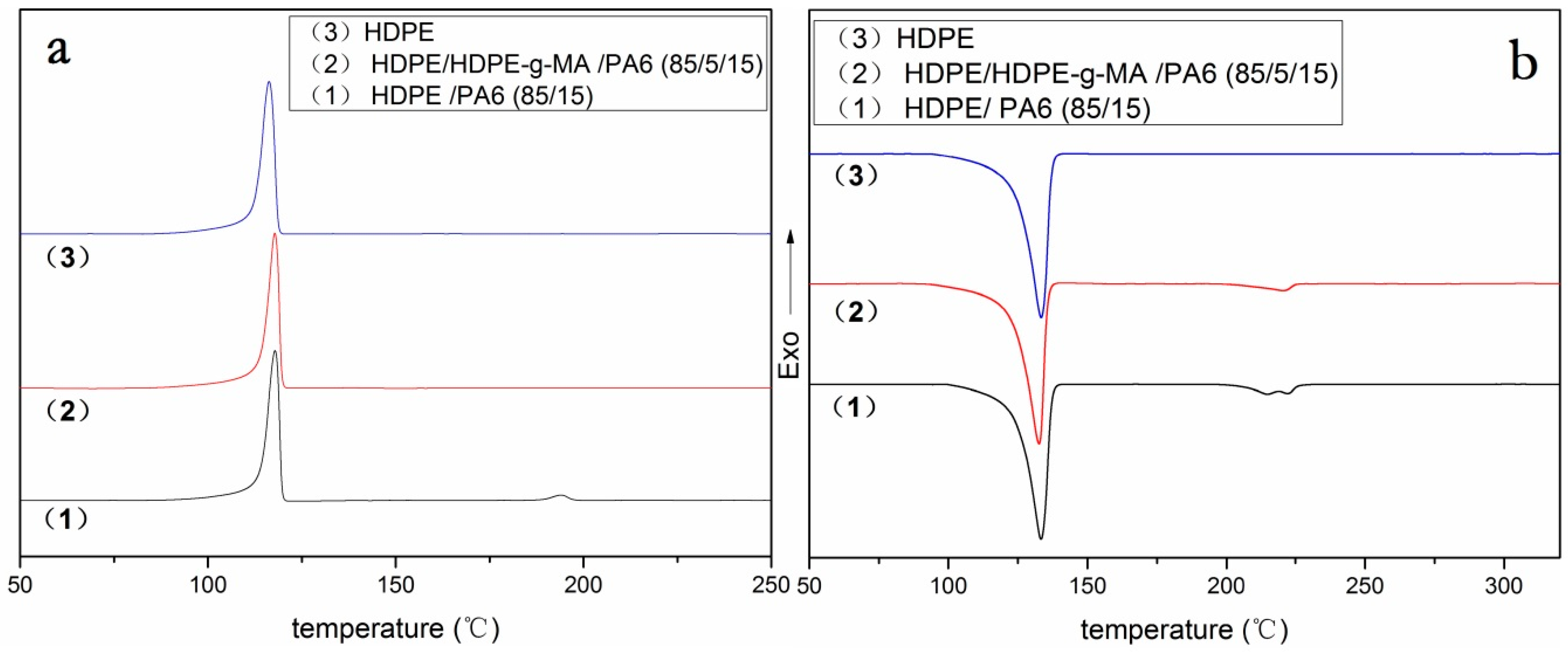
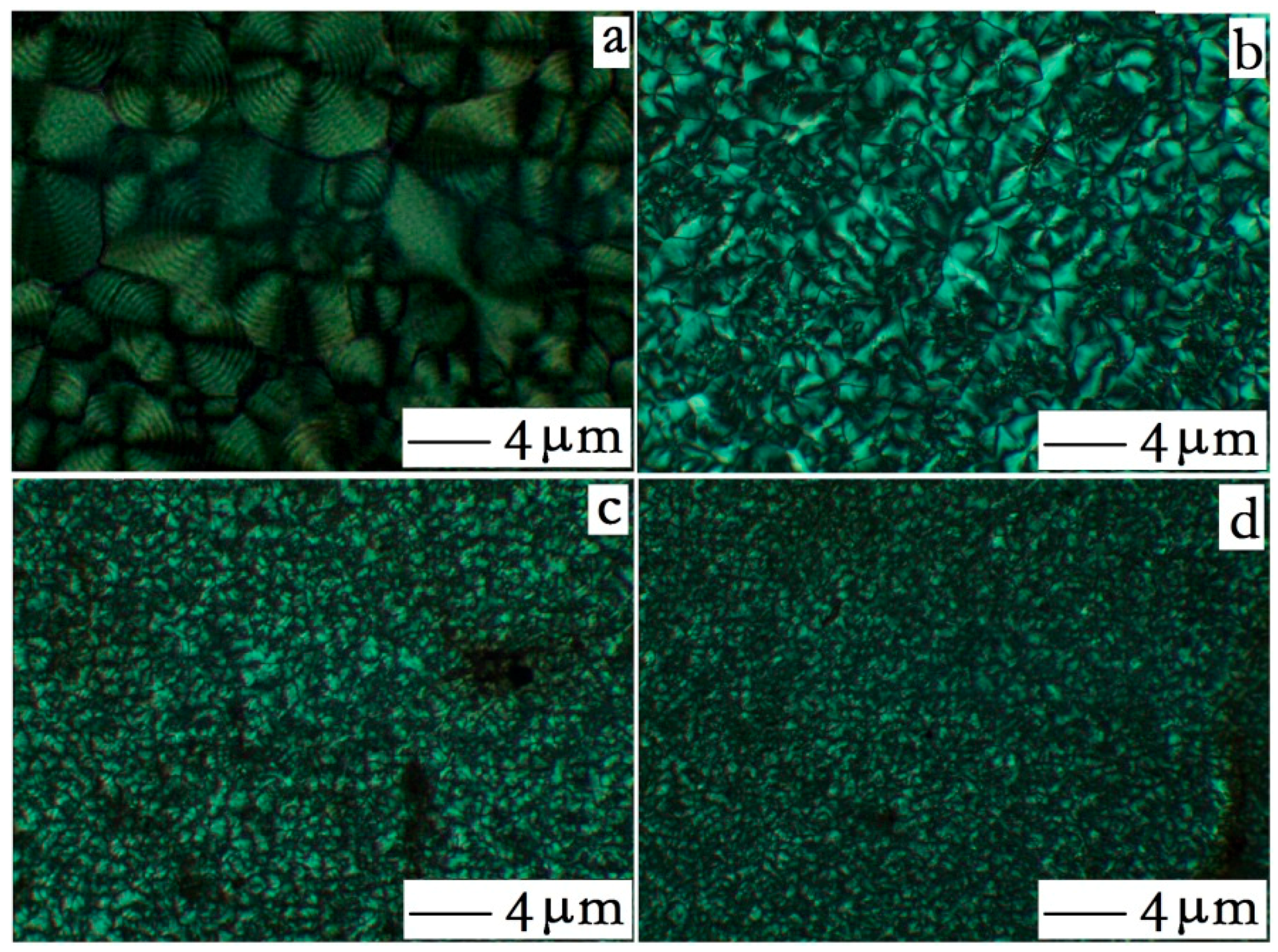
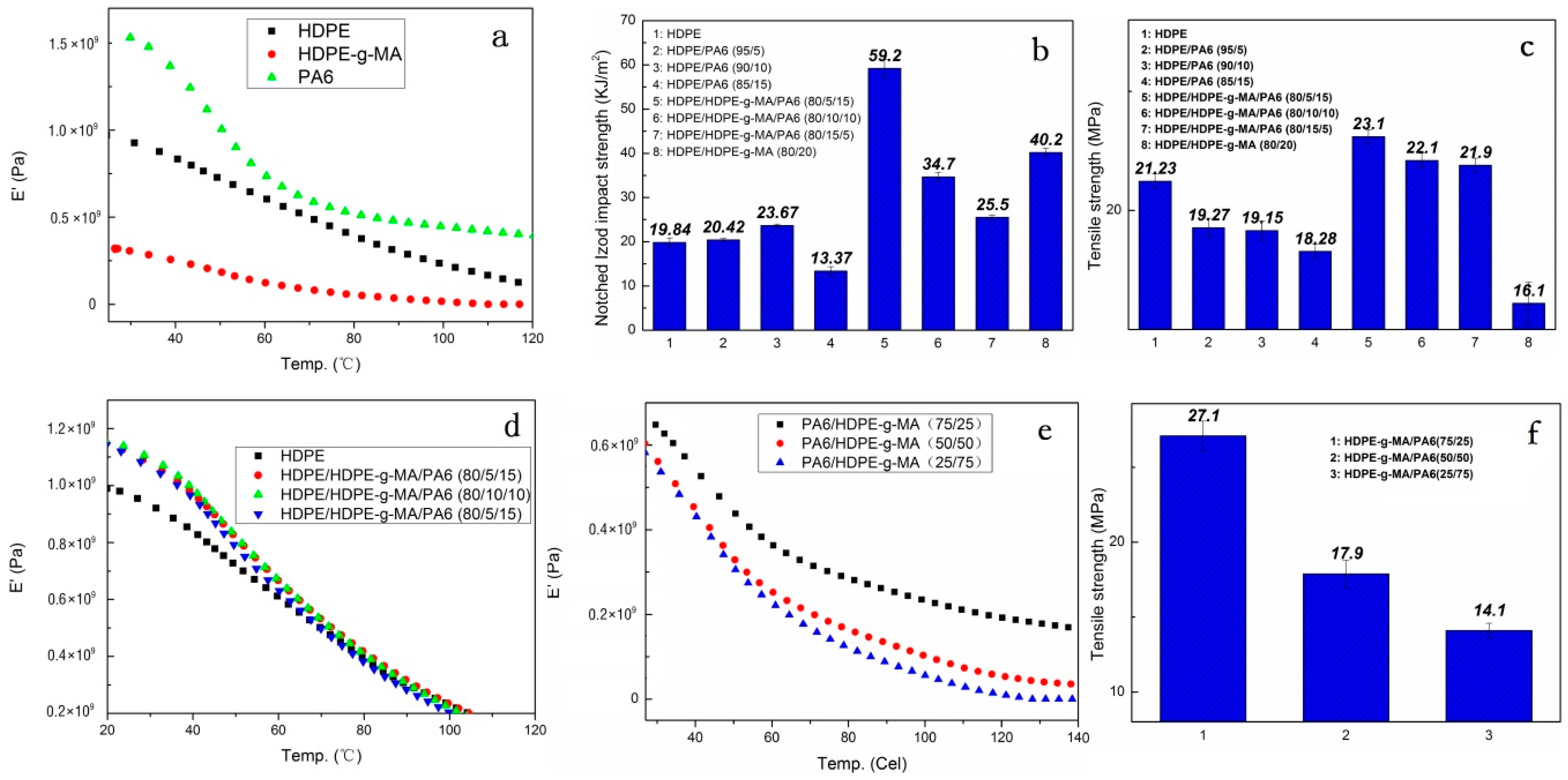
3.2. Crystallization Behavior Analysis
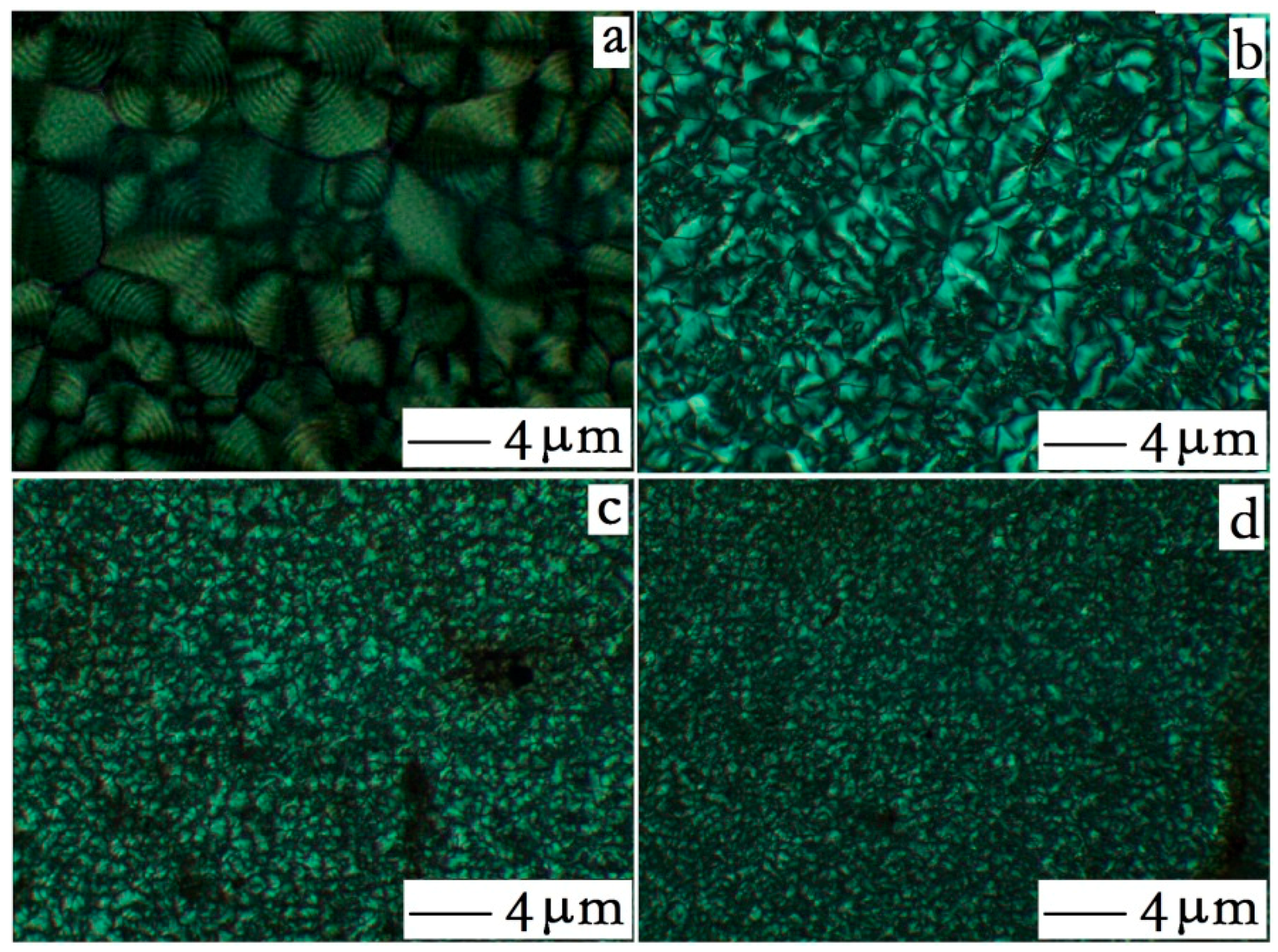
3.2.1. Core-Shell Particle Crystallization Behavior
3.2.2. HDPE Matrix Crystallization Behavior
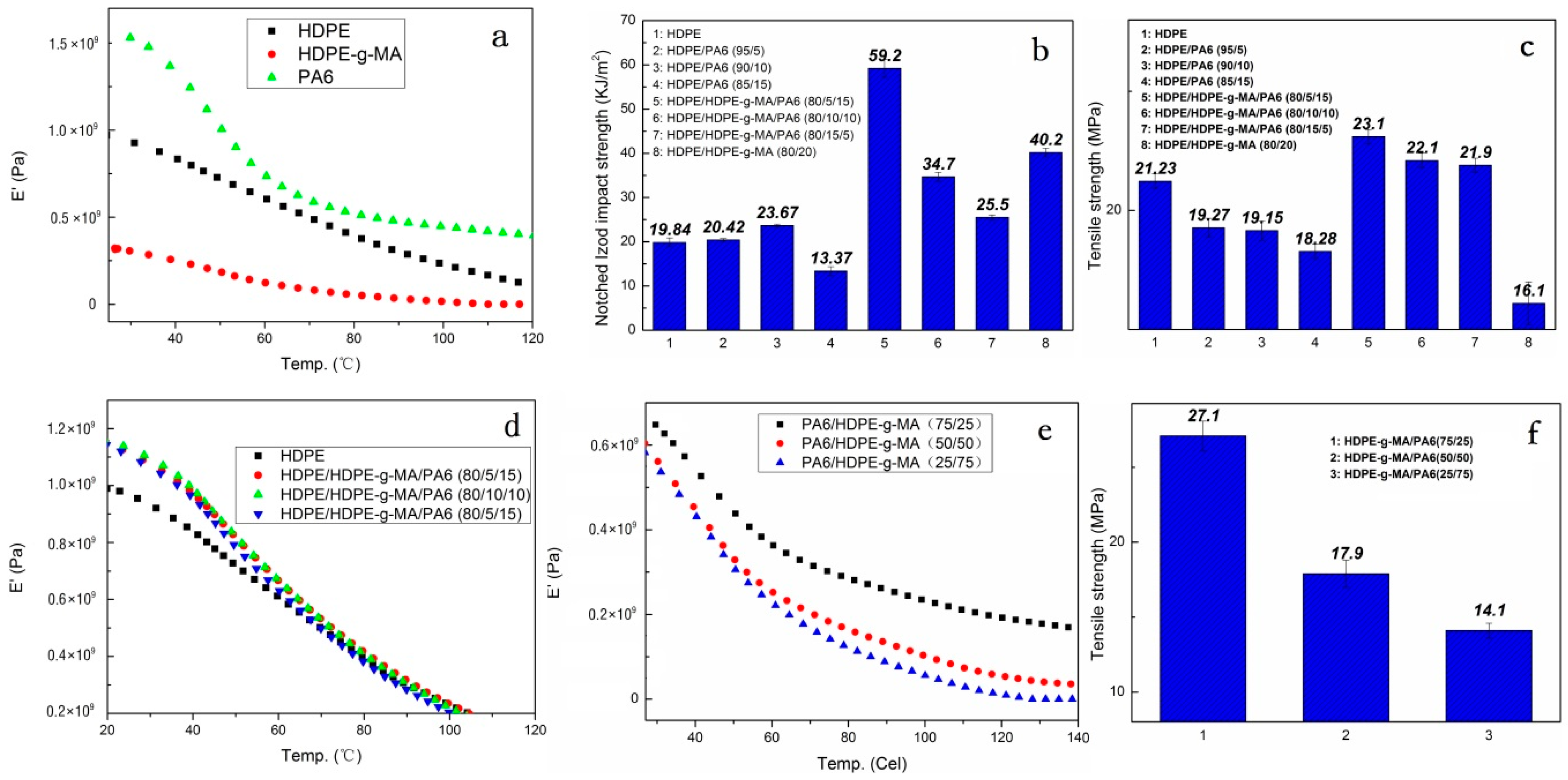
3.3. Mechanical Performance Analysis
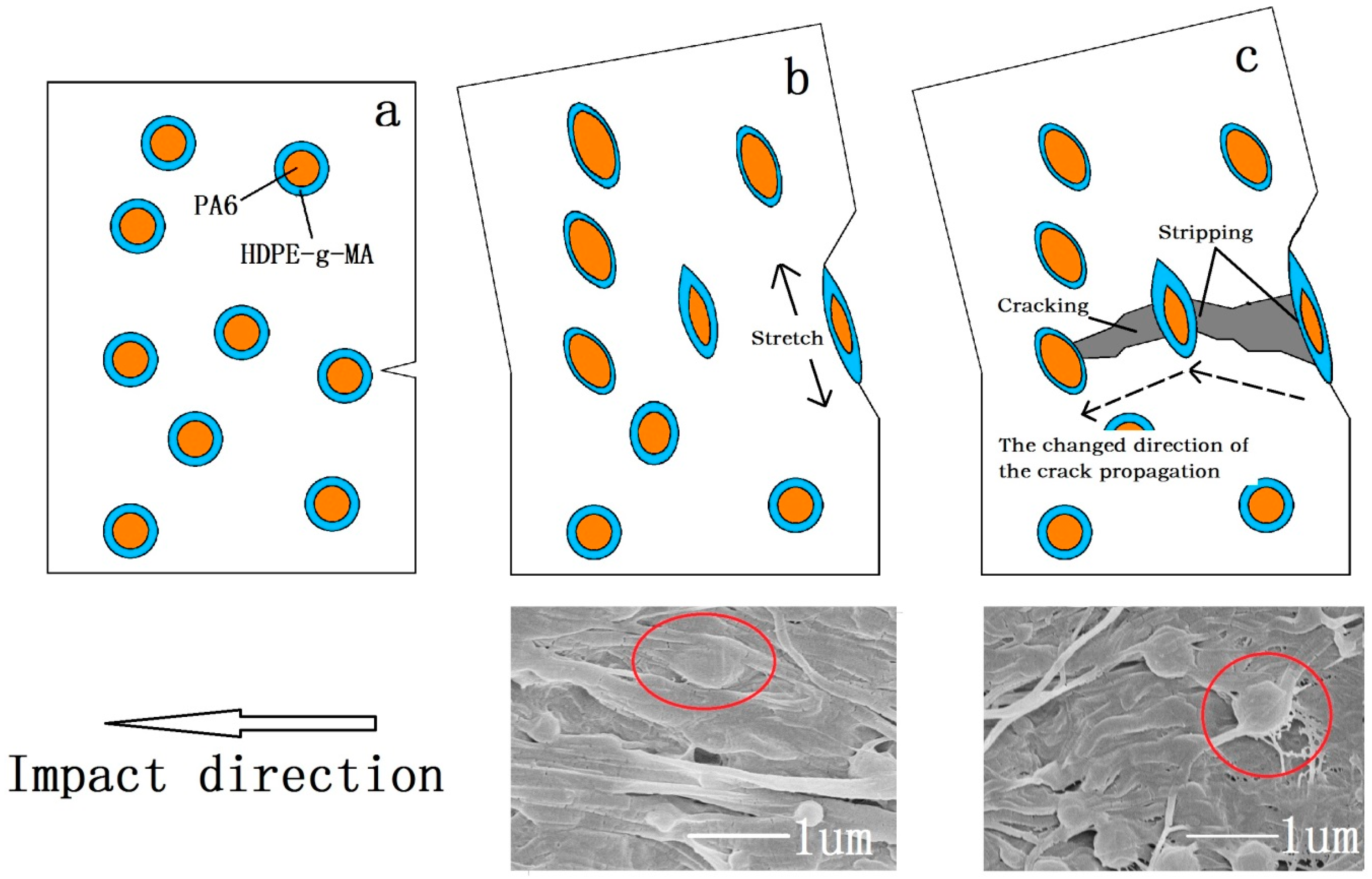
3.3.1. Material Impact Properties
3.3.2. Tensile Strength and Elastic Modulus
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bajracharya, R.M.; Manalo, A.C.; Karunasena, W.; Lau, K.T. Experimental and theoretical studies on the properties of injection molded glass fiber reinforced mixed plastics composites. Compos. Part A 2016, 84, 393–405. [Google Scholar] [CrossRef]
- Lu, X.; Tang, L.; Wang, L.; Zhao, J.; Li, D.; Wu, Z.; Xiao, P. Morphology and properties of bio-based poly(lactic acid)/high-density polyethylene blends and their glass fiber reinforced composites. Polym. Test. 2016, 54, 90–97. [Google Scholar] [CrossRef]
- Perez, C.J.; Alvarez, V.A. Ternary composites based on hdpe and Mater-Bi reinforced with hemp fibers. J. Therm. Anal. Calorim. 2016, 124, 499–508. [Google Scholar] [CrossRef]
- Kuzmanović, M.; Delva, L.; Mi, D.; Martins, C.; Cardon, L.; Ragaert, K. Development of crystalline morphology and its relationship with mechanical properties of PP/PET microfibrillar composites containing POE and POE-g-MA. Polymers 2018, 10, 291. [Google Scholar] [CrossRef]
- Zhang, P.; Wang, B. Positive temperature coefficient effect and mechanism of compatible LLDPE/HDPE composites doping conductive graphite powders. J. Appl. Polym. Sci. 2018, 135, 46453. [Google Scholar] [CrossRef]
- Zhang, Q.; Cai, H.; Ren, X.; Kong, L.; Liu, J.; Jiang, X. The Dynamic Mechanical Analysis of Highly Filled Rice Husk Biochar/High-Density Polyethylene Composites. Polymers 2017, 9, 628. [Google Scholar] [CrossRef]
- Srivastava, P.; Sinha, S. Effect of surface treatment on hair fiber as reinforcement of HDPE composites: Mechanical properties and water absorption kinetics. Korean J. Chem. Eng. 2018, 35, 1209–1218. [Google Scholar] [CrossRef]
- Zhang, L.; Lv, S.; Sun, C.; Wan, L.; Tan, H.; Zhang, Y. Effect of mah-g-pla on the properties of wood fiber/polylactic acid composites. Polymers 2017, 9, 591. [Google Scholar] [CrossRef]
- Lee, S.; Kim, M.S.; Ogale, A.A. Influence of carbon nanofiber structure on properties of linear low density polyethylene composites. Polym. Eng. Sci. 2010, 50, 93–99. [Google Scholar] [CrossRef]
- Jang, S.H.; Kim, Y.H.; Lim, S.; Choi, G.D.; Kim, S.H.; Kim, W.N. Effects of fiber characteristics on the mechanical and rheological properties of poly(butylene terephthalate)/glass fiber composites. J. Appl. Polym. Sci. 2010, 116, 3005–3012. [Google Scholar] [CrossRef]
- Lv, Y.; Peng, C. Chemically grafting carbon nanotubes onto carbon fibers for enhancing interfacial strength in carbon fiber/HDPE composites. Surf. Interface Anal. 2018, 50, 552–557. [Google Scholar]
- Zhang, S.; Ke, Y.; Cao, X.; Ma, Y.; Wang, F. Effect of Al2O3 fibers on the thermal conductivity and mechanical properties of high density polyethylene with the absence and presence of compatibilizer. J. Appl. Polym. Sci. 2012, 124, 4874–4881. [Google Scholar] [CrossRef]
- Liu, W.; Wang, L.; Mai, J.; Zhang, A. Synthesis of EPDM-g-MAN by suspension graft copolymerization and its toughening effect on SAN resin. Polym. Bull. 2014, 71, 3111–3129. [Google Scholar] [CrossRef]
- Mao, Z.; Zhang, J. Largely improved the low temperature toughness of acrylonitrile-styrene-acrylate (ASA) resin: Fabricated a core-shell structure of two elastomers through the differences of interfacial tensions. Appl. Surf. Sci. 2018, 444, 345–354. [Google Scholar] [CrossRef]
- Jung, H. Carbon fiber-reinforced plastics based on epoxy resin toughened with core shell rubber impact modifiers. Polymers. 2015, 15, 369–375. [Google Scholar]
- Sun, D.X.; Yang, C.J.; Qi, X.D.; Yang, J.H.; Wang, Y. Largely enhanced fracture toughness of the PP/EPDM blends induced by adding carbon nanofibers. Compos. Sci. Technol. 2018, 164, 146–152. [Google Scholar] [CrossRef]
- Mansour, G.; Tsongas, K.; Tzetzis, D. Investigation of the dynamic mechanical properties of epoxy resins modified with elastomers. Compos. Part B 2016, 94, 152–159. [Google Scholar] [CrossRef]
- Chi, X.; Cheng, L.; Liu, W.; Zhang, X.; Li, S. Characterization of Polypropylene Modified by Blending Elastomer and Nano-Silica. Materials 2018, 11, 1321. [Google Scholar] [CrossRef] [PubMed]
- Douse, E.; Kopsidas, S.; Jesson, D.; Hamerton, I. Modification of stress-strain behavior in aromatic polybenzoxazines using core shell rubbers. React. Funct. Polym. 2016, 103, 117–130. [Google Scholar] [CrossRef]
- Yuan, Q.; Caillard, J.; Rong, L. Fracture toughness of soft materials with rate-independent hysteresis. J. Mech. Phys. Solids 2018, 118, 341–364. [Google Scholar]
- Wal, A.V.D.; Mulder, J.J.; Oderkerk, J.; Gaymans, R.J. Polypropylene-rubber blends: 1. the effect of the matrix properties on the impact behavior. Polymer 1998, 39, 6781–6787. [Google Scholar]
- Badamshina, E.R.; Goldstein, R.V.; Ustinov, K.B.; Estrin, Y.I. Strength and fracture toughness of polyurethane elastomers modified with carbon nanotubes. Phys. Mesotech. 2018, 21, 187–192. [Google Scholar] [CrossRef]
- Wal, A.V.D.; Nijhof, R.; Gaymans, R.J. Polypropylene-rubber blends: 2. the effect of the rubber content on the deformation and impact behavior. Polymer. 1999, 40, 6031–6044. [Google Scholar]
- Chaudhary, S.; Surekha, P.; Kumar, D.; Rajagopal, C.; Roy, P.K. Amine-functionalized poly(styrene) microspheres as thermoplastic toughened for epoxy resin. Polym. Compos. 2015, 36, 174–183. [Google Scholar] [CrossRef]
- Keivani, M.; Khamesinia, A.; Bagheri, R.; Kouchakzadeh, M.A.; Abadyan, M. Study of synergistic toughening in a bimodal epoxy nanocomposite. J. Reinf. Plast. Compos. 2015, 34, 281–292. [Google Scholar] [CrossRef]
- Kurauchi, T.; Ohta, T. Energy absorption in blends of polycarbonate with abs and san. J. Mater. Sci. 1984, 19, 1699–1709. [Google Scholar] [CrossRef]
- Sprenger, S. Epoxy resins modified with elastomers and surface-modified silica nanoparticles. Polymer 2013, 54, 4790–4797. [Google Scholar] [CrossRef]
- Bucknall, C.B.; Paul, D.R. Notched impact behavior of polymer blends: Part 2: Dependence of critical particle size on rubber particle volume fraction. Polymer 2013, 54, 320–329. [Google Scholar] [CrossRef]
- Zuiderduin, W.C.J.; Westzaan, C.; Huétink, J.; Gaymans, R.J. Toughening of polypropylene with calcium carbonate particles. Polymer 2003, 44, 261–275. [Google Scholar] [CrossRef]
- Argon, A.S.; Cohen, R.E. Toughen ability of polymers. Polymer 2003, 44, 6013–6032. [Google Scholar] [CrossRef]
- Wang, B.; Wu, D.; Zhu, L.; Jin, Z.; Zhao, K. High-density polyethylene-based ternary blends toughened by PA6/PBT core-shell particles. Polym. Plast. Technol. 2017, 56, 1–8. [Google Scholar] [CrossRef]
- Li, L.P.; Yin, B.; Zhou, Y.; Gong, L.; Yang, M.B.; Xie, B.H. Characterization of PA6/EPDM-g-MA/HDPE ternary blends: The role of core-shell structure. Polymer 2012, 53, 3043–3051. [Google Scholar] [CrossRef]
- Shen, C.; Zhou, Y.; Dou, R.; Wang, W.; Yin, B.; Yang, M.B. Effect of the core-forming polymer on phase morphology and mechanical properties of PA6/EPDM-g-MA/HDPE ternary blends. Polymer 2015, 56, 395–405. [Google Scholar] [CrossRef]
- Chen, F.; Shangguan, Y.; Jiang, Y.; Qiu, B.; Luo, G.; Zheng, Q. Toughening with little rigidity loss and mechanism for modified polypropylene by polymer particles with core-shell structure. Polymer 2015, 65, 81–92. [Google Scholar] [CrossRef]
- Yin, B.; Li, L.P.; Zhou, Y.; Gong, L.; Yang, M.B.; Xie, B.H. Largely improved impact toughness of PA6/EPDM-g-MA/HDPE ternary blends: The role of core-shell particles formed in melt processing on preventing micro-crack propagation. Polymer 2013, 54, 1938–1947. [Google Scholar] [CrossRef]
- Valera, T.S.; And, A.T.M.; Demarquette, N.R. Study of morphologies of PMMA/PP/PS ternary blends. Macromolecules 2006, 39, 2663–2675. [Google Scholar] [CrossRef]
- Filippi, S.; Chiono, V.; Polacco, G.; Paci, M.; Minkova, L.I.; Magagnini, P. Reactive compatibilizer precursors for LDPE/PA6 blends, 1. Ethylene/acrylic acid copolymers. Macromol. Chem. Phys. 2002, 203, 1512–1525. [Google Scholar] [CrossRef]
- Jiang, C.; Filippi, S.; Magagnini, P. Reactive compatibilizer precursors for LDPE/PA6 blends. ii: Maleic anhydride grafted polyethylenes. Polymer 2003, 44, 2411–2422. [Google Scholar] [CrossRef]
- Kord, B.; Roohani, M. Water transport kinetics and thickness swelling behavior of natural fiber-reinforced hdpe/cnt nanocomposites. Compos. Part B 2017, 126, 94–99. [Google Scholar] [CrossRef]
- Bian, J.; Wang, G.; Lin, H.L.; Zhou, X.; Wang, Z.J.; Xiao, W.Q. HDPE composites strengthened-toughened synergistically by l-aspartic acid functionalized graphene/carbon nanotubes hybrid nanomaterials. J. Appl. Polym. Sci. 2017, 134, 45055. [Google Scholar] [CrossRef]
- Luzinov, I.; Xi, K.; Pagnoulle, C.; Huynh-Ba, G.; Jérôme, R. Composition effect on the core-shell morphology and mechanical properties of ternary polystyrene/styrene-butadiene rubber/polyethylene blends. Polymer 1999, 40, 2511–2520. [Google Scholar] [CrossRef]
- Shangguan, Y.; Chen, F.; Yang, J.; Jia, E.; Zheng, Q. A new approach to fabricate polypropylene alloy with excellent low-temperature toughness and balanced toughness-rigidity through unmatched thermal expansion coefficients between components. Polymer 2017, 112, 318–324. [Google Scholar] [CrossRef]
- Ohlsson, B.; Hassander, H.; Törnell, B. Effect of the mixing procedure on the morphology and properties of compatibilized polypropylene/polyamide blends. Polymer 1998, 39, 4715–4721. [Google Scholar] [CrossRef]
- Psarski, M.; Pracella, M.; Galeski, A. Crystal phase and crystallinity of polyamide 6/functionalized polyolefin blends. Polymer 2000, 41, 4923–4932. [Google Scholar] [CrossRef]
- Klata, E.; Velde, K.V.D.; Krucińska, I. Dsc investigations of polyamide 6 in hybrid GF/PA6 yarns and composites. Polym. Test. 2003, 22, 929–937. [Google Scholar] [CrossRef]
- Åkesson, D.; Fuchs, T.; Stöss, M.; Root, A.; Stenvall, E.; Skrifvars, M. Recycling of wood fiber-reinforced HDPE by multiple reprocessing. J. Appl. Polym. Sci. 2016, 133. [Google Scholar] [CrossRef]
- Charoenpongpool, S.; Nithitanakul, M.; Grady, B.P. Melt-neutralization of maleic anhydride grafted on high-density polyethylene compatibilizer for polyamide-6/high-density polyethylene blend: Effect of neutralization level on compatibility of the blend. Polym. Bull. 2013, 70, 293–309. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Contact | Angle (°) | Surface Tension | (mN/m) | |
|---|---|---|---|---|---|
| Water | Ethanediol | Total (γ) | Dispersion Component (γd) | Polar Component (γp) | |
| HDPE | 95.7 | 69.9 | 24.3 | 16 | 8.3 |
| HDPE-g-MA | 73.4 | 46.1 | 36.1 | 17.4 | 18.7 |
| PA6 | 64.2 | 60.5 | 41.7 | 7.2 | 34.5 |
| Polymer Pairs | Interfacial Tension (mN/m) |
|---|---|
| HDPE/ HDPE-g-MA | 4.4 |
| HDPE/PA6 | 19.2 |
| HDPE-g-MA/PA6 | 8.8 |
| PA6/HDPE-g-MA (w/w) | (J/g) | Tm (°C) | Tc (°C) | Xc (%) | ||||
|---|---|---|---|---|---|---|---|---|
| PA6 | HDPE-g-MA | PA6 | HDPE-g-MA | PA6 | HDPE-g-MA | PA6 | HDPE-g-MA | |
| 0/100 | - | 120 | - | 110.2 | - | 98.1 | - | 41 |
| 25/75 | 21.2 | 59.2 | 123.5/220.3 | 109.6 | 112.1/189.7 | 99.1 | 37 | 27 |
| 50/50 | 33.3 | 37.8 | 123.5/220.1 | 108.0 | 111.8/120.4 | 98.5 | 29 | 26 |
| 75/25 | 50.2 | 15.0 | 215.1/220.7 | 107.3 | 189.5 | 97.6 | 29 | 21 |
| 100/0 | 69.1 | - | 223 | - | 180.4 | - | 30 | - |
| HDPE/HDPE-g-MA/PA6 (w/w/w) | (J/g) | Tm (HDPE) (°C) | Tc (HDPE) (°C) | Xc (HDPE) (%) | ||||
|---|---|---|---|---|---|---|---|---|
| HDPE | PA6 | HDPE | PA6 | HDPE | PA6 | HDPE | PA6 | |
| 85/0/15 | 153 | 9.3 | 133.3 | 215/220 | 117.8 | 194 | 62 | 30 |
| 80/5/15 | 153 | - | 132.7 | - | 117.9 | - | 65 | - |
| 100/0/0 | 179 | - | 133.3 | - | 116.2 | - | 61 | - |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, L.; Wang, H.; Liu, M.; Jin, Z.; Zhao, K. Effect of Core-Shell Morphology on the Mechanical Properties and Crystallization Behavior of HDPE/HDPE-g-MA/PA6 Ternary Blends. Polymers 2018, 10, 1040. https://doi.org/10.3390/polym10091040
Zhu L, Wang H, Liu M, Jin Z, Zhao K. Effect of Core-Shell Morphology on the Mechanical Properties and Crystallization Behavior of HDPE/HDPE-g-MA/PA6 Ternary Blends. Polymers. 2018; 10(9):1040. https://doi.org/10.3390/polym10091040
Chicago/Turabian StyleZhu, Lien, Haoming Wang, Meihua Liu, Zheng Jin, and Kai Zhao. 2018. "Effect of Core-Shell Morphology on the Mechanical Properties and Crystallization Behavior of HDPE/HDPE-g-MA/PA6 Ternary Blends" Polymers 10, no. 9: 1040. https://doi.org/10.3390/polym10091040
APA StyleZhu, L., Wang, H., Liu, M., Jin, Z., & Zhao, K. (2018). Effect of Core-Shell Morphology on the Mechanical Properties and Crystallization Behavior of HDPE/HDPE-g-MA/PA6 Ternary Blends. Polymers, 10(9), 1040. https://doi.org/10.3390/polym10091040