Efficient Shielding of Polyplexes Using Heterotelechelic Polysarcosines


 and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of Oligomers and DBCO Shielding Agents
2.2.1. Synthesis of Oligomers
2.2.2. Synthesis of Sarcosine-N-Carboxyanhydride
2.2.3. Synthesis of DBCO-pSar
2.2.4. Synthesis of DBCO-pSar-Ac
2.2.5. Synthesis of DBCO-pSar-FolA
2.2.6. Gel Permeation Chromatography
2.2.7. UV-Vis Spectroscopy
2.3. Formation of siRNA Polyplexes
2.4. Functionalization of Polyplexes with DBCO Reagents
2.5. siRNA Binding Assays
2.6. Particle Size and Zeta Potential Measurements
2.7. Cell Culture
2.8. Cell Association and Internalization of siRNA Polyplexes Measured with Flow Cytometry
2.9. Confocal Laser Scanning Microscopy (CLSM)
2.10. Mouse Tumor Model
2.11. Biodistribution Study
3. Results and Discussion
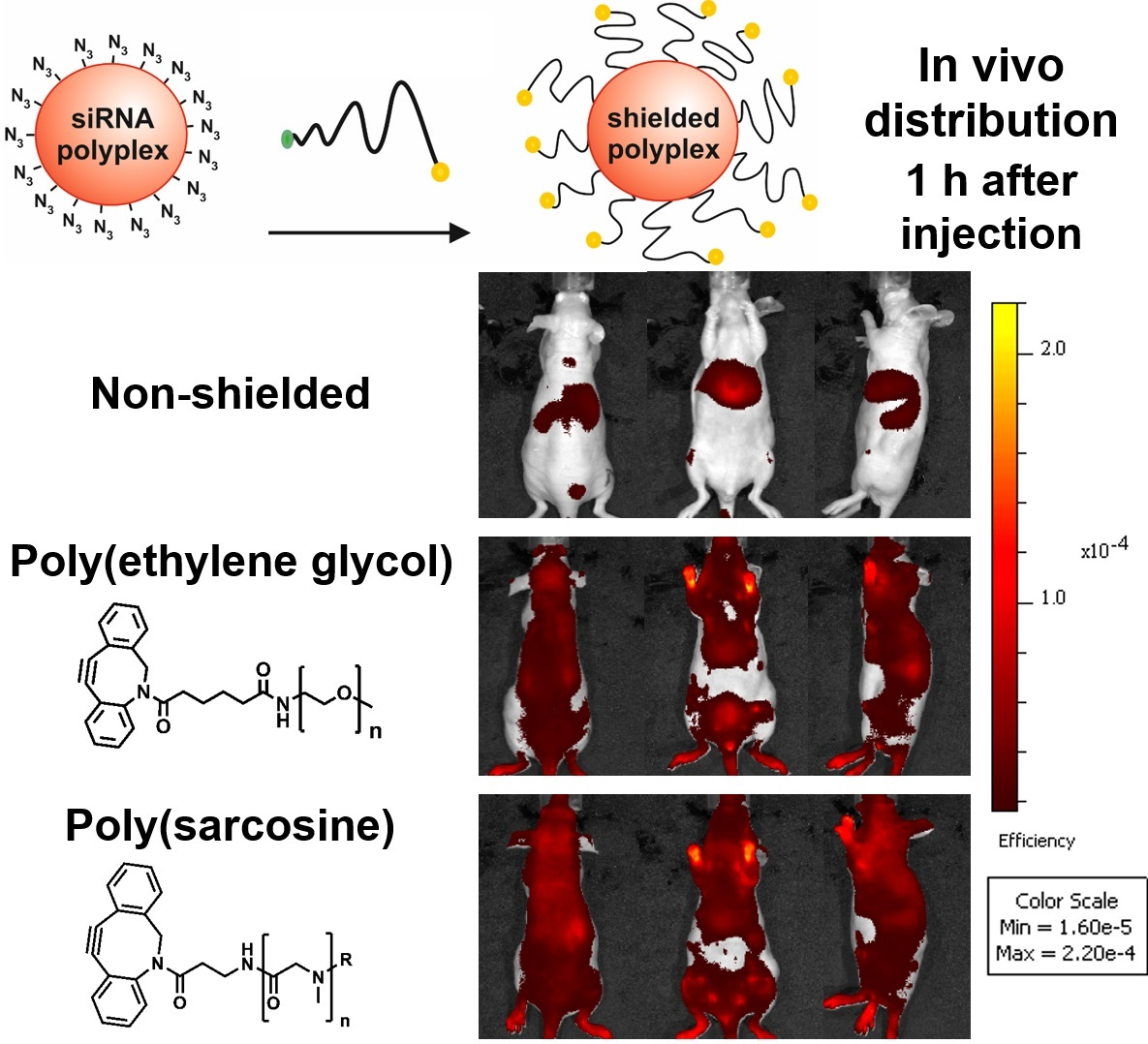
3.1. Design and Synthesis of Lipo-Oligomers for Click Chemistry
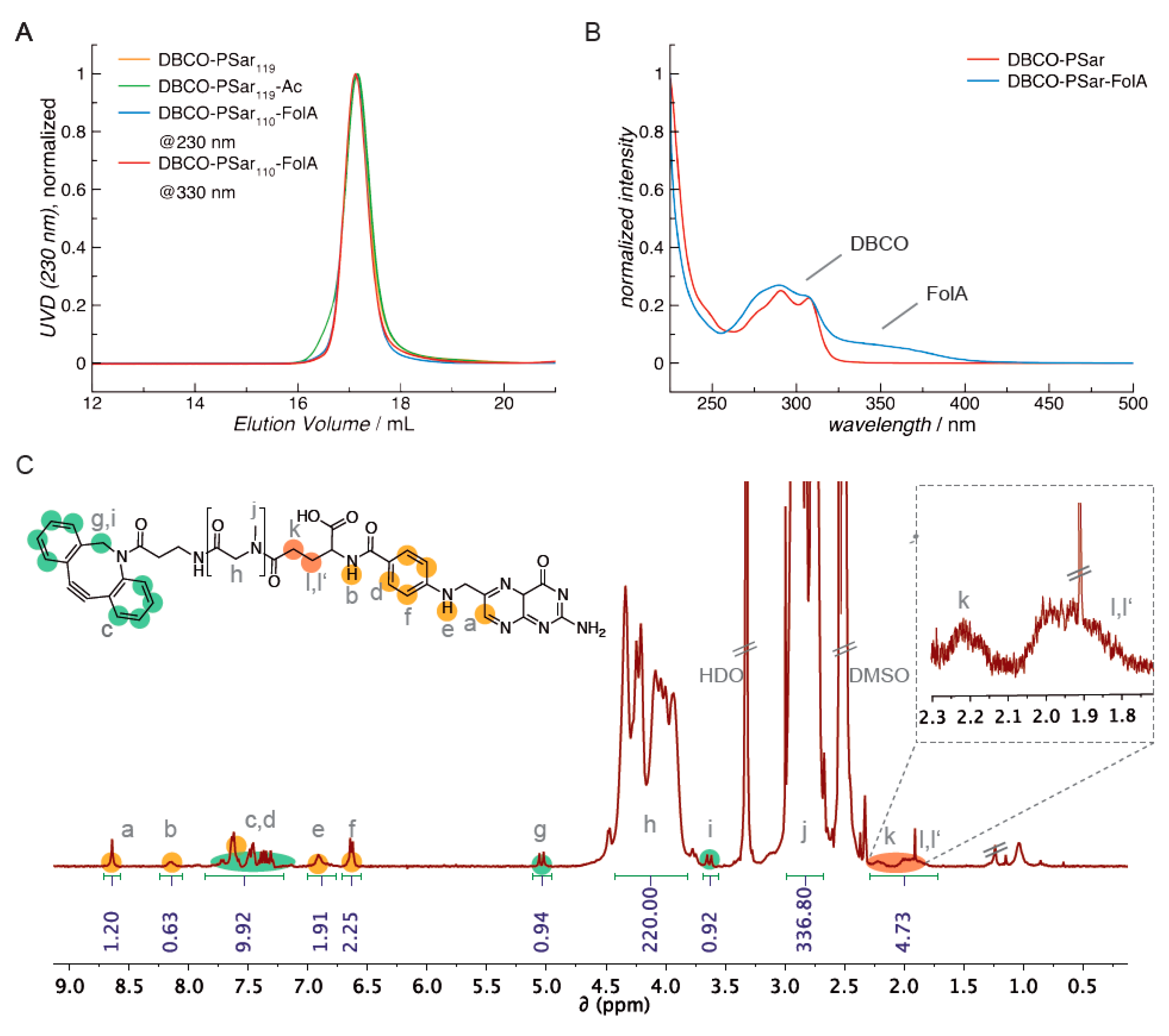
3.2. Synthesis of DBCO-Modified Polysarcosine
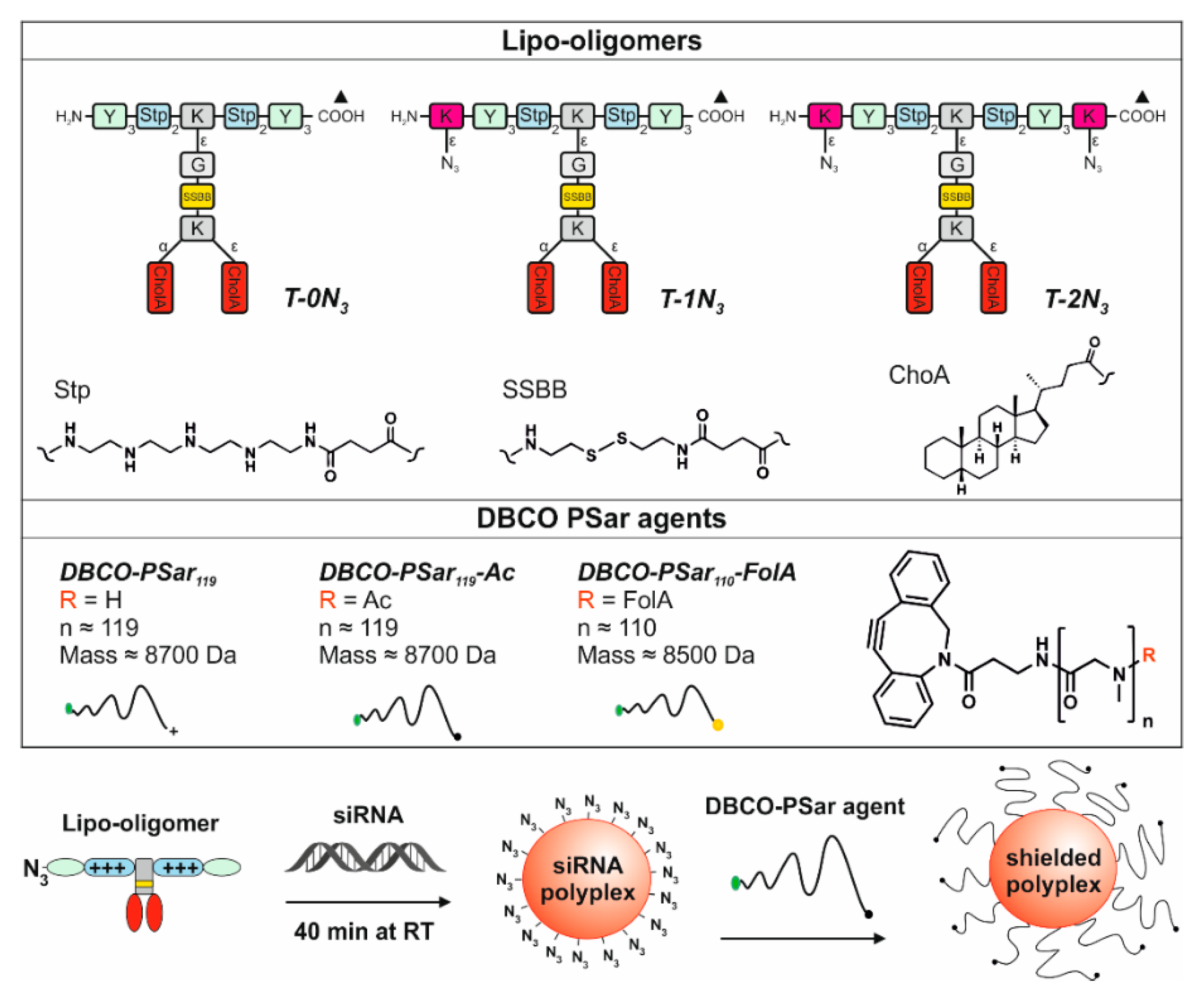
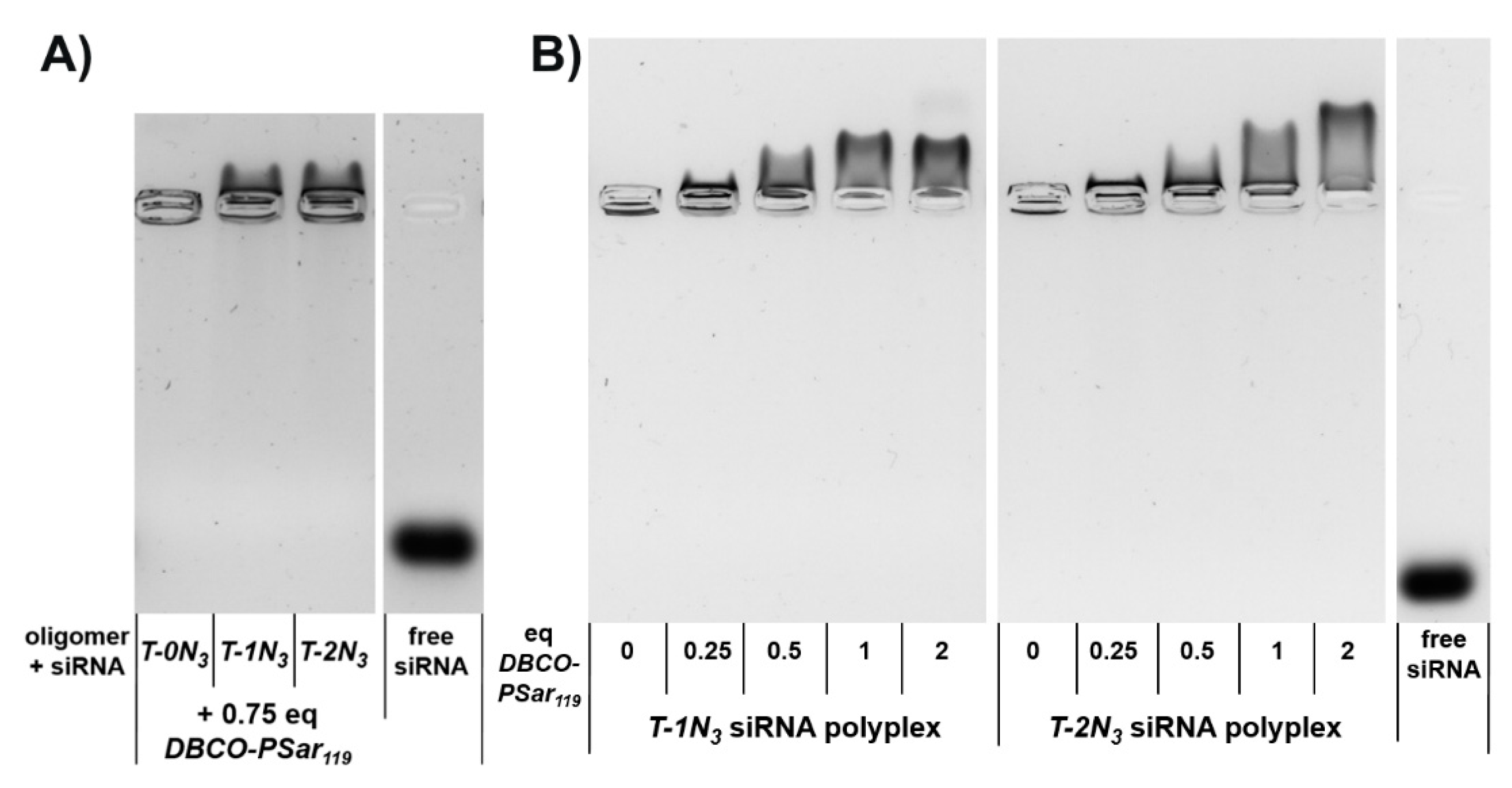
3.3. Polyplex Formation and pSar-Shielding
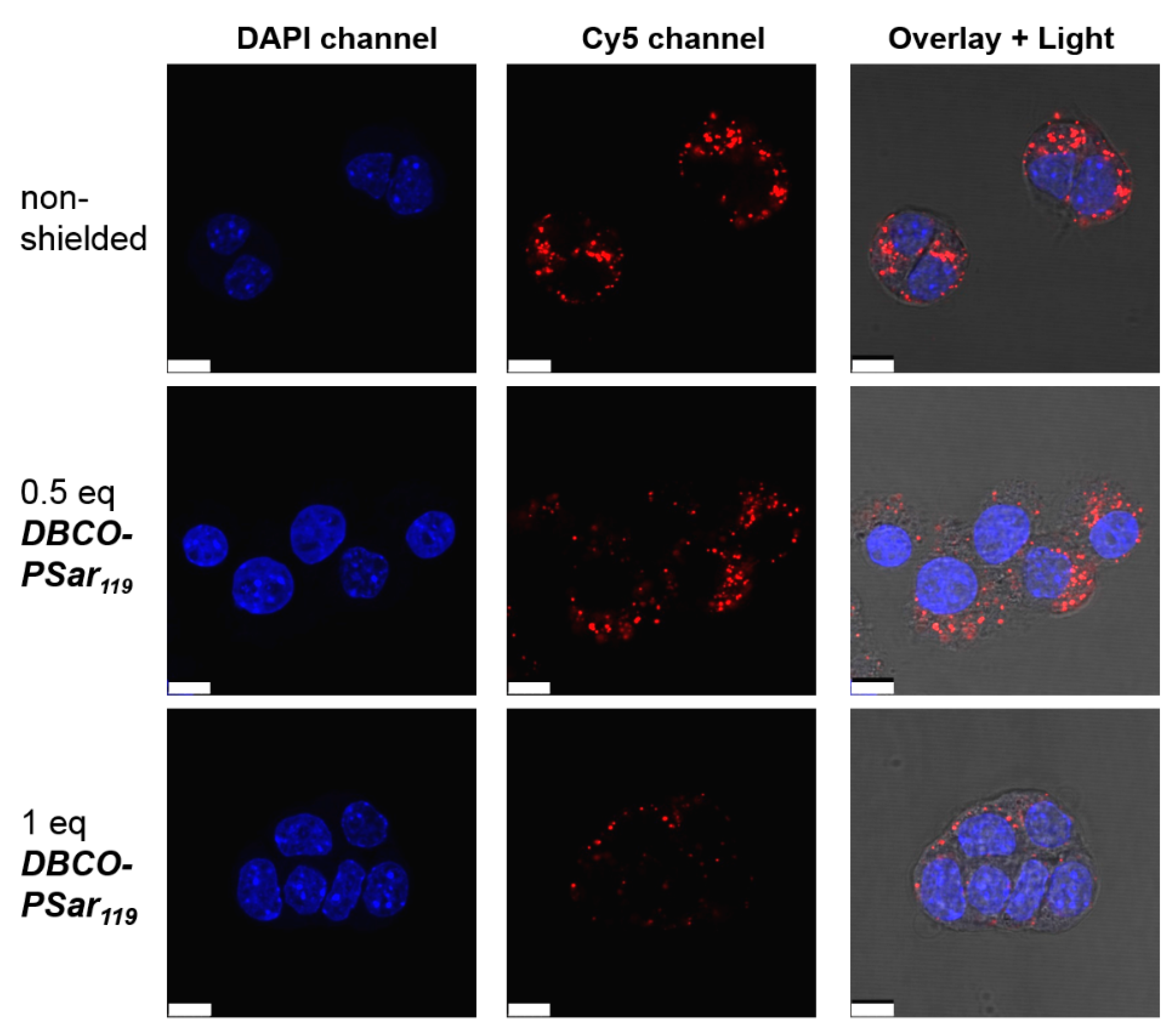
3.4. Evaluation of pSar-Shielding Agents In Vitro
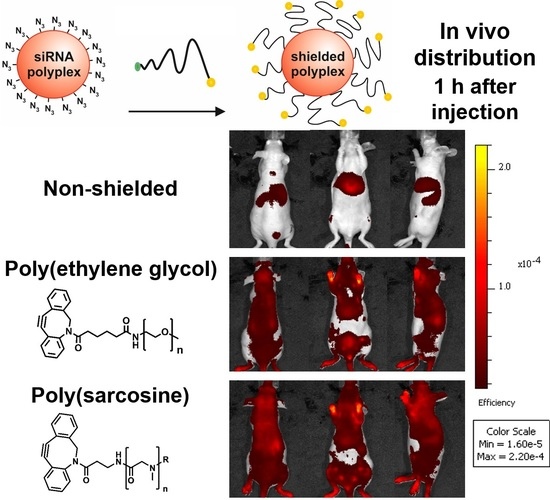
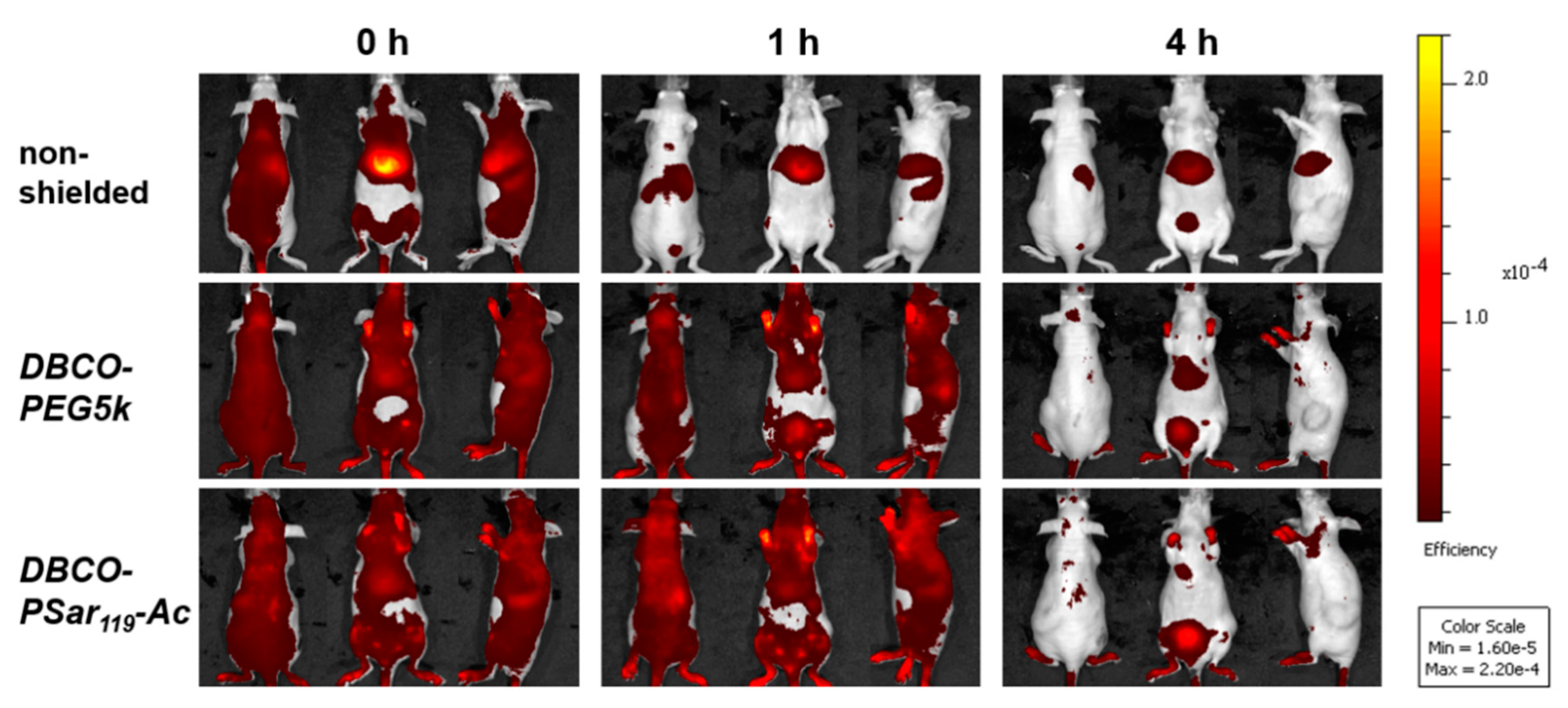
3.5. Distribution of pSar-Functionalized Polyplexes In Vivo
3.6. Attachment of the Targeting Ligand Folate to Polysarcosine
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tabernero, J.; Shapiro, G.I.; LoRusso, P.M.; Cervantes, A.; Schwartz, G.K.; Weiss, G.J.; Paz-Ares, L.; Cho, D.C.; Infante, J.R.; Alsina, M.; et al. First-in-humans trial of an rna interference therapeutic targeting VEGF and KSP in cancer patients with liver involvement. Cancer Discov. 2013, 3, 406–417. [Google Scholar] [CrossRef] [PubMed]
- Kacsinta, A.D.; Dowdy, S.F. Current views on inducing synthetic lethal RNAi responses in the treatment of cancer. Expert Opin. Biol. Ther. 2016, 16, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Haussecker, D. Current issues of RNAi therapeutics delivery and development. J. Control. Release 2014, 195, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.E.; Zuckerman, J.E.; Choi, C.H.; Seligson, D.; Tolcher, A.; Alabi, C.A.; Yen, Y.; Heidel, J.D.; Ribas, A. Evidence of RNAi in humans from systemically administered sirna via targeted nanoparticles. Nature 2010, 464, 1067–1070. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.T.; Mixson, A.J. SiRNA nanoparticles: The future of RNAi therapeutics for oncology? Nanomedicine 2014, 9, 2251–2254. [Google Scholar] [CrossRef] [PubMed]
- Nair, J.K.; Willoughby, J.L.; Chan, A.; Charisse, K.; Alam, M.R.; Wang, Q.; Hoekstra, M.; Kandasamy, P.; Kel’in, A.V.; Milstein, S.; et al. Multivalent n-acetylgalactosamine-conjugated sirna localizes in hepatocytes and elicits robust RNAi-mediated gene silencing. J. Am. Chem. Soc. 2014, 136, 16958–16961. [Google Scholar] [CrossRef] [PubMed]
- Parmar, R.; Willoughby, J.L.; Liu, J.; Foster, D.J.; Brigham, B.; Theile, C.S.; Charisse, K.; Akinc, A.; Guidry, E.; Pei, Y.; et al. 5′-(e)-vinylphosphonate: A stable phosphate mimic can improve the RNAi activity of sirna-galnac conjugates. Chembiochem 2016, 17, 985–989. [Google Scholar] [CrossRef] [PubMed]
- Dohmen, C.; Edinger, D.; Frohlich, T.; Schreiner, L.; Lachelt, U.; Troiber, C.; Radler, J.; Hadwiger, P.; Vornlocher, H.P.; Wagner, E. Nanosized multifunctional polyplexes for receptor-mediated sirna delivery. ACS Nano 2012, 6, 5198–5208. [Google Scholar] [CrossRef] [PubMed]
- van de Water, F.M.; Boerman, O.C.; Wouterse, A.C.; Peters, J.G.; Russel, F.G.; Masereeuw, R. Intravenously administered short interfering rna accumulates in the kidney and selectively suppresses gene function in renal proximal tubules. Drug Metab. Dispos. 2006, 34, 1393–1397. [Google Scholar] [CrossRef] [PubMed]
- Meade, B.R.; Gogoi, K.; Hamil, A.S.; Palm-Apergi, C.; van den Berg, A.; Hagopian, J.C.; Springer, A.D.; Eguchi, A.; Kacsinta, A.D.; Dowdy, C.F.; et al. Efficient delivery of RNAi prodrugs containing reversible charge-neutralizing phosphotriester backbone modifications. Nat. Biotechnol. 2014, 32, 1256–1261. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, Y.; Hatakeyama, H.; Sato, Y.; Hyodo, M.; Akita, H.; Harashima, H. Gene silencing via RNAi and sirna quantification in tumor tissue using mend, a liposomal sirna delivery system. Mol. Ther. 2013, 21, 1195–1203. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.; Philipp, A.; Oskuee, R.; Schmidt, C.; Wagner, E. Breathing life into polycations: Functionalization with ph-responsive endosomolytic peptides and polyethylene glycol enables sirna delivery. J. Am. Chem. Soc. 2008, 130, 3272–3273. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.; Dohmen, C.; Philipp, A.; Kiener, D.; Maiwald, G.; Scheu, C.; Ogris, M.; Wagner, E. Synthesis and biological evaluation of a bioresponsive and endosomolytic sirna-polymer conjugate. Mol. Pharm. 2009, 6, 752–762. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E. Polymers for sirna delivery: Inspired by viruses to be targeted, dynamic, and precise. Acc. Chem. Res. 2012, 45, 1005–1013. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E. Biomaterials in RNAi therapeutics: Quo vadis? Biomater. Sci. 2013, 1, 804–809. [Google Scholar] [CrossRef]
- Pittella, F.; Cabral, H.; Maeda, Y.; Mi, P.; Watanabe, S.; Takemoto, H.; Kim, H.J.; Nishiyama, N.; Miyata, K.; Kataoka, K. Systemic sirna delivery to a spontaneous pancreatic tumor model in transgenic mice by pegylated calcium phosphate hybrid micelles. J. Control. Release 2014, 178, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Lachelt, U.; Wagner, E. Nucleic acid therapeutics using polyplexes: A journey of 50 years (and beyond). Chem. Rev. 2015, 115, 11043–11078. [Google Scholar] [CrossRef] [PubMed]
- Leng, Q.; Chou, S.T.; Scaria, P.V.; Woodle, M.C.; Mixson, A.J. Increased tumor distribution and expression of histidine-rich plasmid polyplexes. J. Gene Med. 2014, 16, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Zintchenko, A.; Philipp, A.; Dehshahri, A.; Wagner, E. Simple modifications of branched pei lead to highly efficient sirna carriers with low toxicity. Bioconjug. Chem. 2008, 19, 1448–1455. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, K.A.; Dorkin, J.R.; Vegas, A.J.; Chang, P.H.; Veiseh, O.; Matthews, J.; Fenton, O.S.; Zhang, Y.; Olejnik, K.T.; Yesilyurt, V.; et al. Degradable lipid nanoparticles with predictable in vivo sirna delivery activity. Nat. Commun. 2014, 5, 4277. [Google Scholar] [CrossRef] [PubMed]
- Love, K.T.; Mahon, K.P.; Levins, C.G.; Whitehead, K.A.; Querbes, W.; Dorkin, J.R.; Qin, J.; Cantley, W.; Qin, L.L.; Racie, T.; et al. Lipid-like materials for low-dose, in vivo gene silencing. Proc. Natl. Acad. Sci. USA 2010, 107, 1864–1869. [Google Scholar] [CrossRef] [PubMed]
- Leng, Q.; Mixson, A.J. Small interfering rna targeting raf-1 inhibits tumor growth in vitro and in vivo. Cancer Gene Ther. 2005, 12, 682–690. [Google Scholar] [CrossRef] [PubMed]
- Miyata, K.; Nishiyama, N.; Kataoka, K. Rational design of smart supramolecular assemblies for gene delivery: Chemical challenges in the creation of artificial viruses. Chem. Soc. Rev. 2012, 41, 2562–2574. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Takemoto, H.; Yi, Y.; Zheng, M.; Maeda, Y.; Chaya, H.; Hayashi, K.; Mi, P.; Pittella, F.; Christie, R.J.; et al. Precise engineering of sirna delivery vehicles to tumors using polyion complexes and gold nanoparticles. ACS Nano 2014, 8, 8979–8991. [Google Scholar] [CrossRef] [PubMed]
- Li, S.D.; Chen, Y.C.; Hackett, M.J.; Huang, L. Tumor-targeted delivery of sirna by self-assembled nanoparticles. Mol. Ther. 2008, 16, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.L.; Ramusovic, S.; Nguyen, T.; Lu, Z.R. Novel polymerizable surfactants with ph-sensitive amphiphilicity and cell membrane disruption for efficient sirna delivery. Bioconjug. Chem. 2007, 18, 2169–2177. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.L.; Xu, R.; Wu, X.; Gillespie, D.; Jensen, R.; Lu, Z.R. Targeted systemic delivery of a therapeutic sirna with a multifunctional carrier controls tumor proliferation in mice. Mol. Pharm. 2009, 6, 738–746. [Google Scholar] [CrossRef] [PubMed]
- Gujrati, M.; Vaidya, A.; Lu, Z.R. Multifunctional ph-sensitive amino lipids for sirna delivery. Bioconjug. Chem. 2016, 27, 19–35. [Google Scholar] [CrossRef] [PubMed]
- Schafer, J.; Hobel, S.; Bakowsky, U.; Aigner, A. Liposome-polyethylenimine complexes for enhanced DNA and sirna delivery. Biomaterials 2010, 31, 6892–6900. [Google Scholar] [CrossRef] [PubMed]
- Siegwart, D.J.; Whitehead, K.A.; Nuhn, L.; Sahay, G.; Cheng, H.; Jiang, S.; Ma, M.; Lytton-Jean, A.; Vegas, A.; Fenton, P.; et al. Combinatorial synthesis of chemically diverse core-shell nanoparticles for intracellular delivery. Proc. Natl. Acad. Sci. USA 2011, 108, 12996–13001. [Google Scholar] [CrossRef] [PubMed]
- Green, J.J.; Langer, R.; Anderson, D.G. A combinatorial polymer library approach yields insight into nonviral gene delivery. Acc. Chem. Res. 2008, 41, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Uchida, H.; Miyata, K.; Oba, M.; Ishii, T.; Suma, T.; Itaka, K.; Nishiyama, N.; Kataoka, K. Odd-even effect of repeating aminoethylene units in the side chain of n-substituted polyaspartamides on gene transfection profiles. J. Am. Chem. Soc. 2011, 133, 15524–15532. [Google Scholar] [CrossRef] [PubMed]
- Frohlich, T.; Edinger, D.; Klager, R.; Troiber, C.; Salcher, E.; Badgujar, N.; Martin, I.; Schaffert, D.; Cengizeroglu, A.; Hadwiger, P.; et al. Structure-activity relationships of sirna carriers based on sequence-defined oligo (ethane amino) amides. J. Control. Release 2012, 160, 532–541. [Google Scholar] [CrossRef] [PubMed]
- Krzyszton, R.; Salem, B.; Lee, D.J.; Schwake, G.; Wagner, E.; Radler, J.O. Microfluidic self-assembly of folate-targeted monomolecular sirna-lipid nanoparticles. Nanoscale 2017, 9, 7442–7453. [Google Scholar] [CrossRef] [PubMed]
- Leng, Q.; Scaria, P.; Zhu, J.; Ambulos, N.; Campbell, P.; Mixson, A.J. Highly branched hk peptides are effective carriers of sirna. J. Gene Med. 2005, 7, 977–986. [Google Scholar] [CrossRef] [PubMed]
- Gilleron, J.; Querbes, W.; Zeigerer, A.; Borodovsky, A.; Marsico, G.; Schubert, U.; Manygoats, K.; Seifert, S.; Andree, C.; Stoter, M.; et al. Image-based analysis of lipid nanoparticle-mediated sirna delivery, intracellular trafficking and endosomal escape. Nat. Biotechnol. 2013, 31, 638–646. [Google Scholar] [CrossRef] [PubMed]
- Sahay, G.; Querbes, W.; Alabi, C.; Eltoukhy, A.; Sarkar, S.; Zurenko, C.; Karagiannis, E.; Love, K.; Chen, D.; Zoncu, R.; et al. Efficiency of sirna delivery by lipid nanoparticles is limited by endocytic recycling. Nat. Biotechnol. 2013, 31, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Wittrup, A.; Lieberman, J. Knocking down disease: A progress report on sirna therapeutics. Nat. Rev. Genet. 2015, 16, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Klein, P.M.; Reinhard, S.; Lee, D.J.; Muller, K.; Ponader, D.; Hartmann, L.; Wagner, E. Precise redox-sensitive cleavage sites for improved bioactivity of sirna lipopolyplexes. Nanoscale 2016, 8, 18098–18104. [Google Scholar] [CrossRef] [PubMed]
- Klibanov, A.L.; Maruyama, K.; Torchilin, V.P.; Huang, L. Amphipathic polyethyleneglycols effectively prolong the circulation time of liposomes. FEBS Lett. 1990, 268, 235–237. [Google Scholar] [CrossRef]
- Senior, J.; Delgado, C.; Fisher, D.; Tilcock, C.; Gregoriadis, G. Influence of surface hydrophilicity of liposomes on their interaction with plasma protein and clearance from the circulation: Studies with poly (ethylene glycol)-coated vesicles. Biochim. Biophys. Acta Biomembr. 1991, 1062, 77–82. [Google Scholar] [CrossRef]
- Mori, A.; Klibanov, A.L.; Torchilin, V.P.; Huang, L. Influence of the steric barrier activity of amphipathic poly (ethyleneglycol) and ganglioside gm1 on the circulation time of liposomes and on the target binding of immunoliposomes in vivo. FEBS Lett. 1991, 284, 263–266. [Google Scholar] [CrossRef]
- Yang, Q.; Lai, S.K. Anti-peg immunity: Emergence, characteristics, and unaddressed questions. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2015, 7, 655–677. [Google Scholar] [CrossRef] [PubMed]
- Plank, C.; Mechtler, K.; Szoka, F.C., Jr.; Wagner, E. Activation of the complement system by synthetic DNA complexes: A potential barrier for intravenous gene delivery. Hum. Gene Ther. 1996, 7, 1437–1446. [Google Scholar] [CrossRef] [PubMed]
- Tockary, T.A.; Osada, K.; Motoda, Y.; Hiki, S.; Chen, Q.; Takeda, K.M.; Dirisala, A.; Osawa, S.; Kataoka, K. Rod-to-globule transition of pdna/peg–poly (l-lysine) polyplex micelles induced by a collapsed balance between DNA rigidity and peg crowdedness. Small 2016, 12, 1193–1200. [Google Scholar] [CrossRef] [PubMed]
- Merkel, O.M.; Librizzi, D.; Pfestroff, A.; Schurrat, T.; Buyens, K.; Sanders, N.N.; De Smedt, S.C.; Béhé, M.; Kissel, T. Stability of sirna polyplexes from poly (ethylenimine) and poly (ethylenimine)-g-poly (ethylene glycol) under in vivo conditions: Effects on pharmacokinetics and biodistribution measured by fluorescence fluctuation spectroscopy and single photon emission computed tomography (spect) imaging. J. Control. Release 2009, 138, 148–159. [Google Scholar]
- Kursa, M.; Walker, G.F.; Roessler, V.; Ogris, M.; Roedl, W.; Kircheis, R.; Wagner, E. Novel shielded transferrin-polyethylene glycol-polyethylenimine/DNA complexes for systemic tumor-targeted gene transfer. Bioconjug. Chem. 2003, 14, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Fella, C.; Walker, G.F.; Ogris, M.; Wagner, E. Amine-reactive pyridylhydrazone-based peg reagents for ph-reversible pei polyplex shielding. Eur. J. Pharm. Sci. 2008, 34, 309–320. [Google Scholar] [CrossRef] [PubMed]
- DeRouchey, J.; Walker, G.F.; Wagner, E.; Rädler, J.O. Decorated rods: A “bottom-up” self-assembly of monomolecular DNA complexes. J. Phys. Chem. B 2006, 110, 4548–4554. [Google Scholar] [CrossRef] [PubMed]
- Moghimi, S.M.; Hunter, A.C.; Dadswell, C.M.; Savay, S.; Alving, C.R.; Szebeni, J. Causative factors behind poloxamer 188 (pluronic f68, flocor™)-induced complement activation in human sera: A protective role against poloxamer-mediated complement activation by elevated serum lipoprotein levels. Biochim. Biophys. Acta Mol. Basis Dis. 2004, 1689, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Schellekens, H.; Hennink, W.E.; Brinks, V. The immunogenicity of polyethylene glycol: Facts and fiction. Pharm. Res. 2013, 30, 1729–1734. [Google Scholar] [CrossRef] [PubMed]
- Wenande, E.; Garvey, L. Immediate-type hypersensitivity to polyethylene glycols: A review. Clin. Exp. Allergy 2016, 46, 907–922. [Google Scholar] [CrossRef] [PubMed]
- Knop, K.; Hoogenboom, R.; Fischer, D.; Schubert, U.S. Poly (ethylene glycol) in drug delivery: Pros and cons as well as potential alternatives. Angew. Chem. Int. Ed. 2010, 49, 6288–6308. [Google Scholar] [CrossRef] [PubMed]
- Hamad, I.; Hunter, A.; Szebeni, J.; Moghimi, S.M. Poly (ethylene glycol) s generate complement activation products in human serum through increased alternative pathway turnover and a masp-2-dependent process. Mol. Immunol. 2008, 46, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Dewachter, P.; Mouton-Faivre, C. Anaphylaxis to macrogol 4000 after a parenteral corticoid injection. Allergy 2005, 60, 705–706. [Google Scholar] [CrossRef] [PubMed]
- Kircheis, R.; Wightman, L.; Schreiber, A.; Robitza, B.; Rössler, V.; Kursa, M.; Wagner, E. Polyethylenimine/DNA complexes shielded by transferrin target gene expression to tumors after systemic application. Gene Ther. 2001, 8, 28. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Tetley, L.; Uchegbu, I.F. The level of hydrophobic substitution and the molecular weight of amphiphilic poly-l-lysine-based polymers strongly affects their assembly into polymeric bilayer vesicles. J. Colloid Interf. Sci. 2001, 237, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Toncheva, V.; Wolfert, M.A.; Dash, P.R.; Oupicky, D.; Ulbrich, K.; Seymour, L.W.; Schacht, E.H. Novel vectors for gene delivery formed by self-assembly of DNA with poly (l-lysine) grafted with hydrophilic polymers. Biochim. Biophys. Acta Gen. Subj. 1998, 1380, 354–368. [Google Scholar] [CrossRef]
- Oupický, D.; Howard, K.A.; Koňák, Č.; Dash, P.R.; Ulbrich, K.; Seymour, L.W. Steric stabilization of poly-l-lysine/DNA complexes by the covalent attachment of semitelechelic poly [n-(2-hydroxypropyl) methacrylamide]. Bioconjug. Chem. 2000, 11, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Lammers, T.; Ulbrich, K. Hpma Copolymers: 30 Years of Advances; Elsevier: New York, NY, USA, 2010. [Google Scholar]
- Noga, M.; Edinger, D.; Kläger, R.; Wegner, S.V.; Spatz, J.P.; Wagner, E.; Winter, G.; Besheer, A. The effect of molar mass and degree of hydroxyethylation on the controlled shielding and deshielding of hydroxyethyl starch-coated polyplexes. Biomaterials 2013, 34, 2530–2538. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wallace, S. Polymer-drug conjugates: Recent development in clinical oncology. Adv. Drug Deliv. Rev. 2008, 60, 886–898. [Google Scholar] [CrossRef] [PubMed]
- Romberg, B.; Metselaar, J.M.; Baranyi, L.; Snel, C.J.; Bunger, R.; Hennink, W.E.; Szebeni, J.; Storm, G. Poly(amino acid)s: Promising enzymatically degradable stealth coatings for liposomes. Int. J. Pharm. 2007, 331, 186–189. [Google Scholar] [CrossRef] [PubMed]
- Schlapschy, M.; Binder, U.; Börger, C.; Theobald, I.; Wachinger, K.; Kisling, S.; Haller, D.; Skerra, A. Pasylation: A biological alternative to pegylation for extending the plasma half-life of pharmaceutically active proteins. Protein Eng. Des. Sel. 2013, 26, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Mendler, C.T.; Friedrich, L.; Laitinen, I.; Schlapschy, M.; Schwaiger, M.; Wester, H.-J.; Skerra, A. High Contrast Tumor Imaging with Radio-Labeled Antibody Fab Fragments Tailored for Optimized Pharmacokinetics via Pasylation. MAbs 2015, 7, 96–109. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.; Birke, A.; Kim, J.; Jang, Y.; Song, S.Y.; Ryu, S.; Kim, B.S.; Kim, B.G.; Barz, M.; Char, K. Cooperative catechol-functionalized polypept(o)ide brushes and ag nanoparticles for combination of protein resistance and antimicrobial activity on metal oxide surfaces. Biomacromolecules 2018, 19, 1602–1613. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.; Fetsch, C.; Amin, I.; Jordan, R.; Luxenhofer, R. Polypeptoid brushes by surface-initiated polymerization of n-substituted glycine n-carboxyanhydrides. Langmuir 2013, 29, 6983–6988. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.; Tang, Z.; Richter, M.; Marschelke, C.; Forster, P.; Wegener, E.; Amin, I.; Zimmermann, H.; Scharnweber, D.; Braun, H.G.; et al. Patterned polypeptoid brushes. Macromol. Biosci. 2016, 16, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Birke, A.; Ling, J.; Barz, M. Polysarcosine-containing copolymers: Synthesis, characterization, self-assembly, and applications. Prog. Polym. Sci. 2018, 81, 163–208. [Google Scholar] [CrossRef]
- Fetsch, C.; Grossmann, A.; Holz, L.; Nawroth, J.F.; Luxenhofer, R. Polypeptoids from n-substituted glycine n-carboxyanhydrides: Hydrophilic, hydrophobic, and amphiphilic polymers with poisson distribution. Macromolecules 2011, 44, 6746–6758. [Google Scholar] [CrossRef]
- Klinker, K.; Barz, M. Polypept(o)ides: Hybrid systems based on polypeptides and polypeptoids. Macromol. Rapid Commun. 2015, 36, 1943–1957. [Google Scholar] [CrossRef] [PubMed]
- Weber, B.; Birke, A.; Fischer, K.; Schmidt, M.; Barz, M. Solution properties of polysarcosine: From absolute and relative molar mass determinations to complement activation. Macromolecules 2018, 51, 2653–2661. [Google Scholar] [CrossRef]
- Wei, Q.; Becherer, T.; Angioletti-Uberti, S.; Dzubiella, J.; Wischke, C.; Neffe, A.T.; Lendlein, A.; Ballauff, M.; Haag, R. Protein interactions with polymer coatings and biomaterials. Angew. Chem. 2014, 53, 8004–8031. [Google Scholar] [CrossRef] [PubMed]
- Hörtz, C.; Birke, A.; Kaps, L.; Decker, S.; Wächtersbach, E.; Fischer, K.; Schuppan, D.; Barz, M.; Schmidt, M. Cylindrical brush polymers with polysarcosine side chains: A novel biocompatible carrier for biomedical applications. Macromolecules 2015, 48, 2074–2086. [Google Scholar] [CrossRef]
- Sela, M. Immunological studies with synthetic polypeptides. Adv. Immunol. 1966, 5, 29–129. [Google Scholar] [PubMed]
- Hara, E.; Ueda, M.; Kim, C.J.; Makino, A.; Hara, I.; Ozeki, E.; Kimura, S. Suppressive immune response of poly-(sarcosine) chains in peptide-nanosheets in contrast to polymeric micelles. J. Pept. Sci. 2014, 20, 570–577. [Google Scholar] [CrossRef] [PubMed]
- Birke, A.; Huesmann, D.; Kelsch, A.; Weilbacher, M.; Xie, J.; Bros, M.; Bopp, T.; Becker, C.; Landfester, K.; Barz, M. Polypeptoid-block-polypeptide copolymers: Synthesis, characterization, and application of amphiphilic block copolypept(o)ides in drug formulations and miniemulsion techniques. Biomacromolecules 2014, 15, 548–557. [Google Scholar] [CrossRef] [PubMed]
- Otter, R.; Klinker, K.; Spitzer, D.; Schinnerer, M.; Barz, M.; Besenius, P. Folding induced supramolecular assembly into ph-responsive nanorods with a protein repellent shell. Chem. Commun. 2018, 54, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Heller, P.; Hobernik, D.; Lachelt, U.; Schinnerer, M.; Weber, B.; Schmidt, M.; Wagner, E.; Bros, M.; Barz, M. Combining reactive triblock copolymers with functional cross-linkers: A versatile pathway to disulfide stabilized-polyplex libraries and their application as pdna vaccines. J. Control. Release 2017, 258, 146–160. [Google Scholar] [CrossRef] [PubMed]
- Klinker, K.; Schafer, O.; Huesmann, D.; Bauer, T.; Capeloa, L.; Braun, L.; Stergiou, N.; Schinnerer, M.; Dirisala, A.; Miyata, K.; et al. Secondary-structure-driven self-assembly of reactive polypept(o)ides: Controlling size, shape, and function of core cross-linked nanostructures. Angew. Chem. 2017, 56, 9608–9613. [Google Scholar] [CrossRef] [PubMed]
- Duro-Castano, A.; Movellan, J.; Vicent, M.J. Smart branched polymer drug conjugates as nano-sized drug delivery systems. Biomater. Sci. 2015, 3, 1321–1334. [Google Scholar] [CrossRef] [PubMed]
- Park, E.K.; Kim, S.Y.; Lee, S.B.; Lee, Y.M. Folate-conjugated methoxy poly(ethylene glycol)/poly(epsilon-caprolactone) amphiphilic block copolymeric micelles for tumor-targeted drug delivery. J. Control. Release 2005, 109, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.; Low, P.S. Folate-targeted therapies for cancer. J. Med. Chem. 2010, 53, 6811–6824. [Google Scholar] [CrossRef] [PubMed]
- Sudimack, J.; Lee, R.J. Targeted drug delivery via the folate receptor. Adv. Drug Deliv. Rev. 2000, 41, 147–162. [Google Scholar] [CrossRef]
- Low, P.S.; Henne, W.A.; Doorneweerd, D.D. Discovery and development of folic-acid-based receptor targeting for imaging and therapy of cancer and inflammatory diseases. Acc. Chem. Res. 2008, 41, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Klein, P.M.; Kern, S.; Lee, D.J.; Schmaus, J.; Hohn, M.; Gorges, J.; Kazmaier, U.; Wagner, E. Folate receptor-directed orthogonal click-functionalization of sirna lipopolyplexes for tumor cell killing in vivo. Biomaterials 2018. [Google Scholar] [CrossRef] [PubMed]
- Shi, B.; Keough, E.; Matter, A.; Leander, K.; Young, S.; Carlini, E.; Sachs, A.B.; Tao, W.; Abrams, M.; Howell, B.; et al. Biodistribution of small interfering rna at the organ and cellular levels after lipid nanoparticle-mediated delivery. J. Histochem. Cytochem. 2011, 59, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Akinc, A.; Querbes, W.; De, S.; Qin, J.; Frank-Kamenetsky, M.; Jayaprakash, K.N.; Jayaraman, M.; Rajeev, K.G.; Cantley, W.L.; Dorkin, J.R.; et al. Targeted delivery of RNAi therapeutics with endogenous and exogenous ligand-based mechanisms. Mol. Ther. 2010, 18, 1357–1364. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Muller, K.; Kessel, E.; Reinhard, S.; He, D.; Klein, P.M.; Hohn, M.; Rodl, W.; Kempter, S.; Wagner, E. Targeted sirna delivery using a lipo-oligoaminoamide nanocore with an influenza peptide and transferrin shell. Adv. Healthc. Mater. 2016, 5, 1493–1504. [Google Scholar] [CrossRef] [PubMed]
- Troiber, C.; Edinger, D.; Kos, P.; Schreiner, L.; Klager, R.; Herrmann, A.; Wagner, E. Stabilizing effect of tyrosine trimers on pdna and sirna polyplexes. Biomaterials 2013, 34, 1624–1633. [Google Scholar] [CrossRef] [PubMed]
- Schottler, S.; Becker, G.; Winzen, S.; Steinbach, T.; Mohr, K.; Landfester, K.; Mailander, V.; Wurm, F.R. Protein adsorption is required for stealth effect of poly(ethylene glycol)- and poly(phosphoester)-coated nanocarriers. Nat. Nanotechnol. 2016, 11, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, D.; Caracciolo, G.; Marchini, C.; Montani, M.; Amici, A.; Callipo, L.; Capriotti, A.L.; Cavaliere, C.; Lagana, A. Surface adsorption of protein corona controls the cell uptake mechanism in efficient cationic liposome/DNA complexes in serum. J. Control. Release 2010, 148, e94–e95. [Google Scholar] [CrossRef] [PubMed]
- Lundqvist, M.; Stigler, J.; Elia, G.; Lynch, I.; Cedervall, T.; Dawson, K.A. Nanoparticle size and surface properties determine the protein corona with possible implications for biological impacts. Proc. Natl. Acad. Sci. USA 2008, 105, 14265–14270. [Google Scholar] [CrossRef] [PubMed]
- Fokina, A.; Klinker, K.; Braun, L.; Jeong, B.G.; Bae, W.K.; Barz, M.; Zentel, R. Multidentate polysarcosine-based ligands for water-soluble quantum dots. Macromolecules 2016, 49, 3663–3671. [Google Scholar] [CrossRef]
- Klinker, K.; Holm, R.; Heller, P.; Barz, M. Evaluating chemical ligation techniques for the synthesis of block copolypeptides, polypeptoids and block copolypept(o)ides: A comparative study. Polym. Chem. 2015, 6, 4612–4623. [Google Scholar] [CrossRef]
- Dommerholt, J.; Rutjes, F.; van Delft, F.L. Strain-promoted 1,3-dipolar cycloaddition of cycloalkynes and organic azides. Top. Curr. Chem. 2016, 374, 16. [Google Scholar] [CrossRef] [PubMed]
- Schafer, O.; Barz, M. Of thiols and disulfides: Methods for chemoselective formation of asymmetric disulfides in synthetic peptides and polymers. Chemistry 2018. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.J.; Kessel, E.; Lehto, T.; Liu, X.; Yoshinaga, N.; Padari, K.; Chen, Y.C.; Kempter, S.; Uchida, S.; Radler, J.O.; et al. Systemic delivery of folate-peg sirna lipopolyplexes with enhanced intracellular stability for in vivo gene silencing in leukemia. Bioconjug. Chem. 2017, 28, 2393–2409. [Google Scholar] [CrossRef] [PubMed]
- Dohmen, C.; Frohlich, T.; Lachelt, U.; Rohl, I.; Vornlocher, H.P.; Hadwiger, P.; Wagner, E. Defined folate-peg-sirna conjugates for receptor-specific gene silencing. Mol. Ther. Nucleic Acids 2012, 1, e7. [Google Scholar] [CrossRef] [PubMed]
- Leamon, C.P.; Low, P.S. Folate-mediated targeting: From diagnostics to drug and gene delivery. Drug Discov. Today 2001, 6, 44–51. [Google Scholar] [CrossRef]
- Leamon, C.P.; DePrince, R.B.; Hendren, R.W. Folate-mediated drug delivery: Effect of alternative conjugation chemistry. J. Drug Target. 1999, 7, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Lee, R.J.; Mathias, C.J.; Green, M.A.; Low, P.S. Synthesis, purification, and tumor cell uptake of 67ga-deferoxamine—Folate, a potential radiopharmaceutical for tumor imaging. Bioconjug. Chem. 1996, 7, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Muller, K.; Kessel, E.; Klein, P.M.; Hohn, M.; Wagner, E. Post-pegylation of sirna lipo-oligoamino amide polyplexes using tetra-glutamylated folic acid as ligand for receptor-targeted delivery. Mol. Pharm. 2016, 13, 2332–2345. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.; Gunther, M.; Gu, Z.; Wagner, E. Pyridylhydrazone-based pegylation for ph-reversible lipopolyplex shielding. Biomaterials 2011, 32, 858–869. [Google Scholar] [CrossRef] [PubMed]
- Maldonado-Baez, L.; Williamson, C.; Donaldson, J.G. Clathrin-independent endocytosis: A cargo-centric view. Exp. Cell Res. 2013, 319, 2759–2769. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.L.; Van Cuong, N.; Hsieh, M.F. Endocytosis pathways of the folate tethered star-shaped peg-pcl micelles in cancer cell lines. Polymers 2014, 6, 634–650. [Google Scholar] [CrossRef]
- Langston Suen, W.L.; Chau, Y. Size-dependent internalisation of folate-decorated nanoparticles via the pathways of clathrin and caveolae-mediated endocytosis in arpe-19 cells. J. Pharm. Pharmacol. 2014, 66, 564–573. [Google Scholar] [CrossRef] [PubMed]
- Dalal, C.; Saha, A.; Jana, N.R. Nanoparticle multivalency directed shifting of cellular uptake mechanism. J. Phys. Chem. C 2016, 120, 6778–6786. [Google Scholar] [CrossRef]
- Sabharanjak, S.; Sharma, P.; Parton, R.G.; Mayor, S. Gpi-anchored proteins are delivered to recycling endosomes via a distinct cdc42-regulated, clathrin-independent pinocytic pathway. Dev. Cell 2002, 2, 411–423. [Google Scholar] [CrossRef]
- Brulisauer, L.; Kathriner, N.; Prenrecaj, M.; Gauthier, M.A.; Leroux, J.C. Tracking the bioreduction of disulfide-containing cationic dendrimers. Angew. Chem. 2012, 51, 12454–12458. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| siRNA Formulation | eq DBCO-pSar119 | z-Average (nm) | PDI | Mean Zeta Potential (mV) |
|---|---|---|---|---|
| T-1N3 | 0 | 81.0 ± 5.0 | 0.26 ± 0.02 | 20.9 ± 0.9 |
| 0.5 | 86.7 ± 2.8 | 0.24 ± 0.02 | 9.4 ± 0.5 | |
| 1 | 91.8 ± 2.9 | 0.26 ± 0.02 | 8.5 ± 0.6 | |
| 2 | 96.8 ± 4.0 | 0.27 ± 0.02 | 6.0 ± 1.1 | |
| T-2N3 | 0 | 90.6 ± 0.9 | 0.15 ± 0.03 | 17.2 ± 0.8 |
| 0.5 | 98.7 ± 1.3 | 0.15 ± 0.01 | 7.7 ± 0.6 | |
| 1 | 102.3 ± 2.1 | 0.19 ± 0.01 | 6.3 ± 1.0 | |
| 2 | 105.1 ± 1.9 | 0.17 ± 0.01 | 2.5 ± 0.3 |
| siRNA Formulation | eq DBCO-pSar119 | MFI |
|---|---|---|
| T-1N3 | 0 | 881.5 ± 25.5 |
| 0.25 | 780.5 ± 2.5 | |
| 0.5 | 715.0 ± 24.0 | |
| 1 | 359.0 ± 14.0 | |
| 2 | 245.5 ± 8.0 | |
| T-2N3 | 0 | 883.0 ± 86.0 |
| 0.25 | 870.5 ± 62.5 | |
| 0.5 | 785.5 ± 38.5 | |
| 1 | 602.5 ± 37.5 | |
| 2 | 263.5 ± 4.5 | |
| untreated cells | 2.4 ± 0.2 | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klein, P.M.; Klinker, K.; Zhang, W.; Kern, S.; Kessel, E.; Wagner, E.; Barz, M. Efficient Shielding of Polyplexes Using Heterotelechelic Polysarcosines. Polymers 2018, 10, 689. https://doi.org/10.3390/polym10060689
Klein PM, Klinker K, Zhang W, Kern S, Kessel E, Wagner E, Barz M. Efficient Shielding of Polyplexes Using Heterotelechelic Polysarcosines. Polymers. 2018; 10(6):689. https://doi.org/10.3390/polym10060689
Chicago/Turabian StyleKlein, Philipp Michael, Kristina Klinker, Wei Zhang, Sarah Kern, Eva Kessel, Ernst Wagner, and Matthias Barz. 2018. "Efficient Shielding of Polyplexes Using Heterotelechelic Polysarcosines" Polymers 10, no. 6: 689. https://doi.org/10.3390/polym10060689
APA StyleKlein, P. M., Klinker, K., Zhang, W., Kern, S., Kessel, E., Wagner, E., & Barz, M. (2018). Efficient Shielding of Polyplexes Using Heterotelechelic Polysarcosines. Polymers, 10(6), 689. https://doi.org/10.3390/polym10060689