Synthesis of Poly(methyl methacrylate-co-butyl acrylate)/Perfluorosilyl Methacrylate Core-Shell Nanoparticles: Novel Approach for Optimization of Coating Process



Abstract
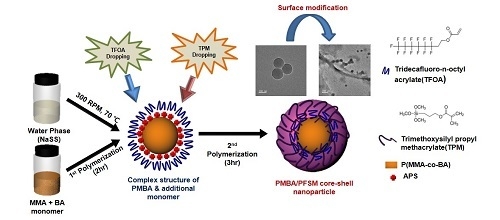
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of Poly(methyl methacrylate-co-butyl acrylate) (PMBA) Nanoparticles
2.3. Synthesis of PMBA/Perfluorosilyl Methacrylate (PMBA-PFSM) Core-Shell Nanoparticles
2.4. Preparation of PMBA-PFSM Cellulose Fiber Nanocomposite by Surface Modification
2.5. Characterization
3. Results and Discussion
3.1. Effects of Change of Input Order on Fluorinated Monomer (TFOA) and Comonomer (TPM)
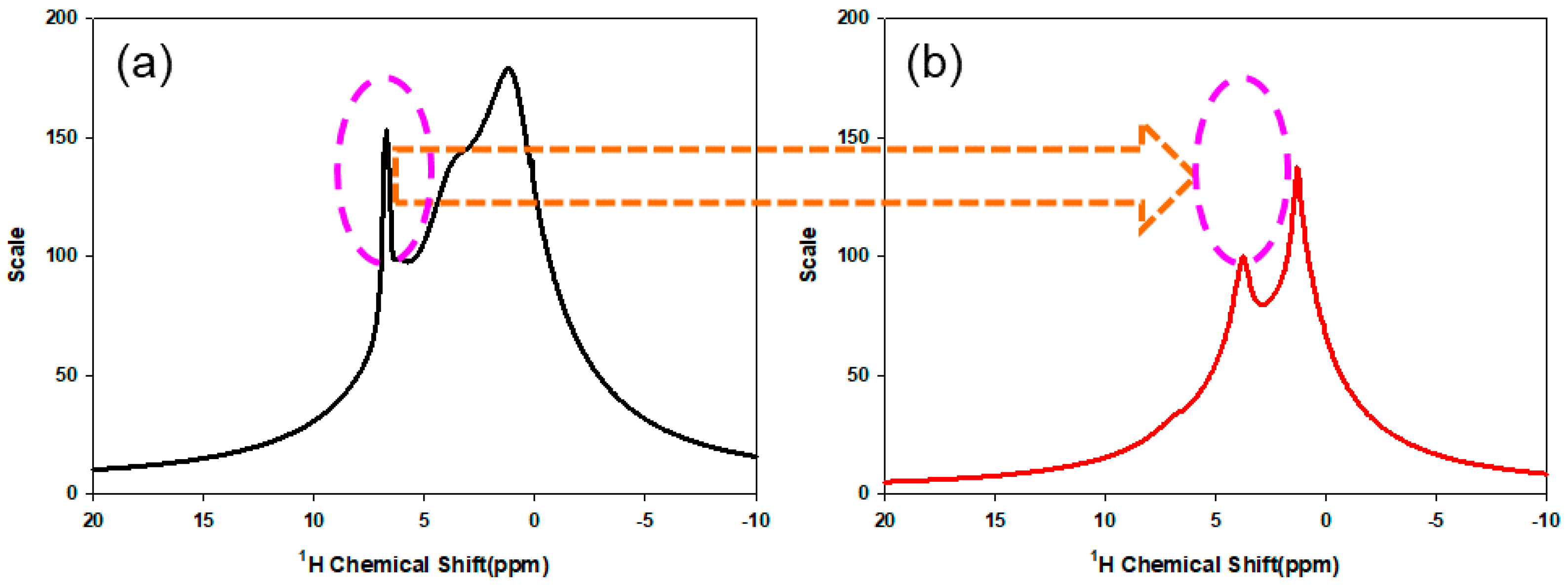
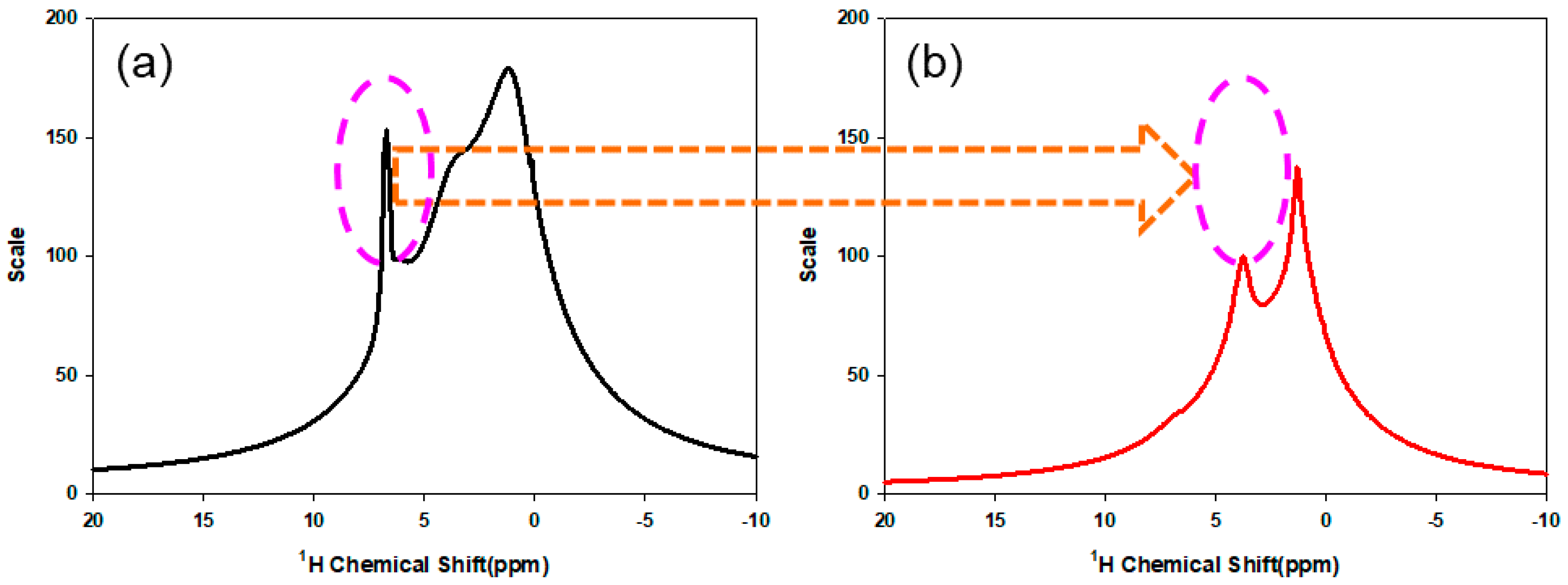
3.2. Chemical Structure of PMBA/PFSM Core-Shell Nanoparticles
3.3. Morphologies of PMBA/PFSM Core-Shell Nanoparticle and Nanocomposite
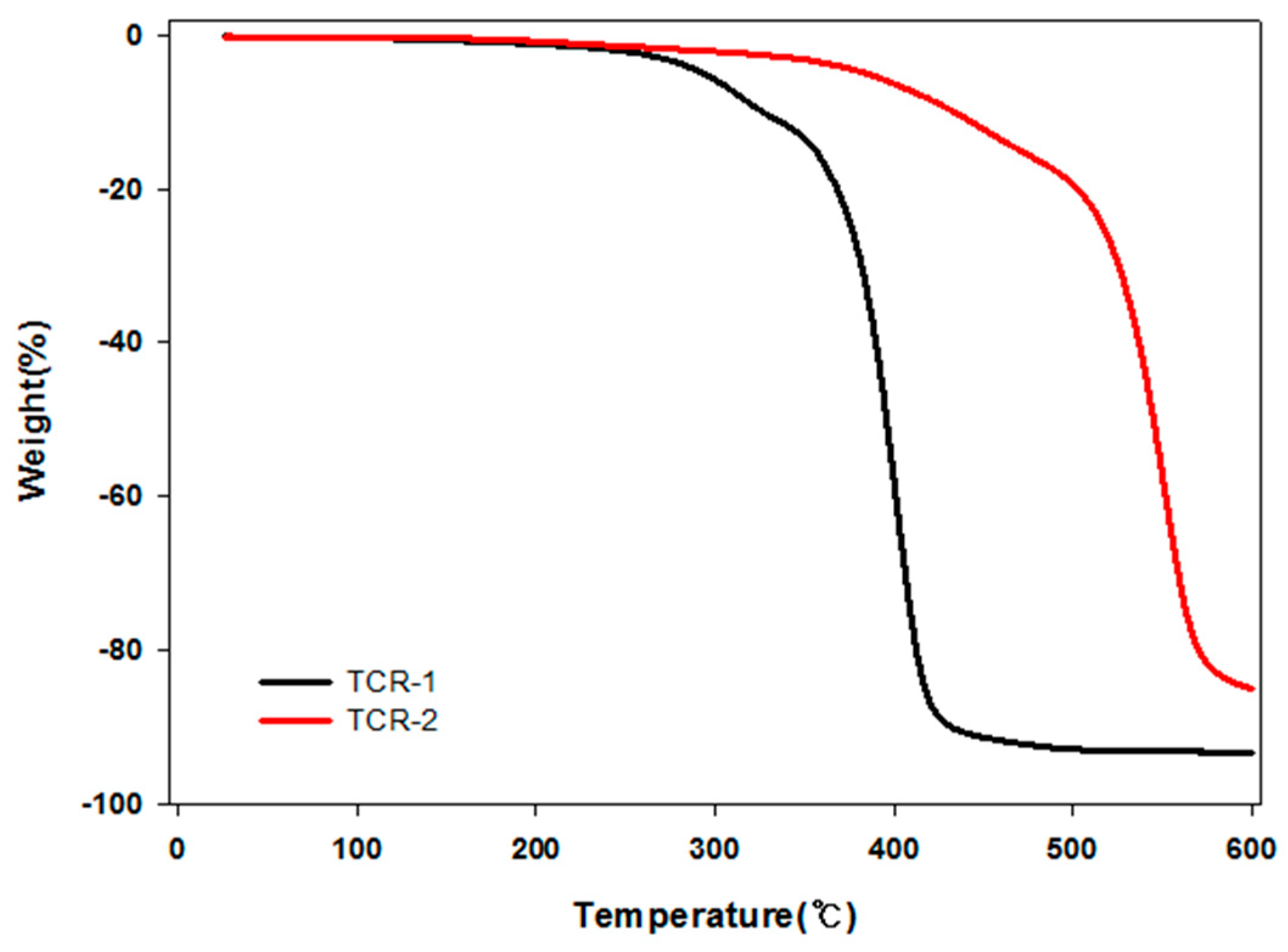
3.4. Physical and Thermal Properties of PMBA/PFSM Core-Shell Nanoparticles
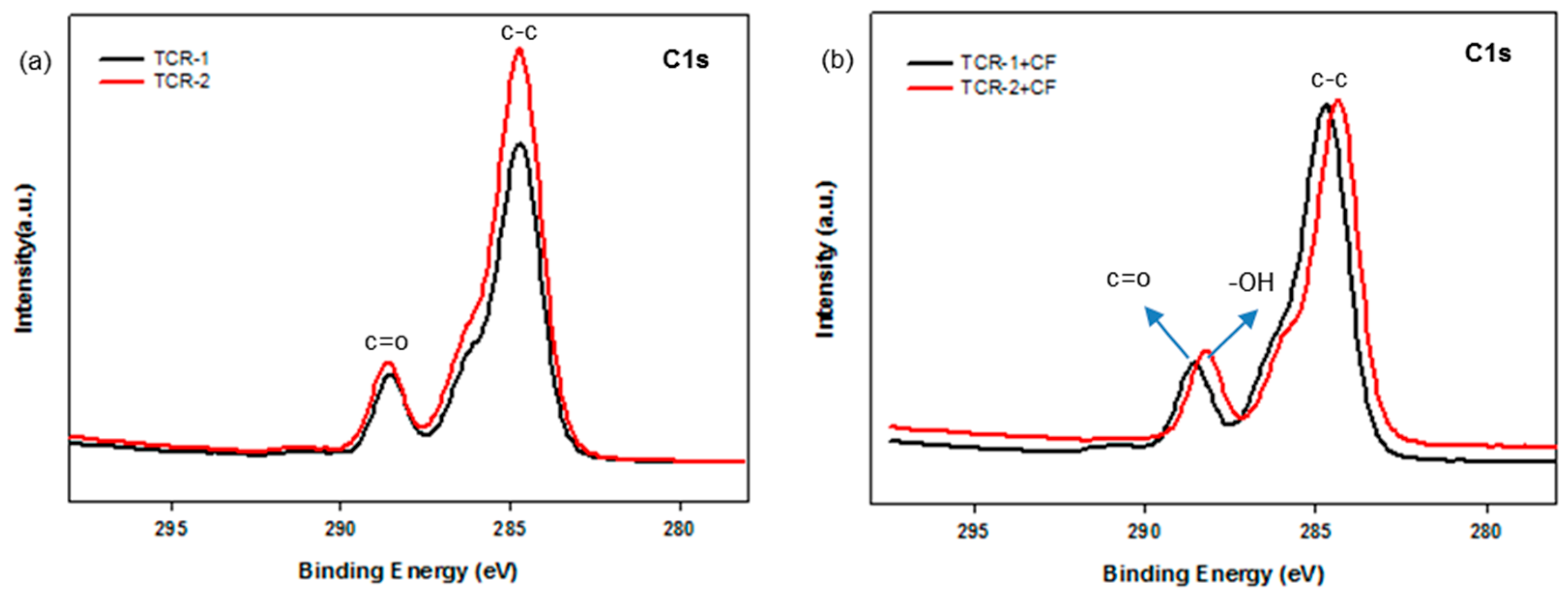
3.5. Surface Chemical Composition of PMBA/PFSM Nanocomposite Coatings
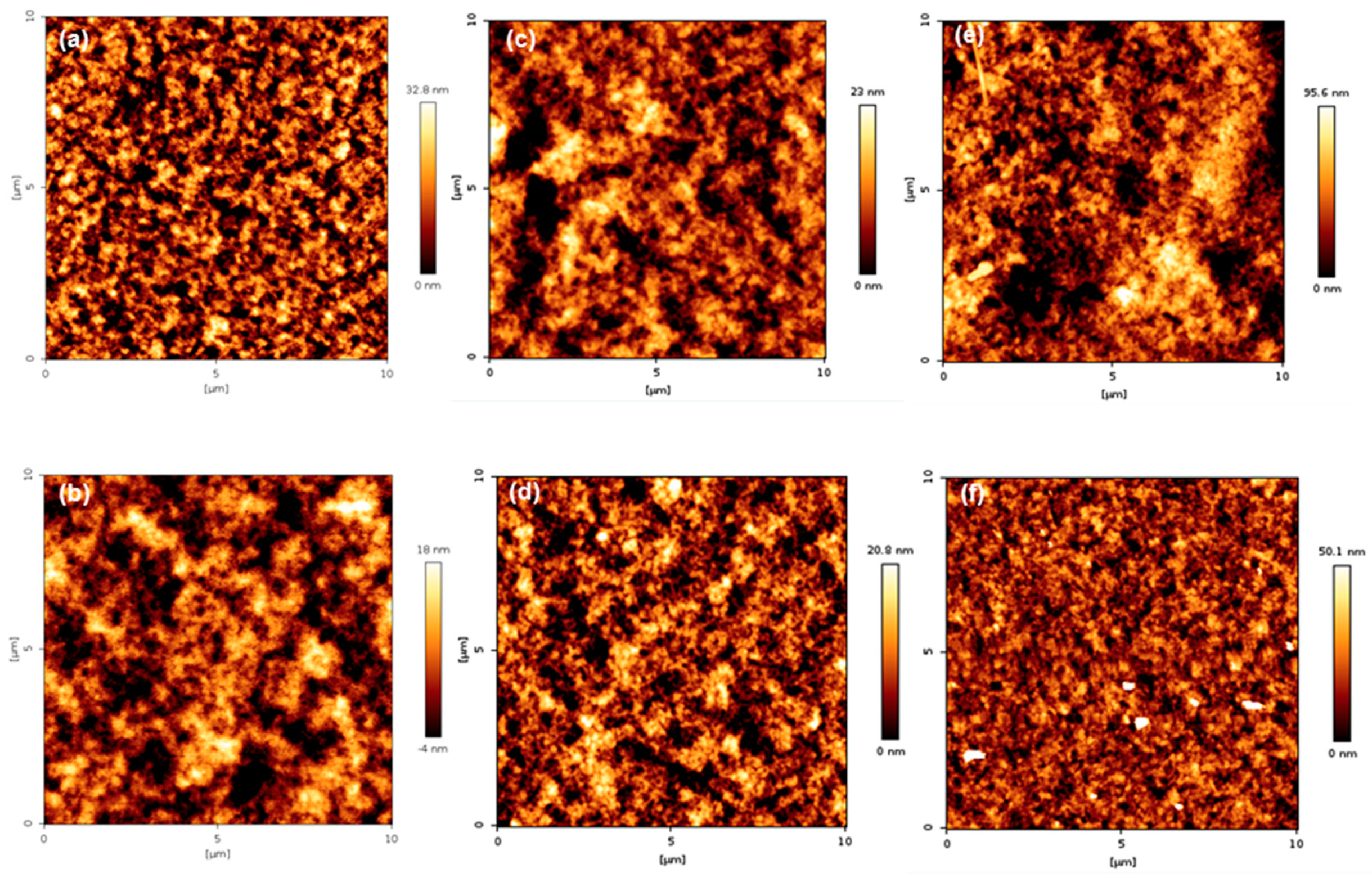
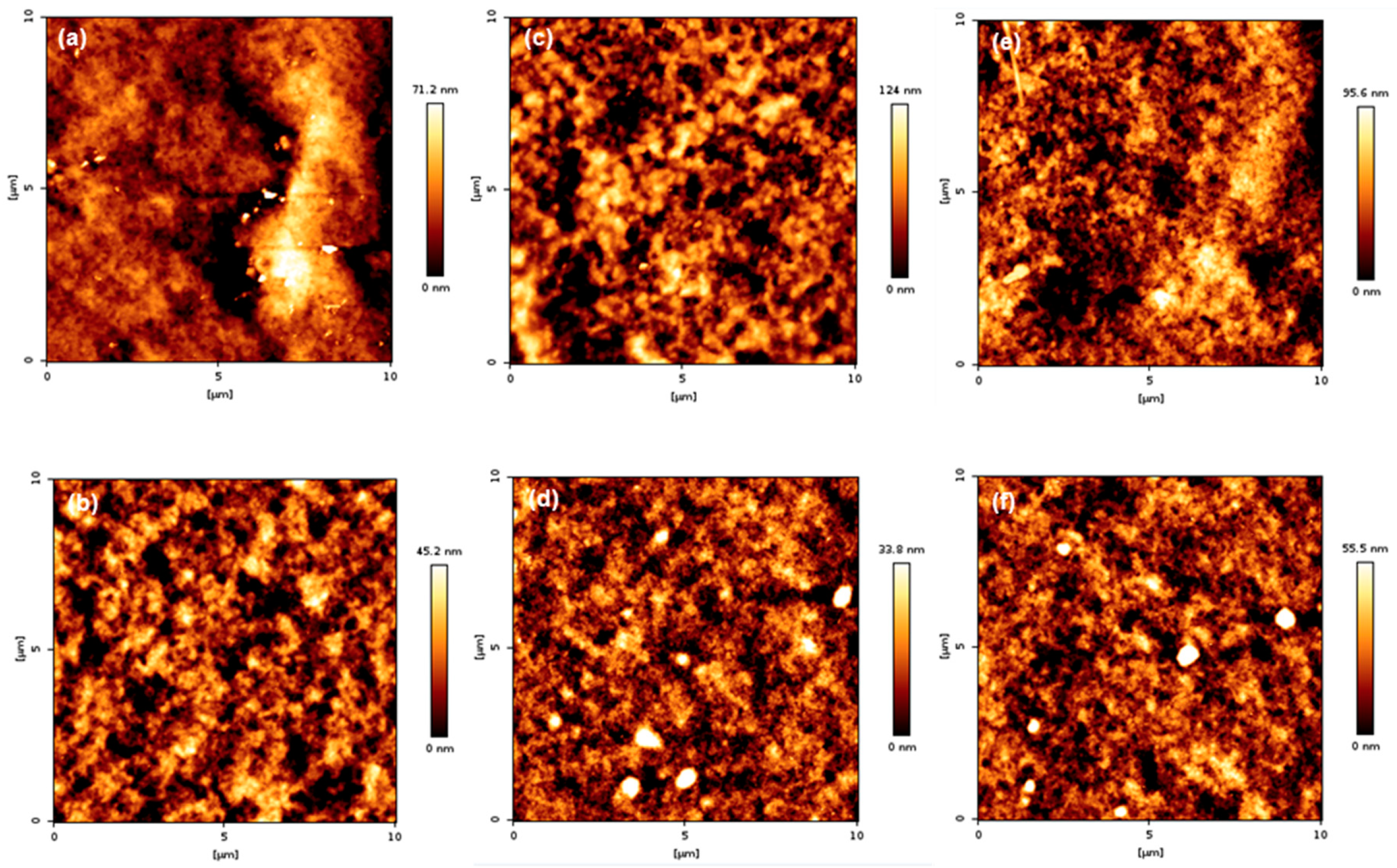
3.6. Surface Topography of PMBA/PFSM Nanocomposite Coatings
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tığlı, R.S.; Evren, V. Synthesis and characterization of pure poly (acrylate) latexes. Prog. Org. Coat. 2005, 52, 144–150. [Google Scholar] [CrossRef]
- Saidi, S.; Guittard, F.; Guimon, C.; Géribaldi, S. Synthesis and characterization of copolymers based on styrene and partially fluorinated acrylates. Eur. Polym. J. 2006, 42, 702–710. [Google Scholar] [CrossRef]
- Timperley, C.M.; Arbon, R.E.; Bird, M.; Brewer, S.A.; Parry, M.W.; Sellers, D.J.; Willis, C.R. Bis (fluoroalkyl) acrylic and methacrylic phosphate monomers, their polymers and some of their properties. J. Fluor. Chem. 2003, 121, 23–31. [Google Scholar] [CrossRef]
- Malshe, V.; Sangaj, N.S. Fluorinated acrylic copolymers: Part I: Study of clear coatings. Prog. Org. Coat. 2005, 53, 207–211. [Google Scholar] [CrossRef]
- Lejars, M.N.; Margaillan, A.; Bressy, C. Fouling release coatings: A nontoxic alternative to biocidal antifouling coatings. Chem. Rev. 2012, 112, 4347–4390. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Morita, M.; Sakata, O.; Sasaki, S.; Takahara, A. Effect of surface molecular aggregation state and surface molecular motion on wetting behavior of water on poly (fluoroalkyl methacrylate) thin films. Macromolecules 2009, 43, 454–460. [Google Scholar] [CrossRef]
- Honda, K.; Morita, M.; Otsuka, H.; Takahara, A. Molecular aggregation structure and surface properties of poly (fluoroalkyl acrylate) thin films. Macromolecules 2005, 38, 5699–5705. [Google Scholar] [CrossRef]
- Morita, M.; Ogisu, H.; Kubo, M. Surface properties of perfluoroalkylethyl acrylate/n-alkyl acrylate copolymers. J. Appl. Polym. Sci. 1999, 73, 1741–1749. [Google Scholar] [CrossRef]
- Parsons, J.R.; Sáez, M.; Dolfing, J.; de Voogt, P. Biodegradation of perfluorinated compounds. In Reviews of Environmental Contamination and Toxicology; Springer: New York, NY, USA, 2008; Volume 196, pp. 53–71. [Google Scholar]
- Frömel, T.; Knepper, T.P. Biodegradation of fluorinated alkyl substances. In Reviews of Environmental Contamination and Toxicology; Springer: New York, NY, USA, 2010; Volume 208, pp. 161–177. [Google Scholar]
- Trudel, D.; Horowitz, L.; Wormuth, M.; Scheringer, M.; Cousins, I.T.; Hungerbühler, K. Estimating consumer exposure to PFOS and PFOA. Risk Anal. 2008, 28, 251–269. [Google Scholar] [CrossRef] [PubMed]
- Nishino, T.; Urushihara, Y.; Meguro, M.; Nakamae, K. Surface properties and structures of diblock copolymer and homopolymer with perfluoroalkyl side chains. J. Colloid Interface Sci. 2005, 283, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Yang, T.; Cheng, S. Synthesis and characterization of poly (fluorinated acrylate)/silica hybrid nanocomposites. J. Appl. Polym. Sci. 2010, 115, 3500–3507. [Google Scholar] [CrossRef]
- Fischer, B.; Autenrieth, T.; Wagner, J. Highly charged inorganic–organic colloidal core-shell particles. Langmuir 2010, 26, 6201–6205. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Ling, H.; Junyan, L.; Gang, C.; Na, W. Preparation and properties of core-shell nanosilica/poly (methyl methacrylate–butyl acrylate–2, 2, 2-trifluoroethyl methacrylate) latex. J. Appl. Polym. Sci. 2011, 120, 1152–1161. [Google Scholar] [CrossRef]
- Xiao, X.; Xu, R. Preparation and surface properties of core-shell polyacrylate latex containing fluorine and silicon in the shell. J. Appl. Polym. Sci. 2011, 119, 1576–1585. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, C.; Chen, X. Emulsifier-free latex of fluorinated acrylate copolymer. Eur. Polym. J. 2006, 42, 694–701. [Google Scholar] [CrossRef]
- Cui, X.; Zhong, S.; Wang, H. Emulsifier-free core-shell polyacrylate latex nanoparticles containing fluorine and silicon in shell. Polymer 2007, 48, 7241–7248. [Google Scholar] [CrossRef]
- Cui, X.; Zhong, S.; Gao, Y.; Wang, H. Preparation and characterization of emulsifier-free core-shell interpenetrating polymer network-fluorinated polyacrylate latex particles. Coll. Surf. A Physicochem. Eng. Asp. 2008, 324, 14–21. [Google Scholar] [CrossRef]
- Tingting, Y.; Hui, P.; Shiyuan, C.; Park, I.J. Soap-free emulsion copolymerization of perfluoroalkyl acrylates in the presence of a reactive surfactant. J. Appl. Polym. Sci. 2007, 104, 2438–2444. [Google Scholar]
- Xiao, X.; Wang, Y. Emulsion copolymerization of fluorinated acrylate in the presence of a polymerizable emulsifier. Coll. Surf. A Physicochem. Eng. Asp. 2009, 348, 151–156. [Google Scholar] [CrossRef]
- Xu, G.; Deng, L.; Wen, X.; Pi, P.; Zheng, D.; Cheng, J.; Yang, Z. Synthesis and characterization of fluorine-containing poly-styrene-acrylate latex with core-shell structure using a reactive surfactant. J. Coat. Technol. Res. 2011, 8, 401–407. [Google Scholar] [CrossRef]
- Wang, J.; Zeng, X.-R.; Li, H.-Q. Preparation and characterization of soap-free fluorine-containing acrylate latex. J. Coat. Technol. Res. 2010, 7, 469–476. [Google Scholar] [CrossRef]
- Chen, L.; Wu, F. BA-MMA-POMA copolymer latexes prepared by using HMPS polymerizable emulsifier. J. Appl. Polym. Sci. 2011, 122, 819–826. [Google Scholar] [CrossRef]
- Chen, L.; Wu, F. Structure and properties of novel fluorinated polyacrylate latex prepared with reactive surfactant. Polym. Sci. Ser. B 2011, 53, 606–611. [Google Scholar] [CrossRef]
- Chen, L.; Wu, F. Properties of polyacrylate latex prepared under different emulsified systems. J. Wuhan Univ. Technol. Mater. Sci. Ed. 2012, 27, 134–137. [Google Scholar] [CrossRef]
- Kobayashi, T.; Singappuli-Arachchige, D.; Wang, Z.; Slowing, I.I.; Pruski, M. Spatial distribution of organic functional groups supported on mesoporous silica nanoparticles: A study by conventional and DNP-enhanced 29 Si solid-state NMR. Phys. Chem. Chem. Phys. 2017, 19, 1781–1789. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | TFOA/TPM Weight Ratio (g) | Diameter (nm) | Contact Angle (°) [Water/HD] | PDI |
|---|---|---|---|---|
| TCR-1 | 2(15%)/1.35(10%) | 123.7 | 130.5/72.8 | 0.201 |
| TCR-2 | 2(15%)/1.35(10%) | 130.1 | 135.1/74.9 | 0.123 |
| TCR-3 | 2.7(20%)/1.35(10%) | 186.3 | 138.6/77.6 | 0.103 |
| TCR-4 | 2.7(20%)/1.35(10%) | 190.1 | 135.1/76.3 | 0.006 |
| TCR-5 | 4.87(30%)/1.35(10%) | 270.5 | 135.1/92.1 | 0.101 |
| TCR-6 | 4.87(30%)/1.35(10%) | 256.3 | 142.4/101.4 | 0.088 |
| TCR-A1 | 2.7(20%)/4.05(30%) | - | 102.1/- | - |
| TCR-A2 | 2.7(20%)/2.7(20%) | - | 106.2/- | - |
| TCR-A3 | 2.7(20%)/0.675(5%) | 96.91 | 141.4/86.7 | 0.05 |
| TCR-B1 | 4.87(30%)/4.05(30%) | - | 107.1/- | - |
| TCR-B2 | 4.87(30%)/2.7(20%) | - | 123.1/- | - |
| TCR-B3 | 4.87(30%)/0.675(5%) | 106.3 | 140.3/97.9 | 0.034 |
| Sample | Concentration, % at wt | |||
|---|---|---|---|---|
| C1s | O1s | F1s | Si2p | |
| TCR-1 | 73.37 | 23.06 | 2.59 | 0.99 |
| TCR-2 | 73.19 | 21.75 | 2.67 | 2.38 |
| TCR-1+CF | 72.14 | 23.07 | 3.52 | 1.26 |
| TCR-2+CF | 62.72 | 29.45 | 1.35 | 6.49 |
| Roughness (nm) | TCR1 | TCR2 | TCR3 | TCR4 | TCR5 | TCR6 |
| Ra | 5.93 | 4.01 | 4.01 | 3.75 | 17.17 | 6.86 |
| Rq | 7.45 | 5.01 | 5.23 | 4.73 | 21.74 | 11.39 |
| Roughness (nm) | TCR1+CF | TCR2+CF | TCR3+CF | TCR4+CF | TCR5+CF | TCR6+CF |
| Ra | 11.38 | 8.16 | 22.40 | 5.54 | 23.95 | 9.06 |
| Rq | 16.19 | 10.28 | 28.22 | 7.68 | 31.73 | 12.62 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kook, J.-W.; Kim, Y.; Hwang, K.; Kim, J.H.; Lee, J.-Y. Synthesis of Poly(methyl methacrylate-co-butyl acrylate)/Perfluorosilyl Methacrylate Core-Shell Nanoparticles: Novel Approach for Optimization of Coating Process. Polymers 2018, 10, 1186. https://doi.org/10.3390/polym10111186
Kook J-W, Kim Y, Hwang K, Kim JH, Lee J-Y. Synthesis of Poly(methyl methacrylate-co-butyl acrylate)/Perfluorosilyl Methacrylate Core-Shell Nanoparticles: Novel Approach for Optimization of Coating Process. Polymers. 2018; 10(11):1186. https://doi.org/10.3390/polym10111186
Chicago/Turabian StyleKook, Jun-Won, Yongsoo Kim, Kiseob Hwang, Jung Hyun Kim, and Jun-Young Lee. 2018. "Synthesis of Poly(methyl methacrylate-co-butyl acrylate)/Perfluorosilyl Methacrylate Core-Shell Nanoparticles: Novel Approach for Optimization of Coating Process" Polymers 10, no. 11: 1186. https://doi.org/10.3390/polym10111186
APA StyleKook, J.-W., Kim, Y., Hwang, K., Kim, J. H., & Lee, J.-Y. (2018). Synthesis of Poly(methyl methacrylate-co-butyl acrylate)/Perfluorosilyl Methacrylate Core-Shell Nanoparticles: Novel Approach for Optimization of Coating Process. Polymers, 10(11), 1186. https://doi.org/10.3390/polym10111186