Influence of Polycation Composition on Electrochemical Film Formation

 ,
, 
Abstract
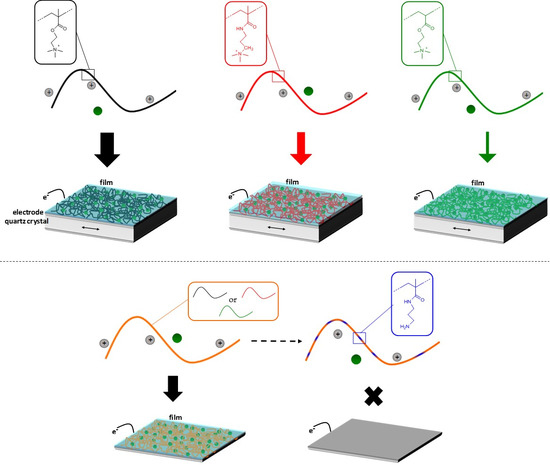
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
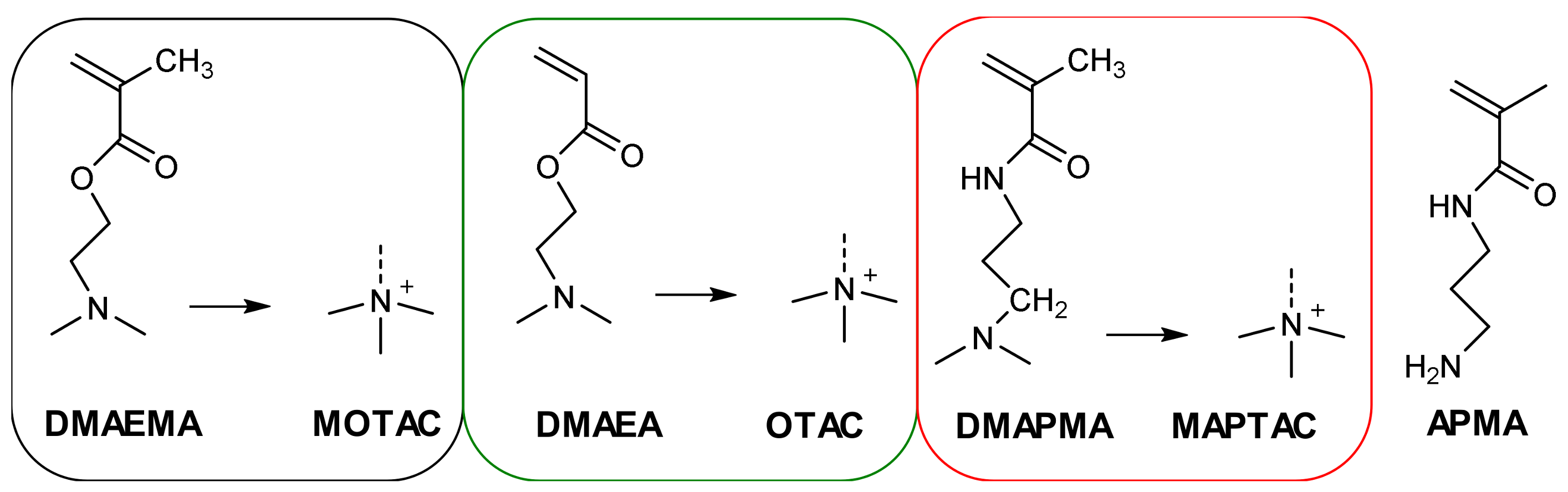
2. Materials and Methods
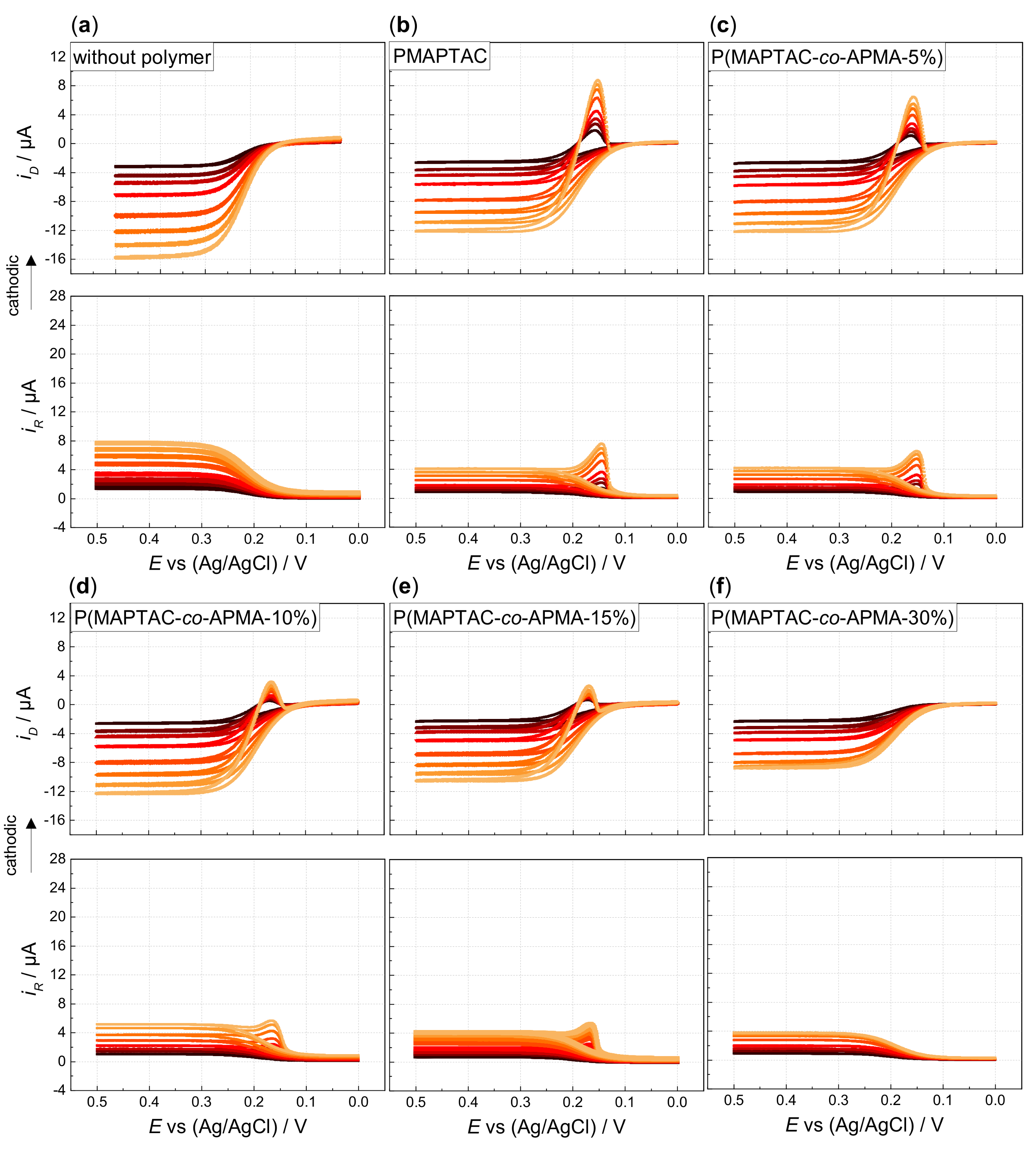
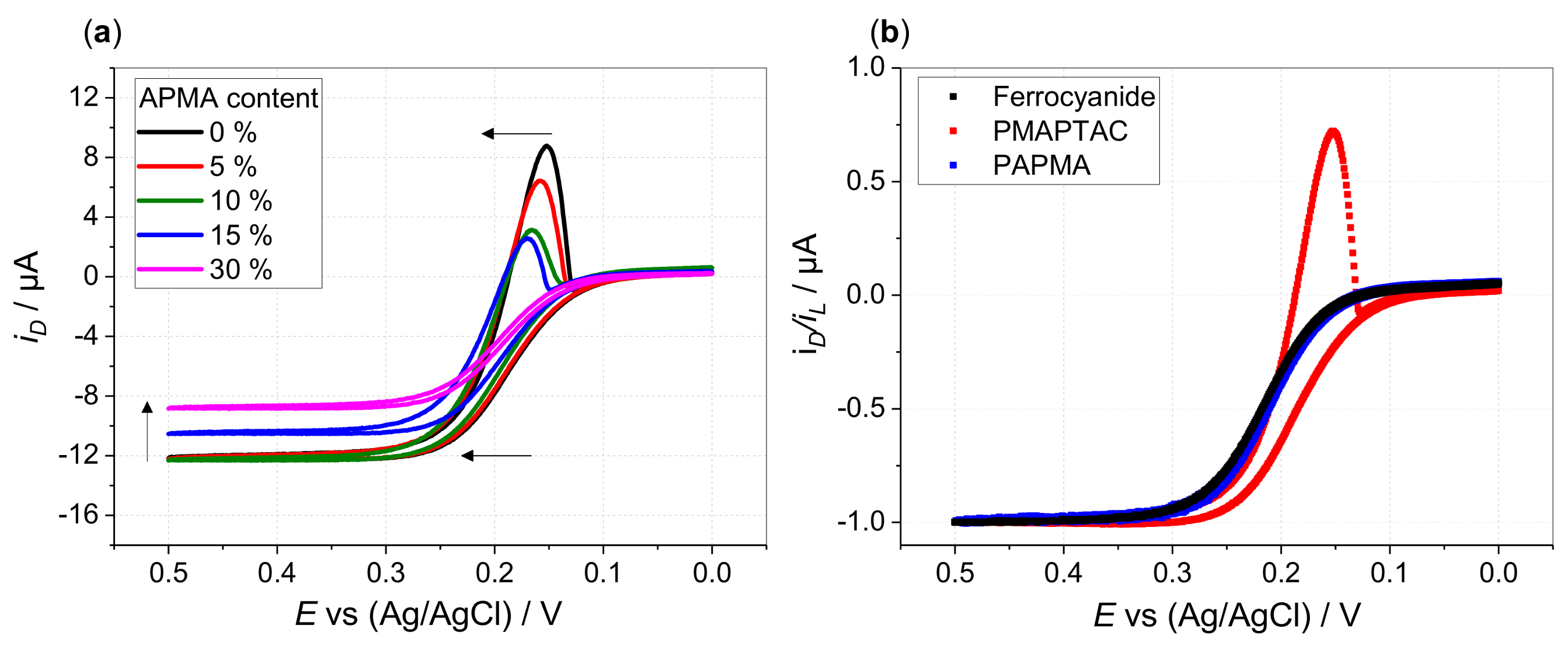
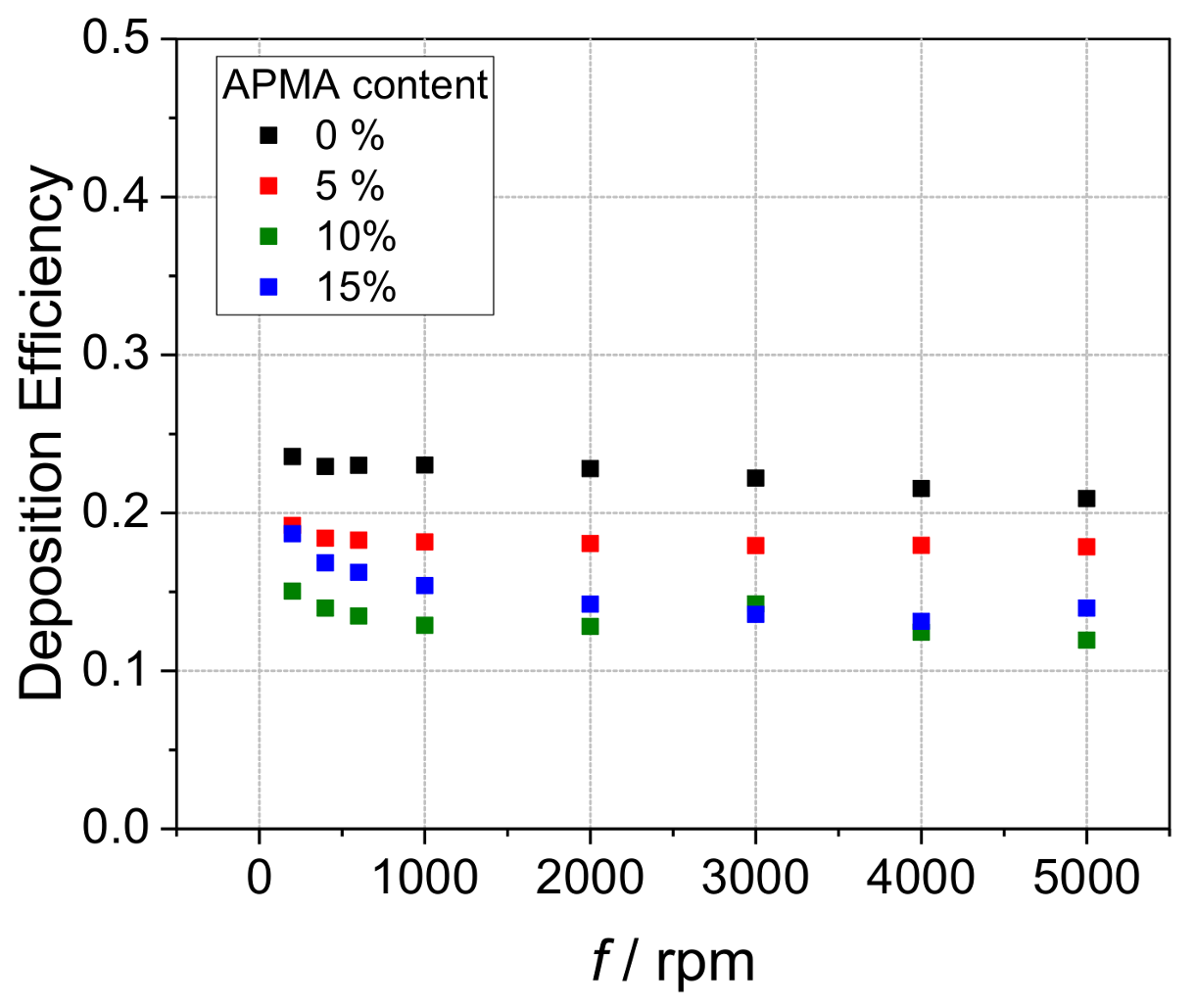
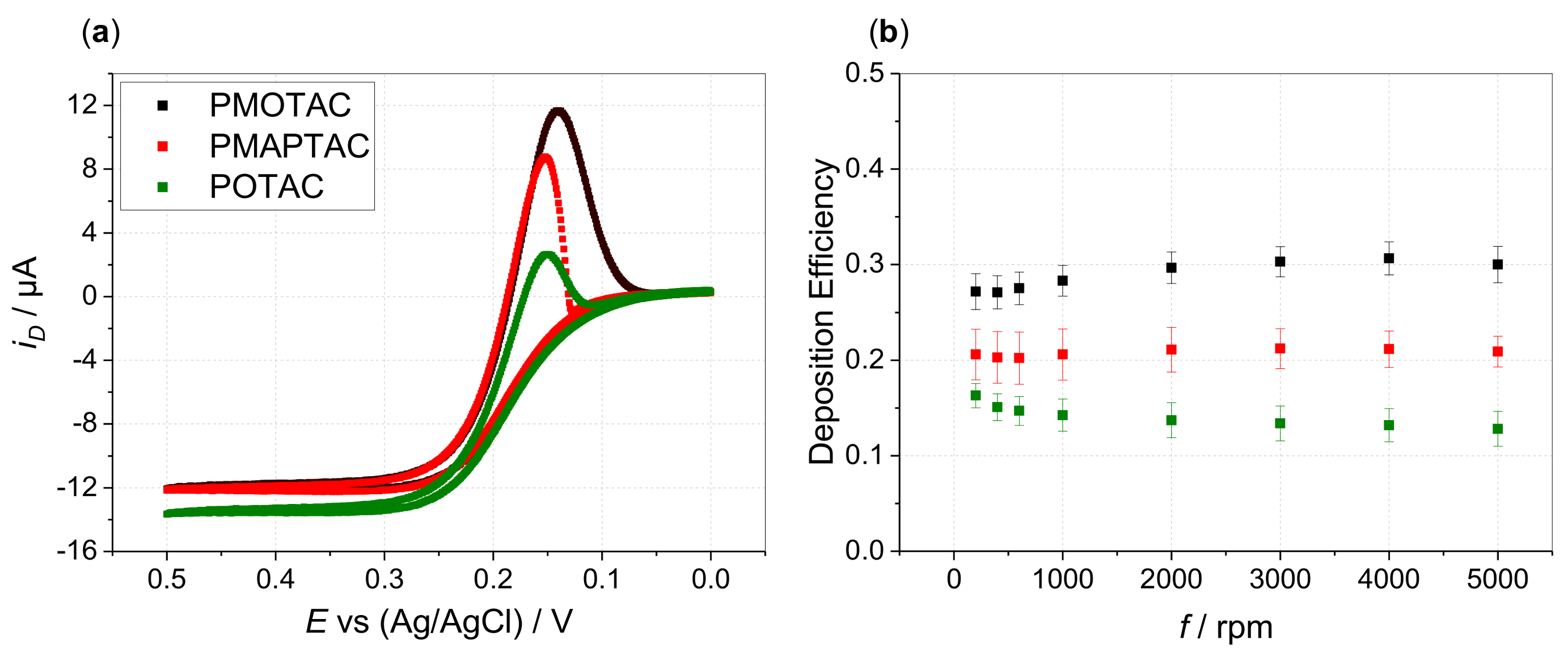
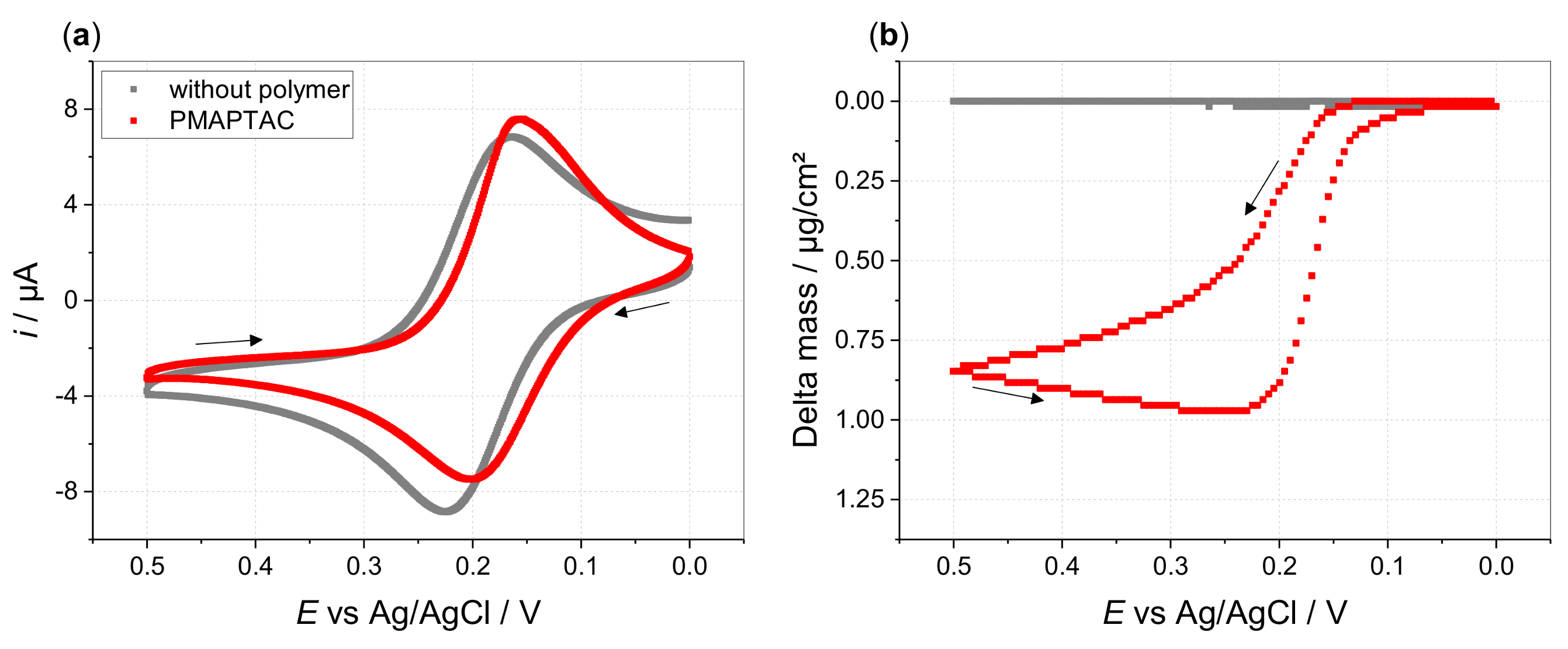
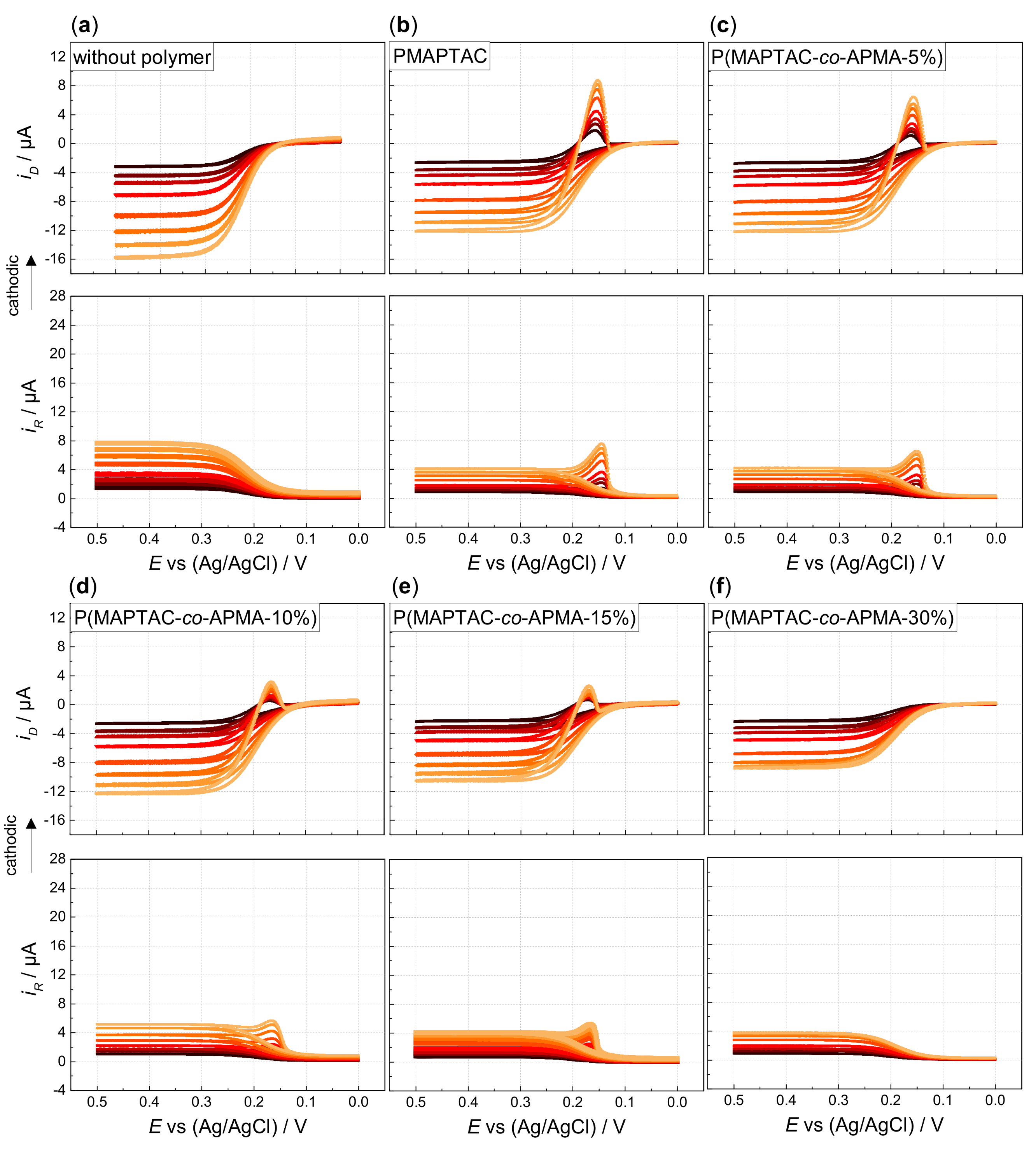
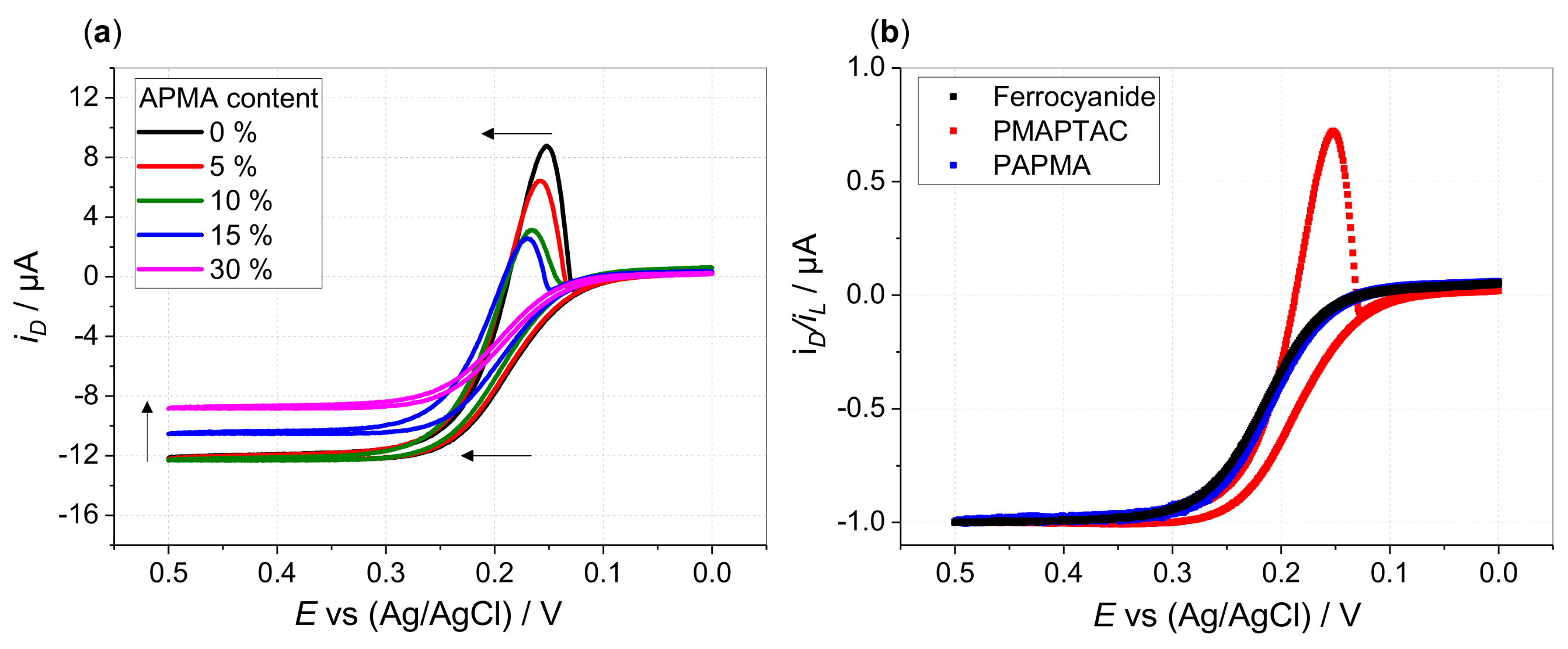
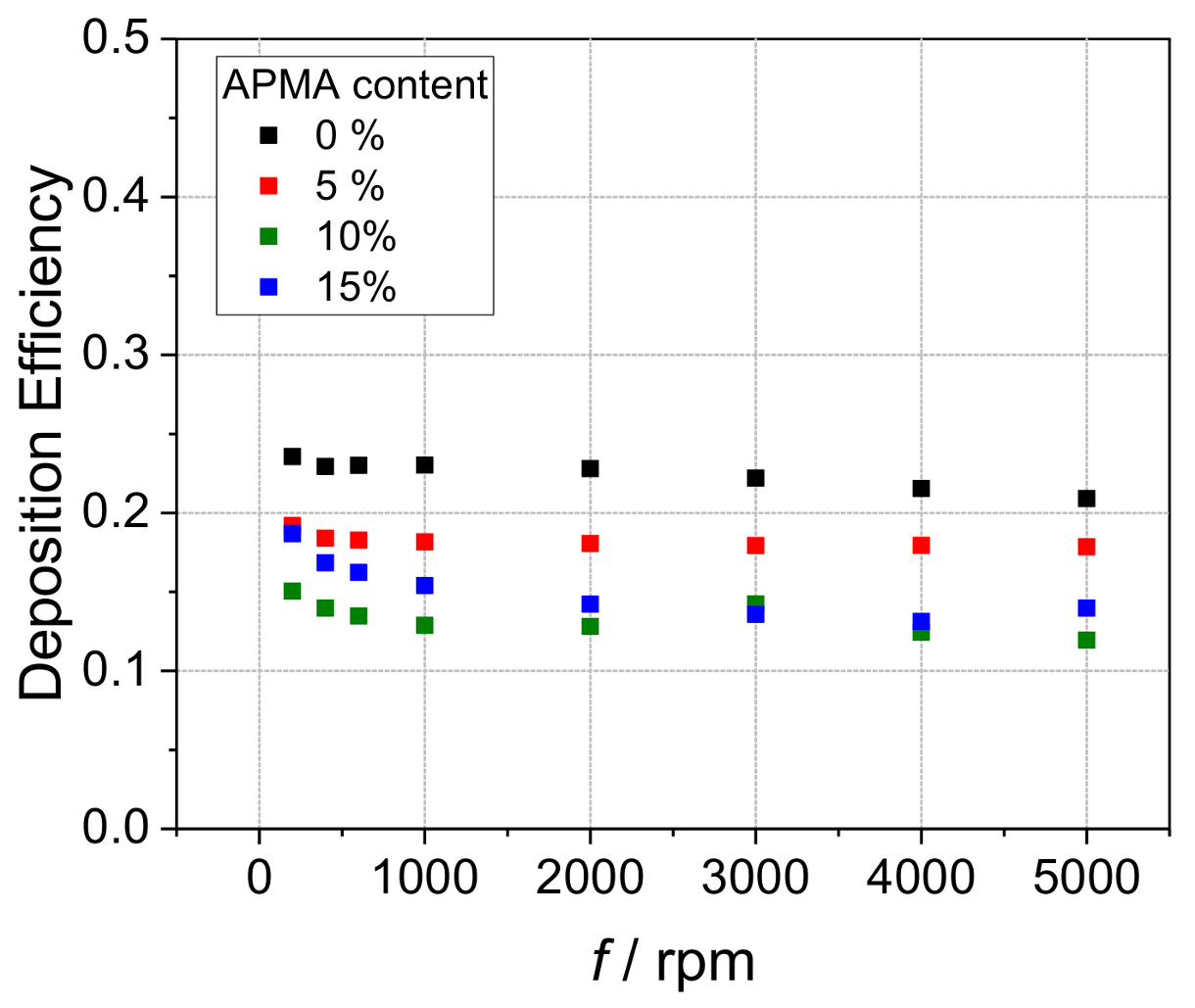
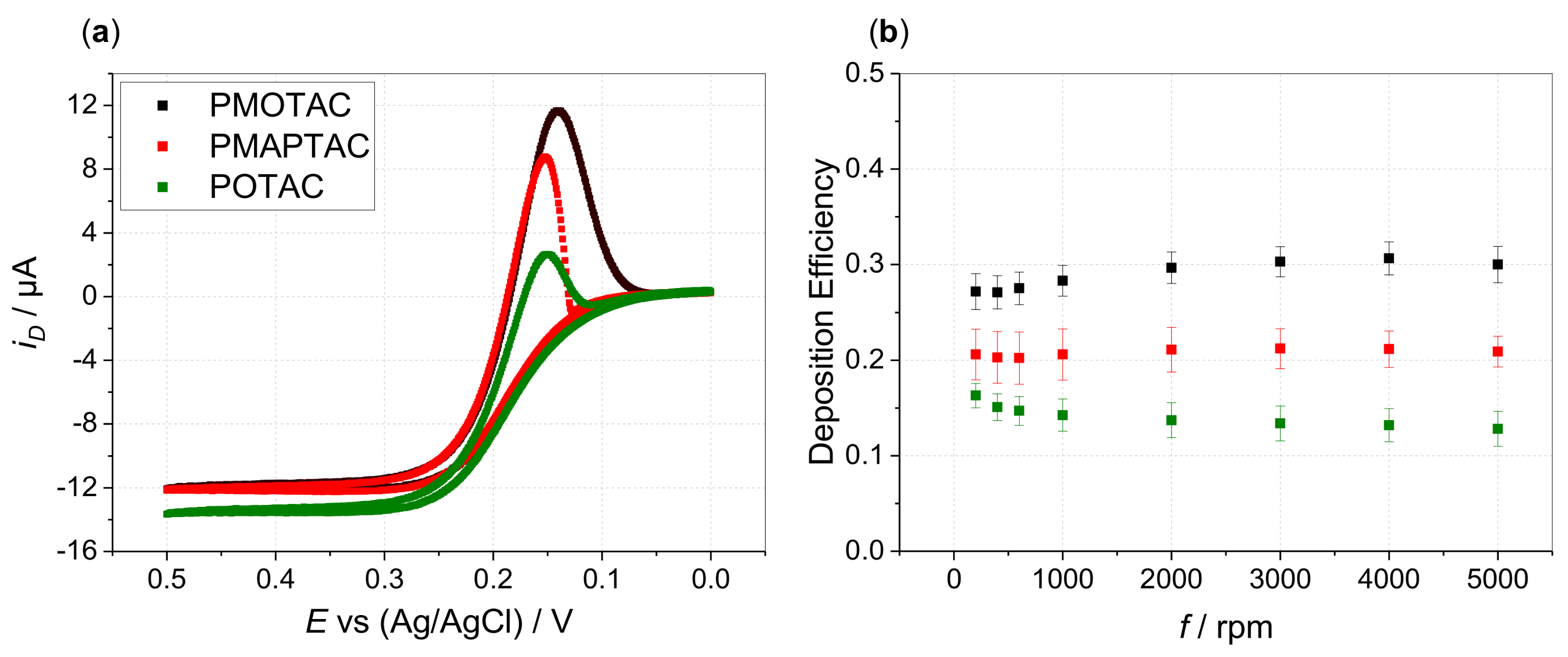
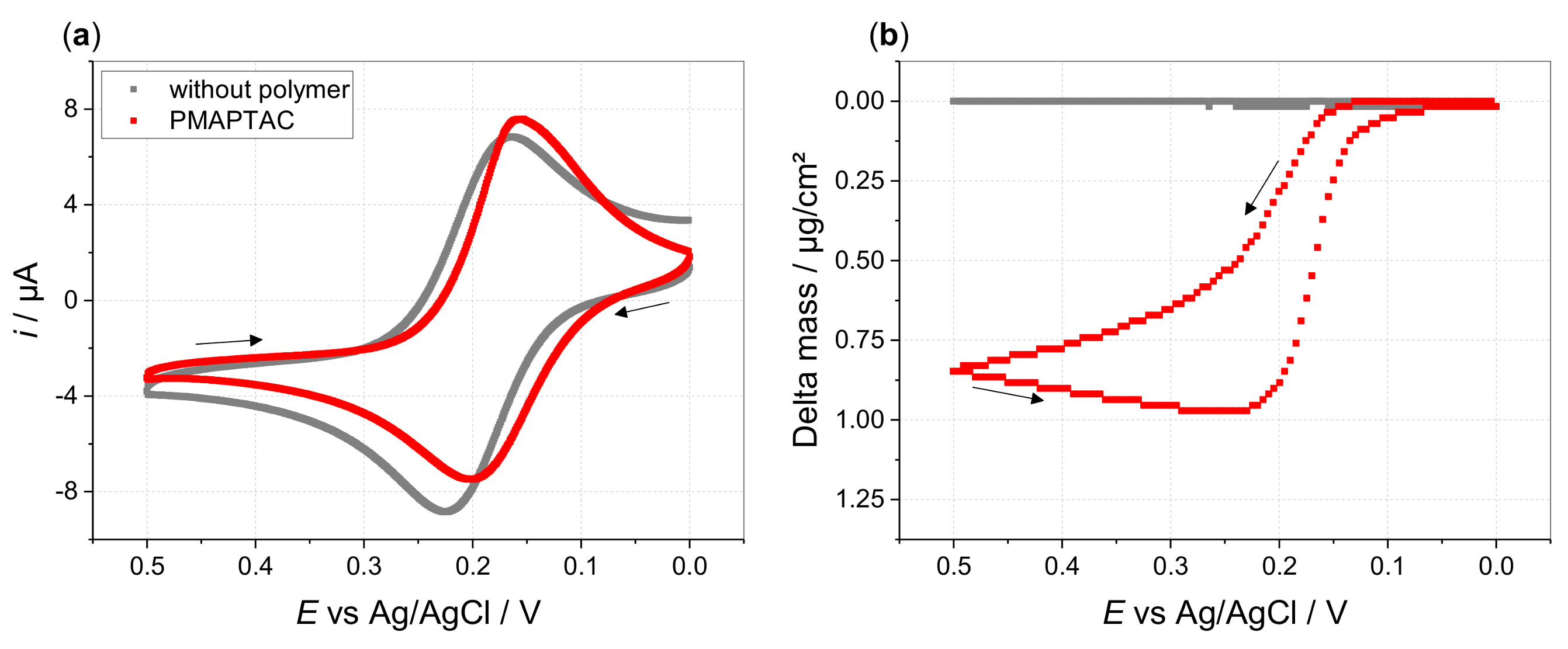
3. Results
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sadman, K.; Wang, Q.; Chen, S.H.; Delgado, D.E.; Shull, K.R. pH-Controlled Electrochemical Deposition of Polyelectrolyte Complex Films. Langmuir 2017, 33, 1834–1844. [Google Scholar] [CrossRef] [PubMed]
- Martin, E.J.; Sadman, K.; Shull, K.R. Anodic Electrodeposition of a Cationic Polyelectrolyte in the Presence of Multivalent Anions. Langmuir 2016, 32, 7747–7756. [Google Scholar] [CrossRef] [PubMed]
- Ozel, T.; Zhang, B.A.; Gao, R.; Day, R.W.; Lieber, C.M.; Nocera, D.G. Electrochemical Deposition of Conformal and Functional Layers on High Aspect Ratio Silicon Micro/Nanowires. Nano Lett. 2017, 17, 4502–4507. [Google Scholar] [CrossRef] [PubMed]
- Abdul Amir Al-Mokaram, A.M.A.; Yahya, R.; Abdi, M.M.; Muhammad Ekramul Mahmud, H.N. One-step electrochemical deposition of Polypyrrole-Chitosan-Iron oxide nanocomposite films for non-enzymatic glucose biosensor. Mater. Lett. 2016, 183, 90–93. [Google Scholar] [CrossRef]
- Bozzini, B.; Bocchetta, P.; Kourousias, G.; Gianoncelli, A. Electrodeposition of Mn-Co/Polypyrrole Nanocomposites: An Electrochemical and In Situ Soft-X-ray Microspectroscopic Investigation. Polymers 2017, 9, 17. [Google Scholar] [CrossRef]
- Chandaluri, C.G.; Pelossof, G.; Tel-Vered, R.; Shenhar, R.; Willner, I. Block Copolymer Patterns as Templates for the Electrocatalyzed Deposition of Nanostructures on Electrodes and for the Generation of Surfaces of Controlled Wettability. ACS Appl. Mater. Interfaces 2016, 8, 1440–1446. [Google Scholar] [CrossRef] [PubMed]
- Sebaa, M.A.; Dhillon, S.; Liu, H. Electrochemical deposition and evaluation of electrically conductive polymer coating on biodegradable magnesium implants for neural applications. J. Mater. Sci. Mater. Med. 2013, 24, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Redepenning, J.; Venkataraman, G.; Chen, J.; Stafford, N. Electrochemical preparation of chitosan/hydroxyapatite composite coatings on titanium substrates. J. Biomed. Mater. Res. A 2003, 66, 411–416. [Google Scholar] [CrossRef] [PubMed]
- Gray, K.M.; Liba, B.D.; Wang, Y.; Cheng, Y.; Rubloff, G.W.; Bentley, W.E.; Montembault, A.; Royaud, I.; David, L.; Payne, G.F. Electrodeposition of a Biopolymeric Hydrogel: Potential for One-Step Protein Electroaddressing. Biomacromolecules 2012, 13, 1181–1189. [Google Scholar] [CrossRef] [PubMed]
- Braglia, M.; Ferrari, I.V.; Djenizian, T.; Kaciulis, S.; Soltani, P.; Di Vona, M.L.; Knauth, P. Bottom-Up Electrochemical Deposition of Poly(styrene sulfonate) on Nanoarchitectured Electrodes. ACS Appl. Mater. Interfaces 2017, 9, 22902–22910. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Zhan, X.; Yao, L.; Chen, Q.; Xie, Z.; Ma, Y. Electrochemical Deposition of Azobenzene-Containing Network Films with High-Contrast and Stable Photoresponse. Macromol. Rapid Commun. 2016, 37, 610–615. [Google Scholar] [CrossRef] [PubMed]
- Oskam, G.; Long, J.G.; Natarajan, A.; Searson, P.C. Electrochemical deposition of metals onto silicon. J. Phys. D Appl. Phys. 1998, 31, 1927. [Google Scholar] [CrossRef]
- Karthikeyan, P.; Mitu, L.; Pandian, K.; Anbarasu, G.; Rajavel, R. Electrochemical deposition of a Zn-HNT/p(EDOT-co-EDOP) nanocomposite on LN SS for anti-bacterial and anti-corrosive applications. New J. Chem. 2017, 41, 4758–4762. [Google Scholar] [CrossRef]
- Wu, L.-Q.; Gadre, A.P.; Yi, H.; Kastantin, M.J.; Rubloff, G.W.; Bentley, W.E.; Payne, G.F.; Ghodssi, R. Voltage-Dependent Assembly of the Polysaccharide Chitosan onto an Electrode Surface. Langmuir 2002, 18, 8620–8625. [Google Scholar] [CrossRef]
- Maerten, C.; Jierry, L.; Schaaf, P.; Boulmedais, F. Review of Electrochemically Triggered Macromolecular Film Buildup Processes and Their Biomedical Applications. ACS Appl. Mater. Interfaces 2017, 9, 28117–28138. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Xiong, Y.; Cheng, Y.; Wu, H.-C.; Liu, Y.; Morrow, B.; Ben-Yoav, H.; Ghodssi, R.; Rubloff, G.; Shen, J.; et al. Chitosan to Connect Biology to Electronics: Fabricating the Bio-Device Interface and Communicating Across This Interface. Polymers 2015, 7, 1–46. [Google Scholar] [CrossRef]
- Xu, G.; Liang, S.; Fan, J.; Sheng, G.; Luo, X. Amperometric sensing of nitrite using a glassy carbon electrode modified with a multilayer consisting of carboxylated nanocrystalline cellulose and poly(diallyldimethyl ammonium) ions in a PEDOT host. Microchim. Acta 2016, 183, 2031–2037. [Google Scholar] [CrossRef]
- Aldalbahi, A.; Rahaman, M.; Almoiqli, M. A Strategy to Enhance the Electrode Performance of Novel Three-Dimensional PEDOT/RVC Composites by Electrochemical Deposition Method. Polymers 2017, 9, 157. [Google Scholar] [CrossRef]
- Plamper, F.A.; Murtomäki, L.; Walther, A.; Kontturi, K.; Tenhu, H. e-Micellization: Electrochemical, Reversible Switching of Polymer Aggregation. Macromolecules 2009, 42, 7254–7257. [Google Scholar] [CrossRef]
- Plamper, F.A.; Walther, A.; Müller, A.H.E.; Ballauff, M. Nanoblossoms: Light-Induced Conformational Changes of Cationic Polyelectrolyte Stars in the Presence of Multivalent Counterions. Nano Lett. 2007, 7, 167–171. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Choi, H. Stimuli-Responsive Polymers and Colloids under Electric and Magnetic Fields. Polymers 2014, 6, 2803. [Google Scholar] [CrossRef]
- Hsiao, S.-H.; Lu, H.-Y. Electrosynthesis of Aromatic Poly(amide-amine) Films from Triphenylamine-Based Electroactive Compounds for Electrochromic Applications. Polymers 2017, 9, 708. [Google Scholar] [CrossRef]
- Guarino, V.; Zuppolini, S.; Borriello, A.; Ambrosio, L. Electro-Active Polymers (EAPs): A Promising Route to Design Bio-Organic/Bioinspired Platforms with on Demand Functionalities. Polymers 2016, 8, 185. [Google Scholar] [CrossRef]
- Bardetsky, D.; Zhitomirsky, I. Electrochemical preparation of composite films containing cationic polyelectrolytes and cobalt hydroxide. Surf. Eng. 2005, 21, 125–130. [Google Scholar] [CrossRef]
- Cheng, Y.; Luo, X.; Betz, J.; Buckhout-White, S.; Bekdash, O.; Payne, G.F.; Bentley, W.E.; Rubloff, G.W. In situ quantitative visualization and characterization of chitosan electrodeposition with paired sidewall electrodes. Soft Matter 2010, 6, 3177–3183. [Google Scholar] [CrossRef]
- Martinez, M.V.; Bruno, M.M.; Miras, M.C.; Barbero, C.A. Electroactive polymers made by loading redox ions inside crosslinked polymeric hydrogels. Effects of hydrophobic interactions and solvent dynamics. Electrochim. Acta 2016, 219, 363–376. [Google Scholar] [CrossRef]
- Ohyanagi, M.; Anson, F.C. Electrochemical behavior of electroactive counterions in solutions of polyelectrolytes. J. Phys. Chem. A 1989, 93, 8377–8382. [Google Scholar] [CrossRef]
- Mergel, O.; Kühn, P.T.; Schneider, S.; Simon, U.; Plamper, F.A. Influence of Polymer Architecture on the Electrochemical Deposition of Polyelectrolytes. Electrochim. Acta 2017, 232, 98–105. [Google Scholar] [CrossRef]
- Ohyanagi, M.; Anson, F.C. Electrodeposition of polyelectrolyte-metal complexes. J. Electroanal. Chem. Interfacial Electrochem. 1989, 258, 469–477. [Google Scholar] [CrossRef]
- Zhuang, X.; Wang, D.; Lin, Y.; Yang, L.; Yu, P.; Jiang, W.; Mao, L. Strong Interaction between Imidazolium-Based Polycationic Polymer and Ferricyanide: Toward Redox Potential Regulation for Selective In Vivo Electrochemical Measurements. Anal. Chem. 2012, 84, 1900–1906. [Google Scholar] [CrossRef] [PubMed]
- Mergel, O.; Gelissen, A.P.H.; Wünnemann, P.; Böker, A.; Simon, U.; Plamper, F.A. Selective Packaging of Ferricyanide within Thermoresponsive Microgels. J. Phys. Chem. C 2014, 118, 26199–26211. [Google Scholar] [CrossRef]
- Spruijt, E.; Choi, E.-Y.; Huck, W.T.S. Reversible Electrochemical Switching of Polyelectrolyte Brush Surface Energy Using Electroactive Counterions. Langmuir 2008, 24, 11253–11260. [Google Scholar] [CrossRef] [PubMed]
- Gelissen, A.P.H.; Schmid, A.J.; Plamper, F.A.; Pergushov, D.V.; Richtering, W. Quaternized microgels as soft templates for polyelectrolyte layer-by-layer assemblies. Polymer 2014, 55, 1991–1999. [Google Scholar] [CrossRef]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods: Fundamentals and Applications, 2nd ed.; Wiley: New York, NY, USA, 2000; pp. 348–360. ISBN 9780471043720. [Google Scholar]
- Yu-Min Tsou, F.C.A. Shifts in Redox Formal Potentials Accompanying the Incorporation of Cationic Complexes in Perfluoro Polycarboxylate and Polysulfonate Coatings on Graphite Electrodes. J. Electrochem. Soc. 1984, 131, 595–601. [Google Scholar] [CrossRef]
- Grieshaber, D.; Vörös, J.; Zambelli, T.; Ball, V.; Schaaf, P.; Voegel, J.-C.; Boulmedais, F. Swelling and Contraction of Ferrocyanide-Containing Polyelectrolyte Multilayers upon Application of an Electric Potential. Langmuir 2008, 24, 13668–13676. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Elliot, M.C.; Cammarata, V. Polyallylammonium Ferrocyanide Films for Trace Water Detection in Halogenated Solvents. Electroanalysis 2009, 21, 2542–2548. [Google Scholar] [CrossRef]
- Barlow, R.B.; Bremner, J.B.; Soh, K.S. The Effects of Replacing Ester by Amide on the Biological Properties of Compounds Related to Acetylcholine. Br. J. Pharmacol. 1978, 62, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Sauerbrey, G. Verwendung von Schwingquarzen zur Wägung dünner Schichten und zur Mikrowägung. Z. Phys. 1959, 155, 206–222. [Google Scholar] [CrossRef]
- Johannsmann, D. Viscoelastic, mechanical, and dielectric measurements on complex samples with the quartz crystal microbalance. Phys. Chem. Chem. Phys. 2008, 10, 4516–4534. [Google Scholar] [CrossRef] [PubMed]
- Buttry, D.A.; Ward, M.D. Measurement of interfacial processes at electrode surfaces with the electrochemical quartz crystal microbalance. Chem. Rev. 1992, 92, 1355–1379. [Google Scholar] [CrossRef]
- Hillman, A.R. The Electrochemical Quartz Crystal Microbalance. In Encyclopedia of Electrochemistry; Wiley-VCH Verlag GmbH & Co., KGaA: Weinheim, Germany, 2007; ISBN 9783527610426. [Google Scholar]
- Sigolaeva, L.V.; Gladyr, S.Y.; Gelissen, A.P.H.; Mergel, O.; Pergushov, D.V.; Kurochkin, I.N.; Plamper, F.A.; Richtering, W. Dual-Stimuli-Sensitive Microgels as a Tool for Stimulated Spongelike Adsorption of Biomaterials for Biosensor Applications. Biomacromolecules 2014, 15, 3735–3745. [Google Scholar] [CrossRef] [PubMed]
- White, E.M.; Yatvin, J.; Grubbs, J.B.; Bilbrey, J.A.; Locklin, J. Advances in smart materials: Stimuli-responsive hydrogel thin films. J. Polym. Sci. B Polym. Phys. 2013, 51, 1084–1099. [Google Scholar] [CrossRef]
- Plamper, F.A. Changing Polymer Solvation by Electrochemical Means: Basics and Applications. Adv. Polym. Sci. 2015, 266, 125–212. [Google Scholar] [CrossRef]
- Plamper, F.A. Polymerizations under electrochemical control. Colloid Polym. Sci. 2014, 292, 777–783. [Google Scholar] [CrossRef]
- Mergel, O.; Wünnemann, P.; Simon, U.; Böker, A.; Plamper, F.A. Microgel Size Modulation by Electrochemical Switching. Chem. Mater. 2015, 27, 7306–7312. [Google Scholar] [CrossRef]
- Maccarrone, S.; Mergel, O.; Plamper, F.A.; Holderer, O.; Richter, D. Electrostatic Effects on the Internal Dynamics of Redox-Sensitive Microgel Systems. Macromolecules 2016, 49, 1911–1917. [Google Scholar] [CrossRef]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schneider, S.; Janssen, C.; Klindtworth, E.; Mergel, O.; Möller, M.; Plamper, F. Influence of Polycation Composition on Electrochemical Film Formation. Polymers 2018, 10, 429. https://doi.org/10.3390/polym10040429
Schneider S, Janssen C, Klindtworth E, Mergel O, Möller M, Plamper F. Influence of Polycation Composition on Electrochemical Film Formation. Polymers. 2018; 10(4):429. https://doi.org/10.3390/polym10040429
Chicago/Turabian StyleSchneider, Sabine, Corinna Janssen, Elisabeth Klindtworth, Olga Mergel, Martin Möller, and Felix Plamper. 2018. "Influence of Polycation Composition on Electrochemical Film Formation" Polymers 10, no. 4: 429. https://doi.org/10.3390/polym10040429
APA StyleSchneider, S., Janssen, C., Klindtworth, E., Mergel, O., Möller, M., & Plamper, F. (2018). Influence of Polycation Composition on Electrochemical Film Formation. Polymers, 10(4), 429. https://doi.org/10.3390/polym10040429