Surface Modification of Wood Flour via ARGET ATRP and Its Application as Filler in Thermoplastics



Abstract
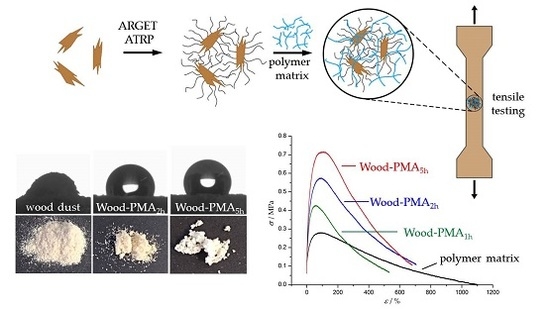
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Pretreatment of Wood Flour
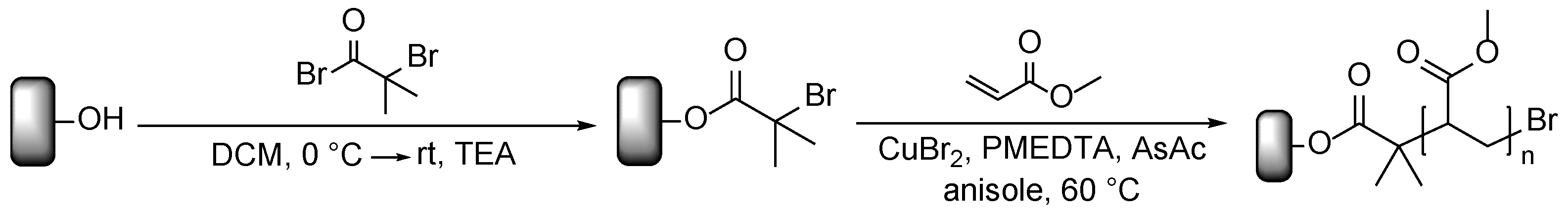
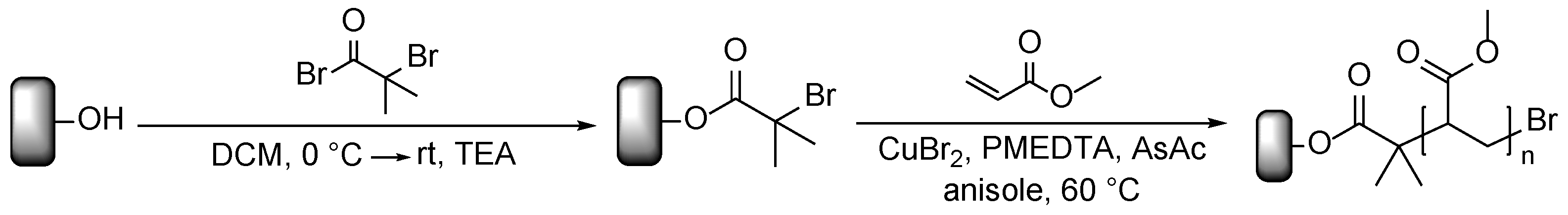
2.3. Immobilization of the ATRP Initiators
2.4. Synthesis of Poly(Methyl Acrylate) (PMA) as Polymer Matrix
2.5. Polymerization on Wood Surfaces via ARGET ATRP
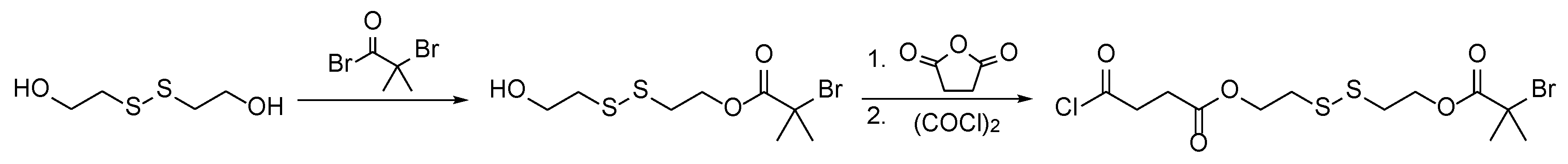
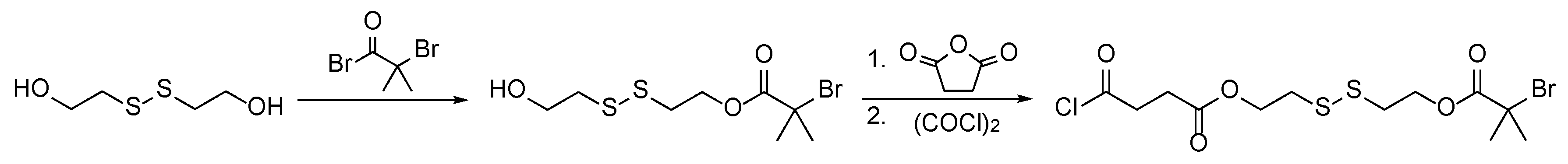
2.6. Synthesis and Cleavage of the Disulfide Initiator
2.7. Preparation of Composite Materials
2.8. Methods
2.8.1. Size-Exclusion Chromatography (SEC)
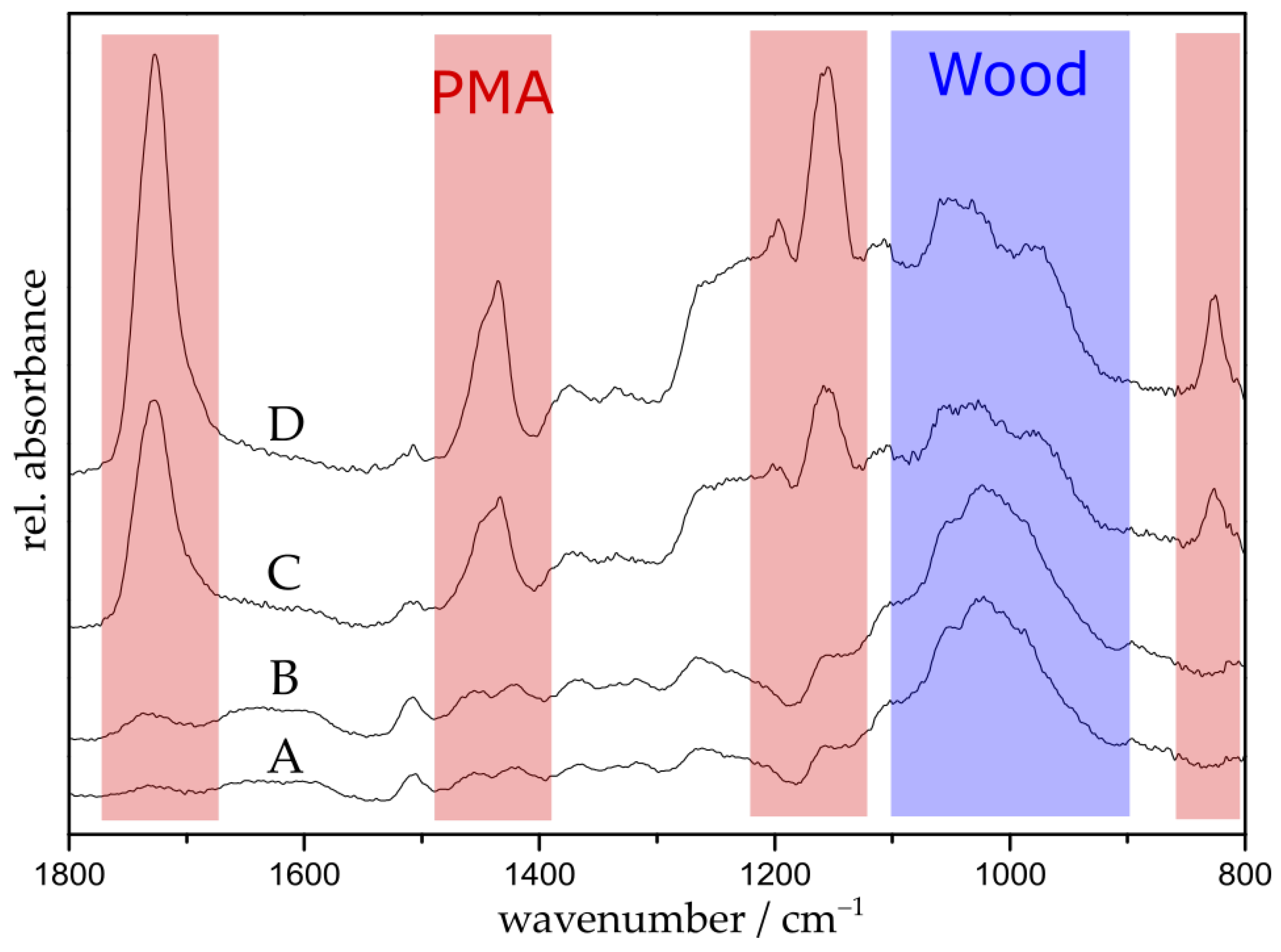
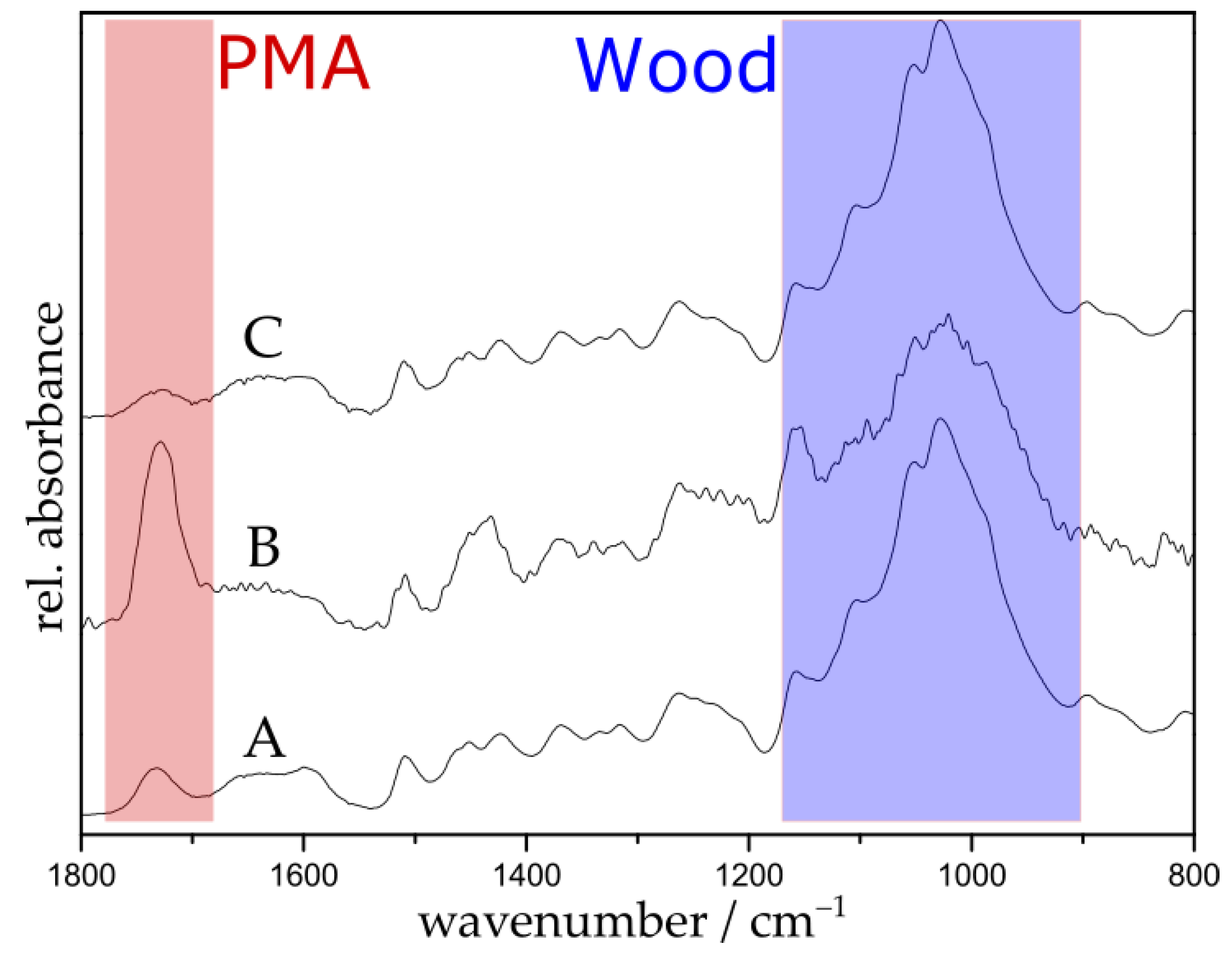
2.8.2. Attenuated Total Reflectance Fourier-Transform Infrared Spectroscopy (ATR-FTIR)
2.8.3. Scanning Electron Microscopy (SEM) and Energy-Dispersive X-ray Spectroscopy (EDX)
2.8.4. Differential Scanning Calorimetry (DSC)
2.8.5. Tensile Testing
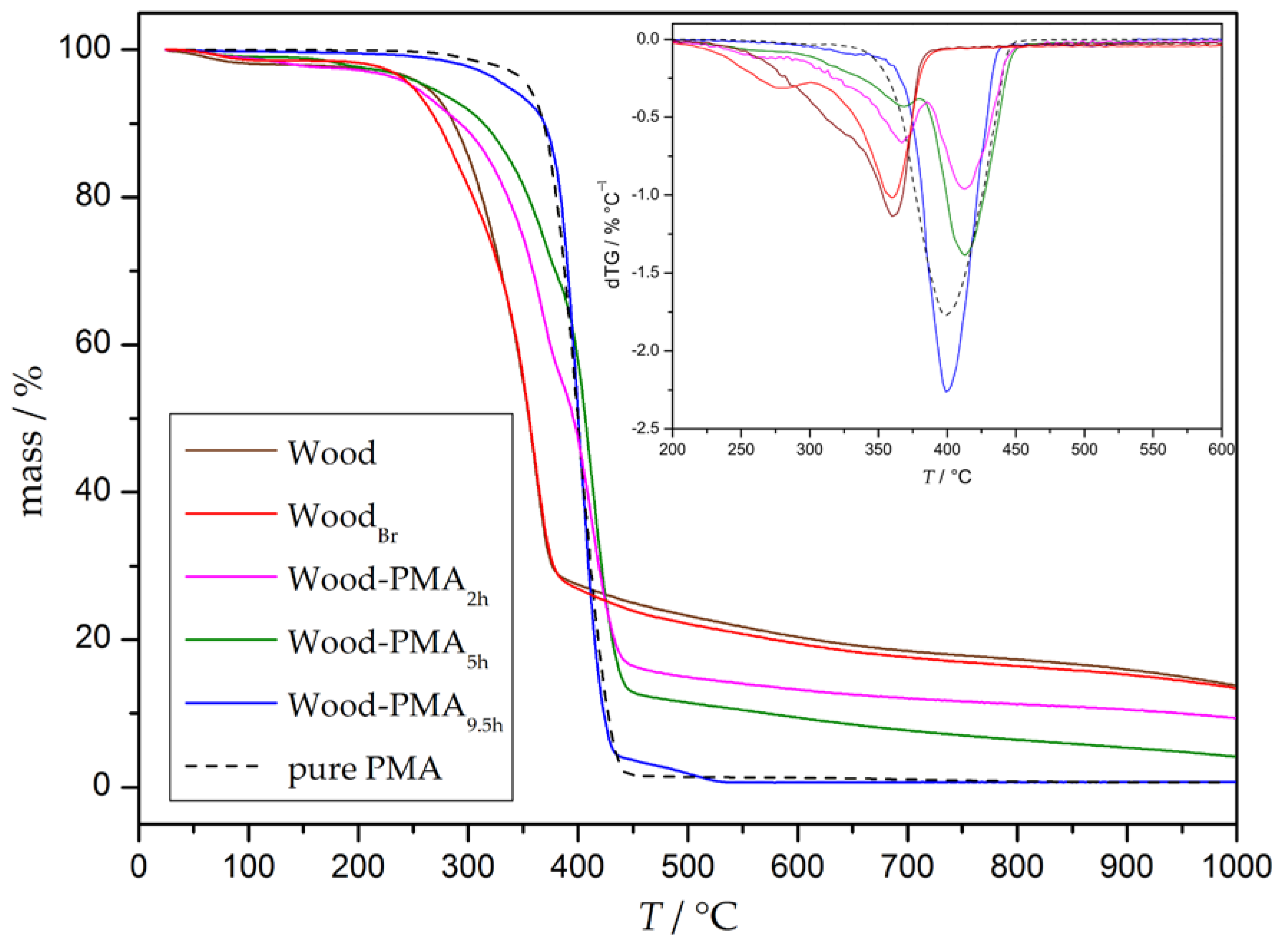
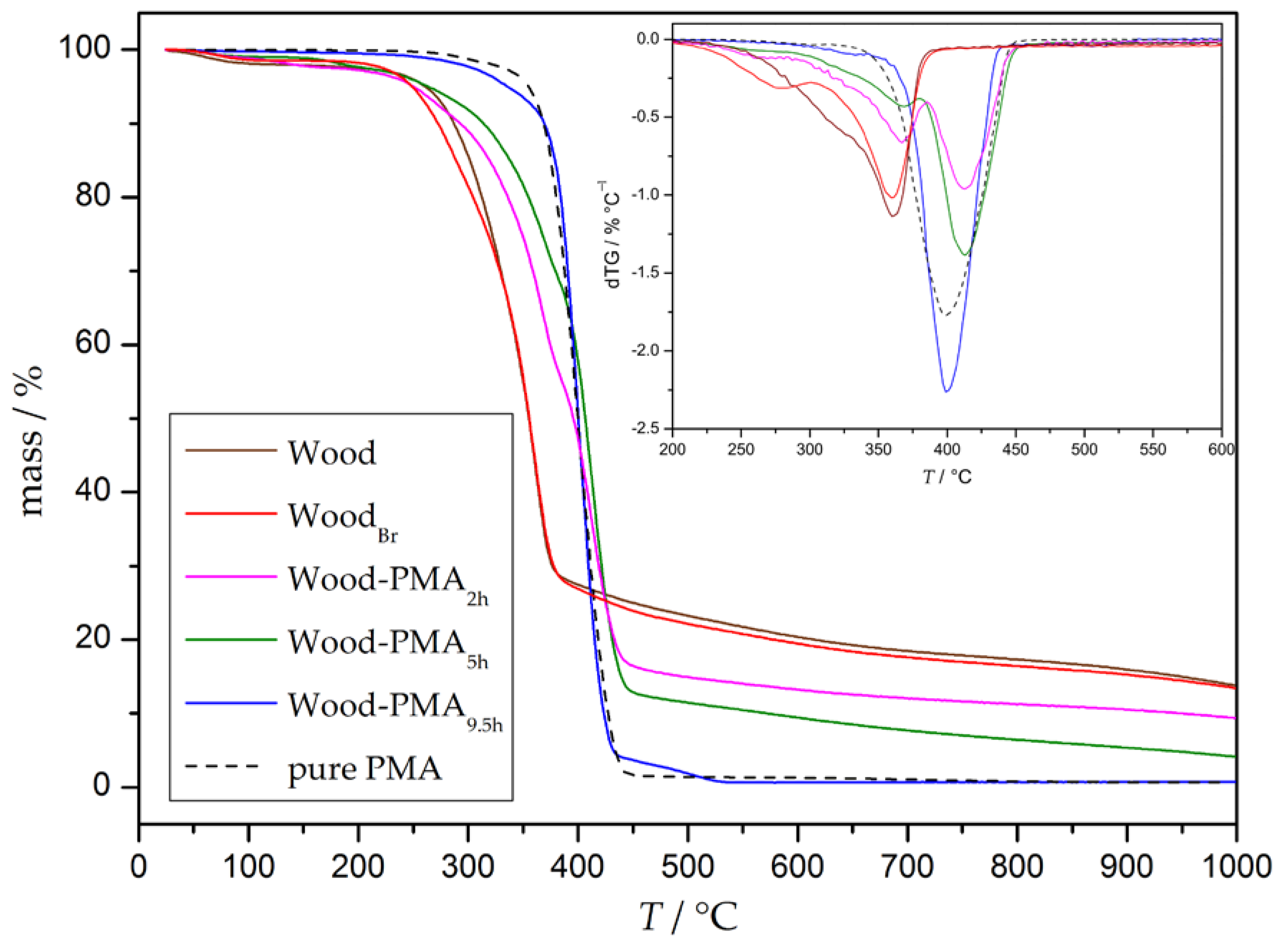
2.8.6. Thermogravimetric Analysis (TGA)
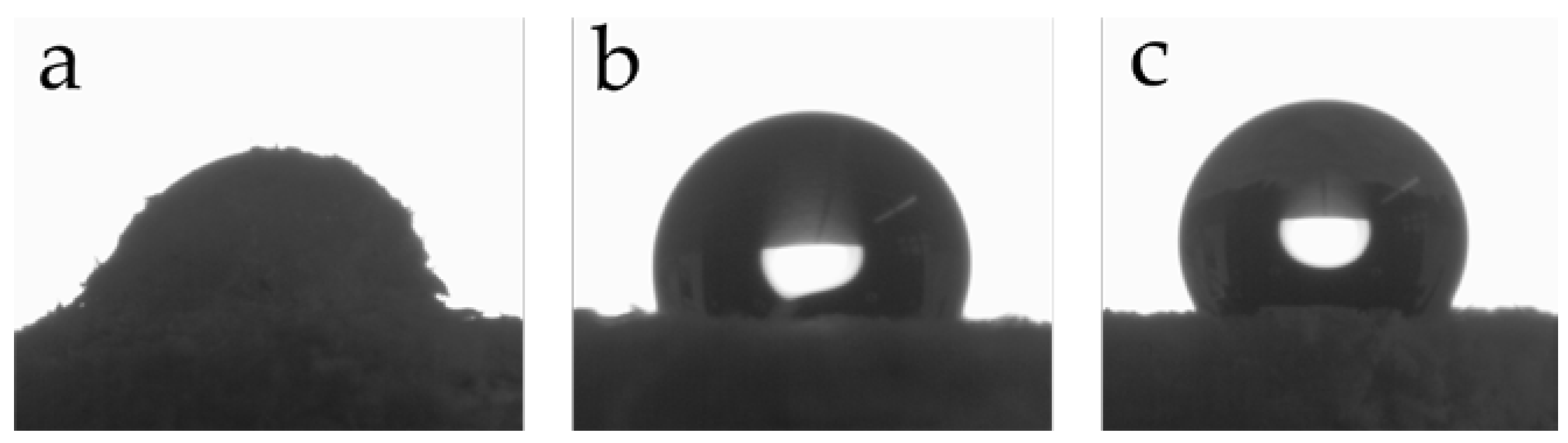
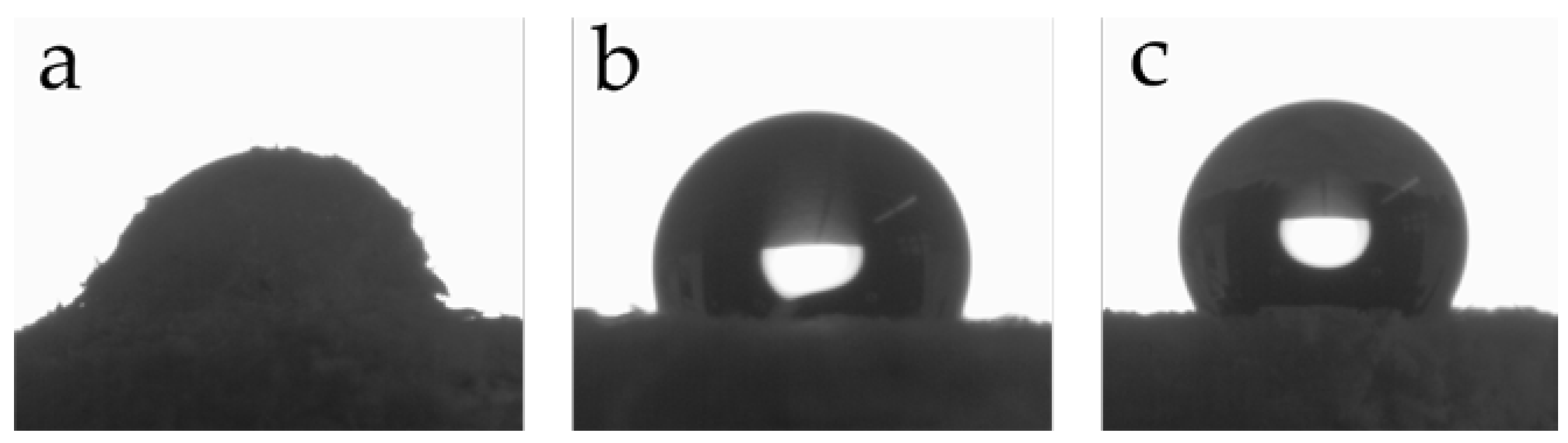
2.8.7. Water Contact Angle (WCA) Measurements
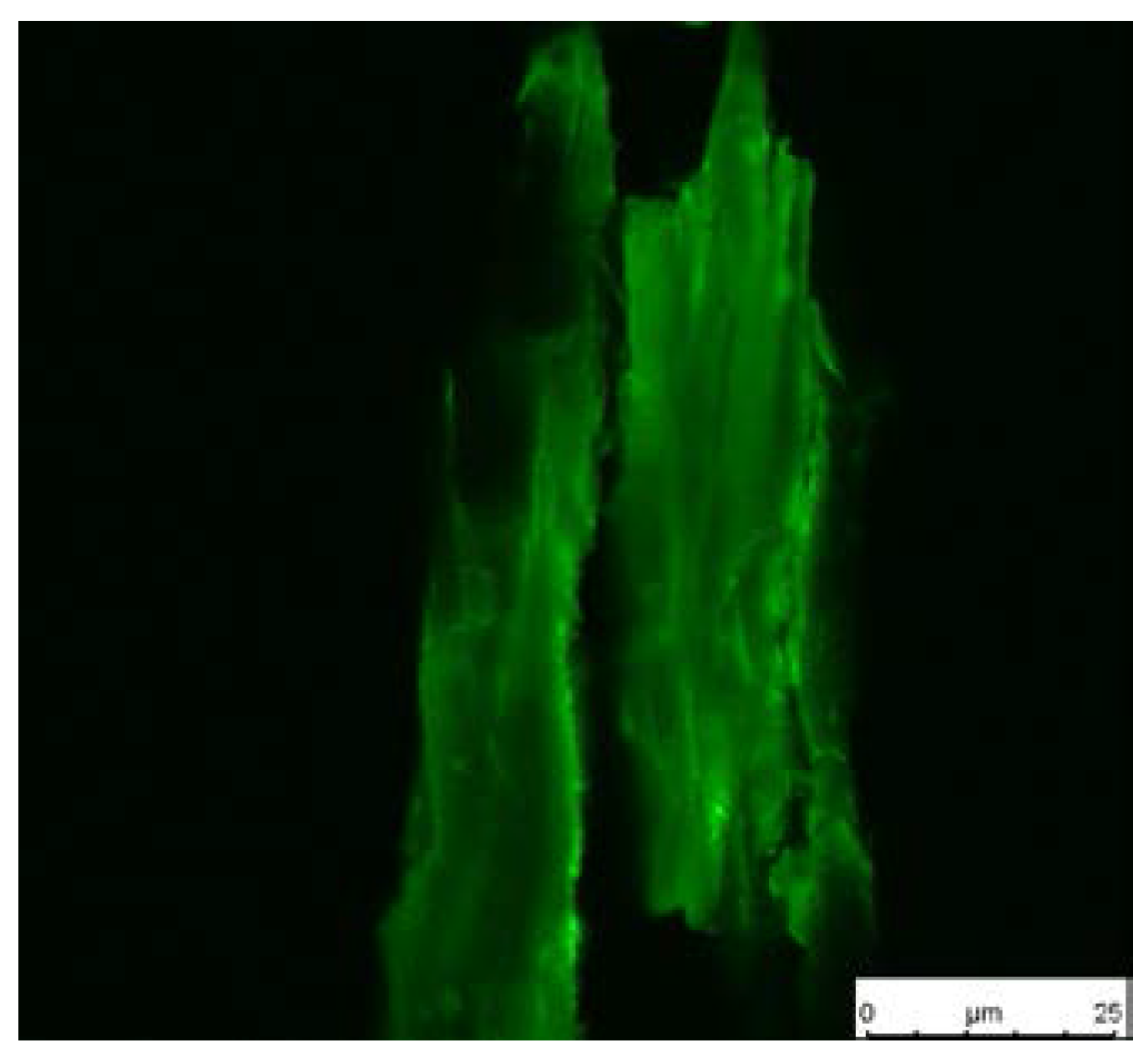
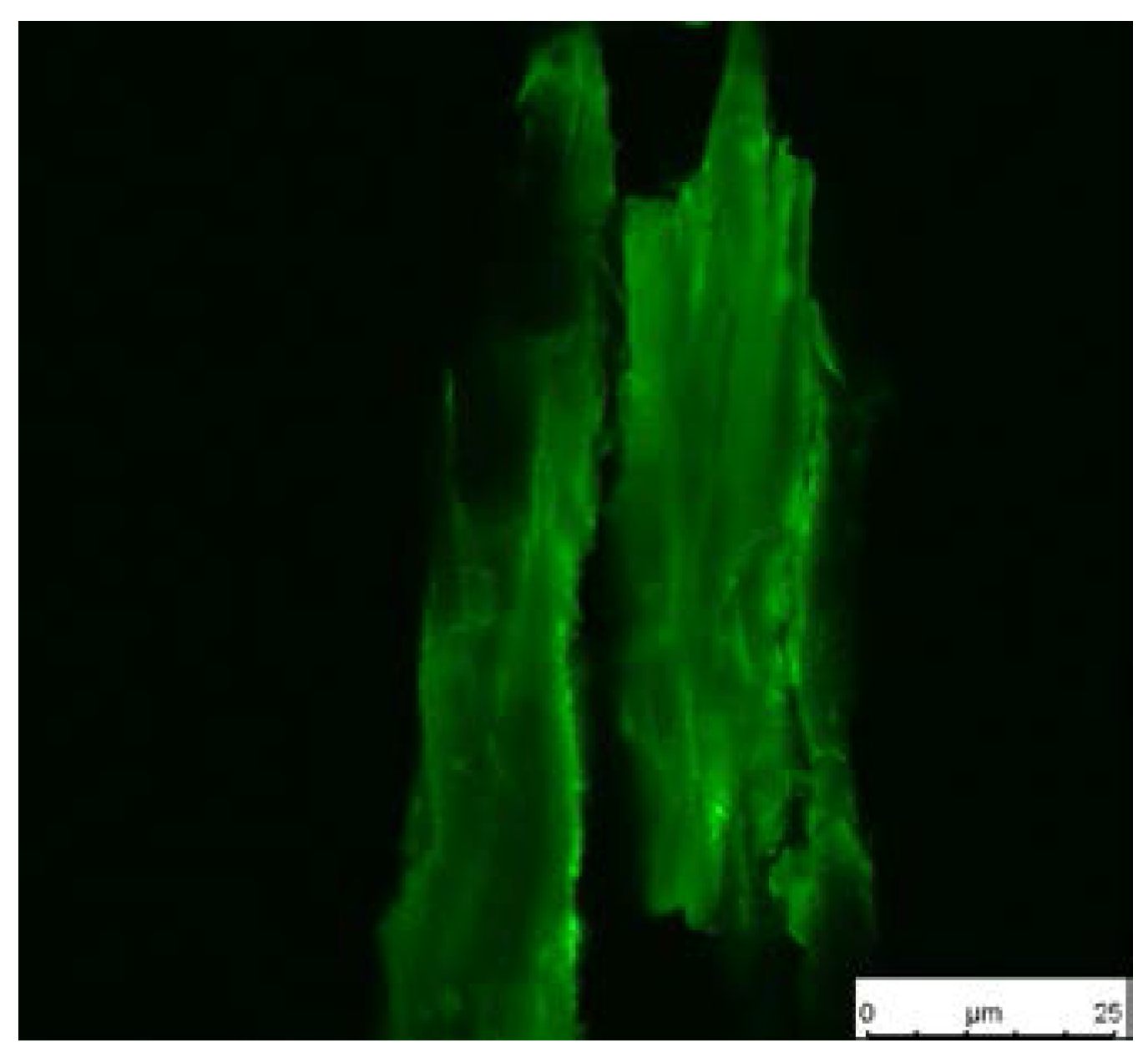
2.8.8. Fluorescence Microscopy
3. Results and Discussion
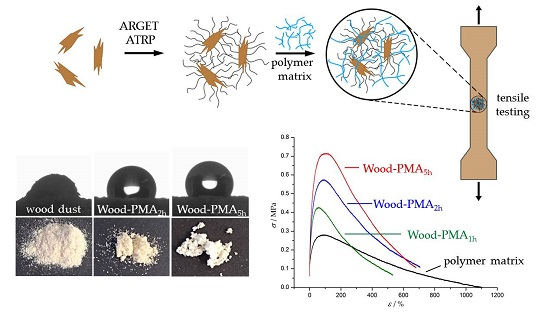
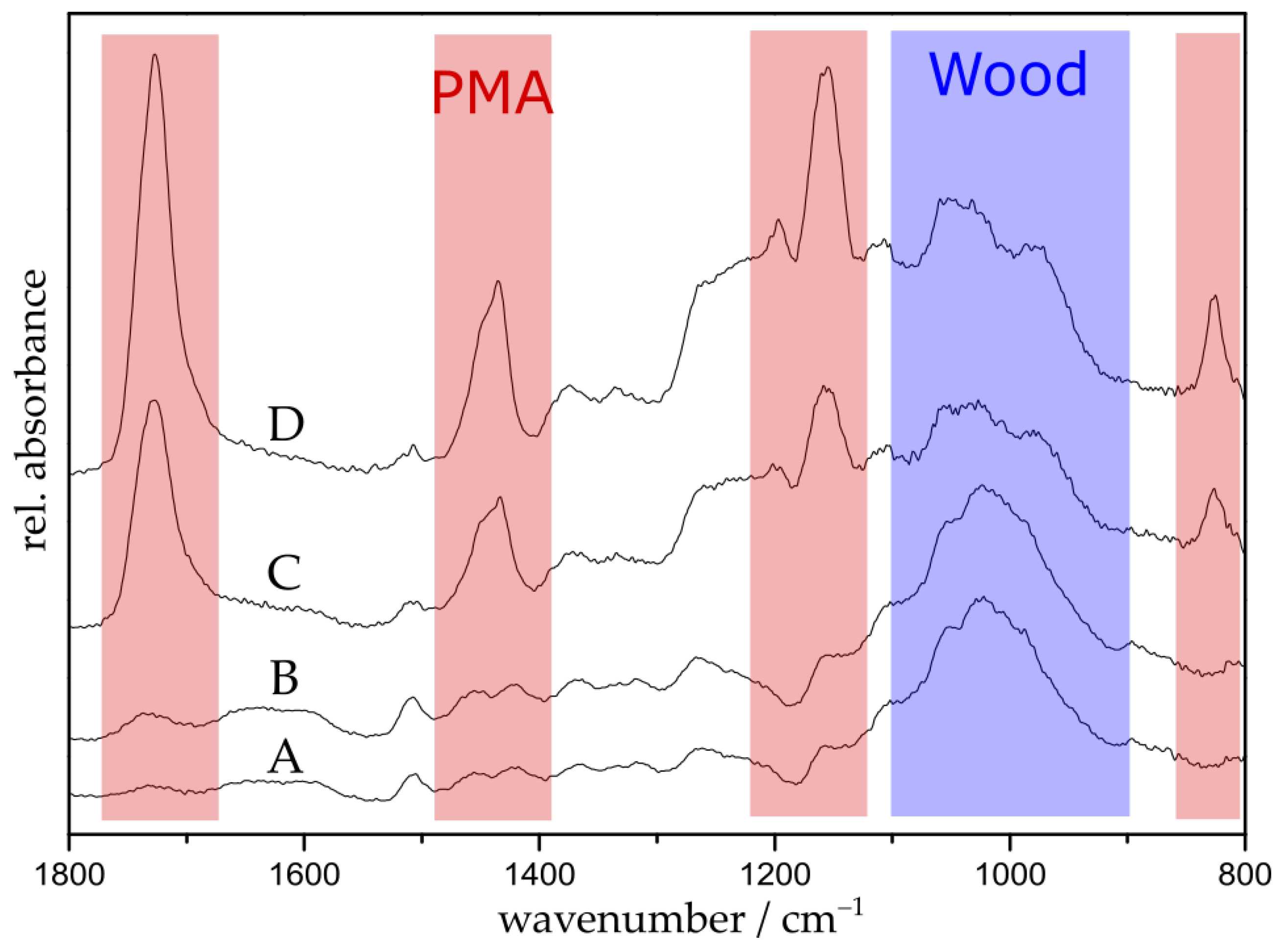
3.1. Polymerization of Wood Surfaces via ARGET ATRP
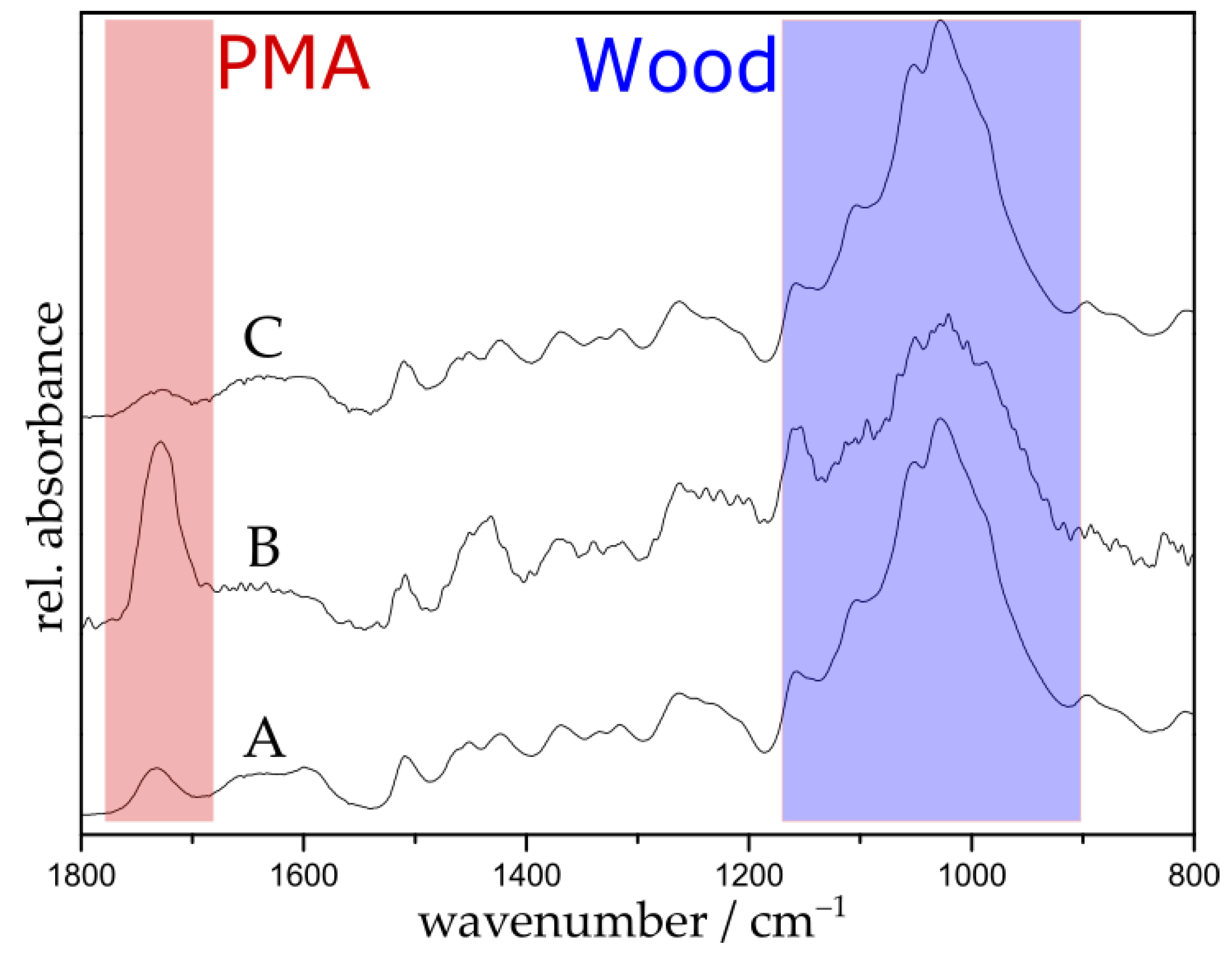
3.2. Selective Cleavage of the Grafted Polymer under Mild Conditions
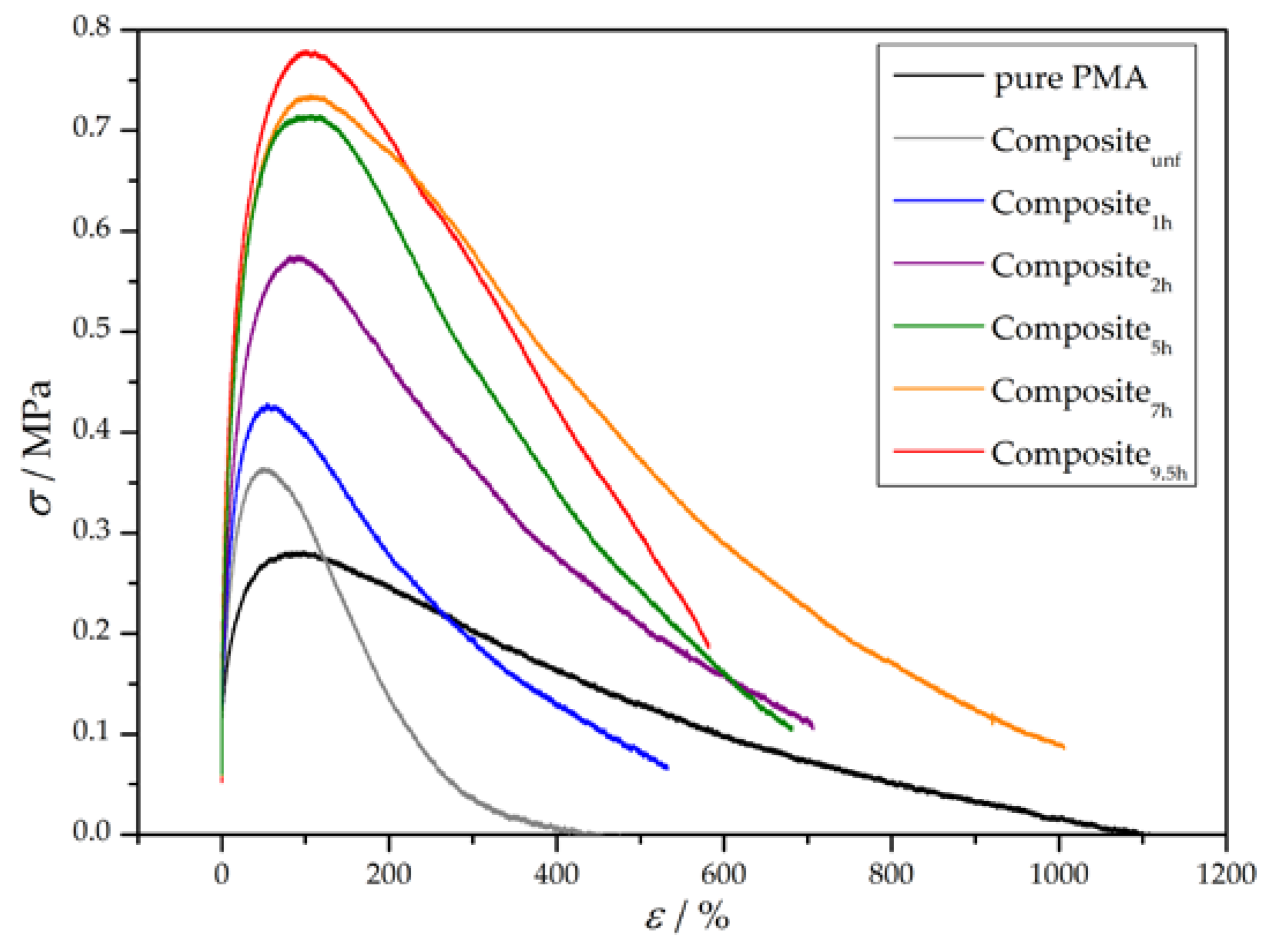
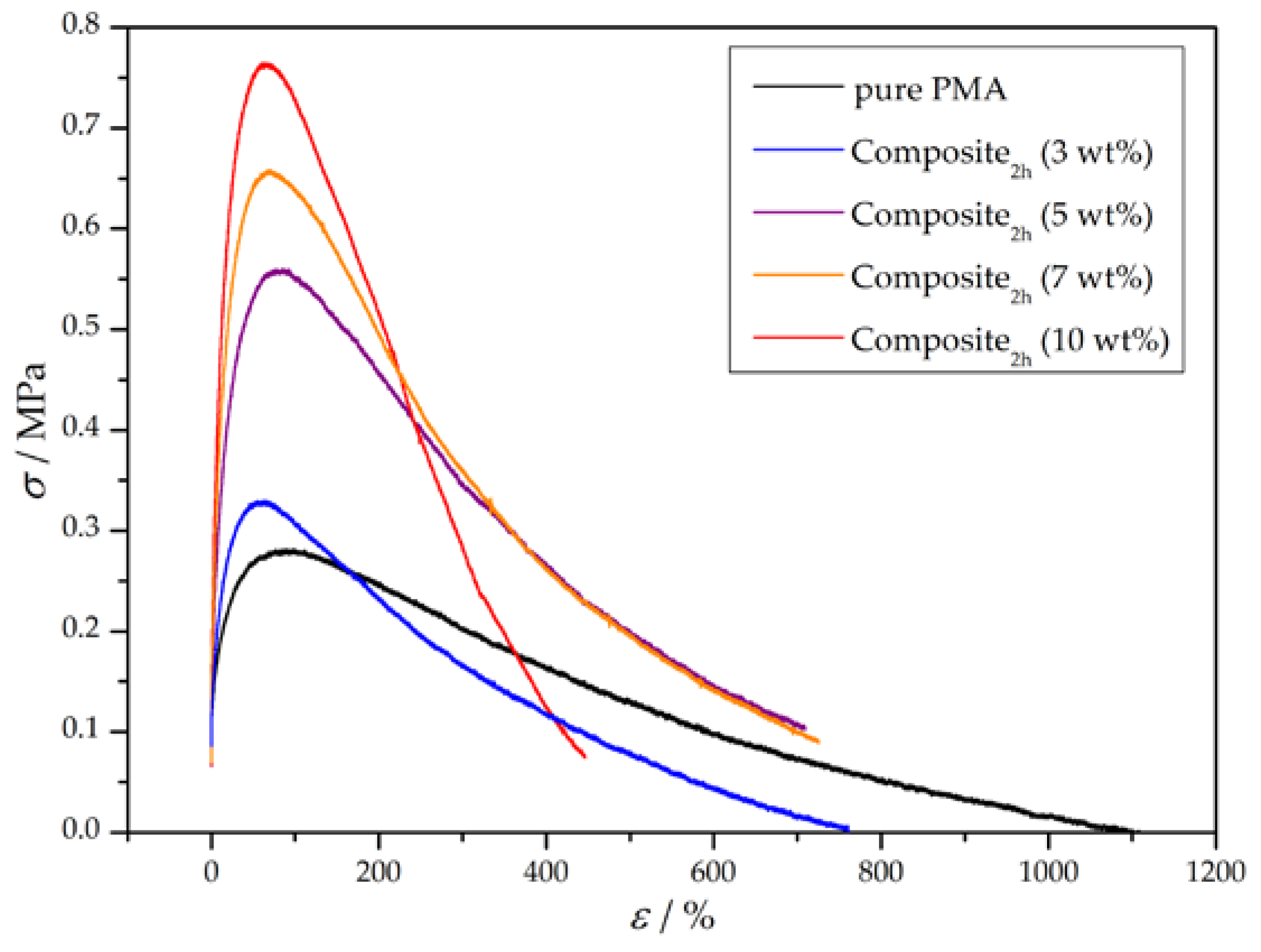
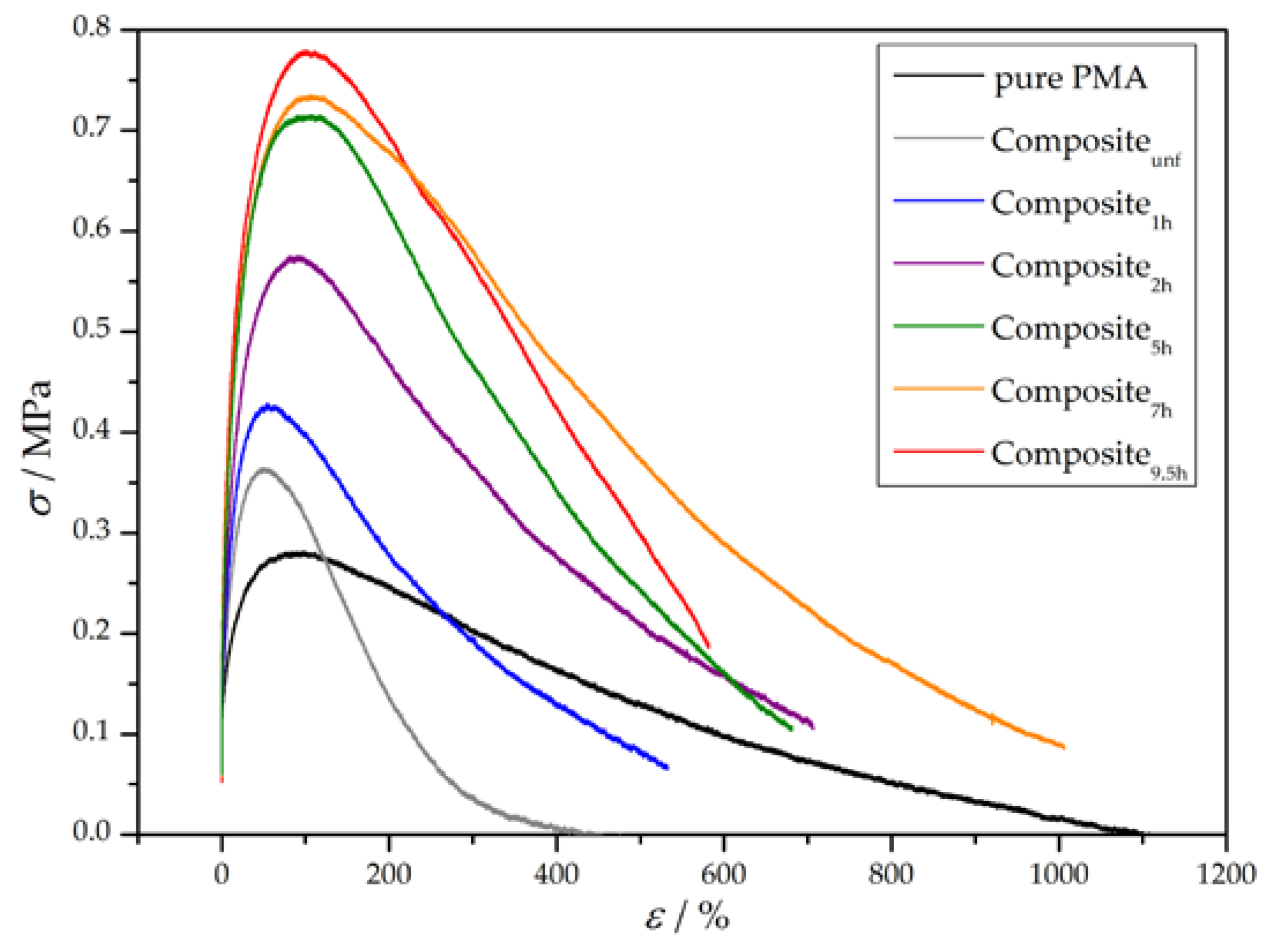
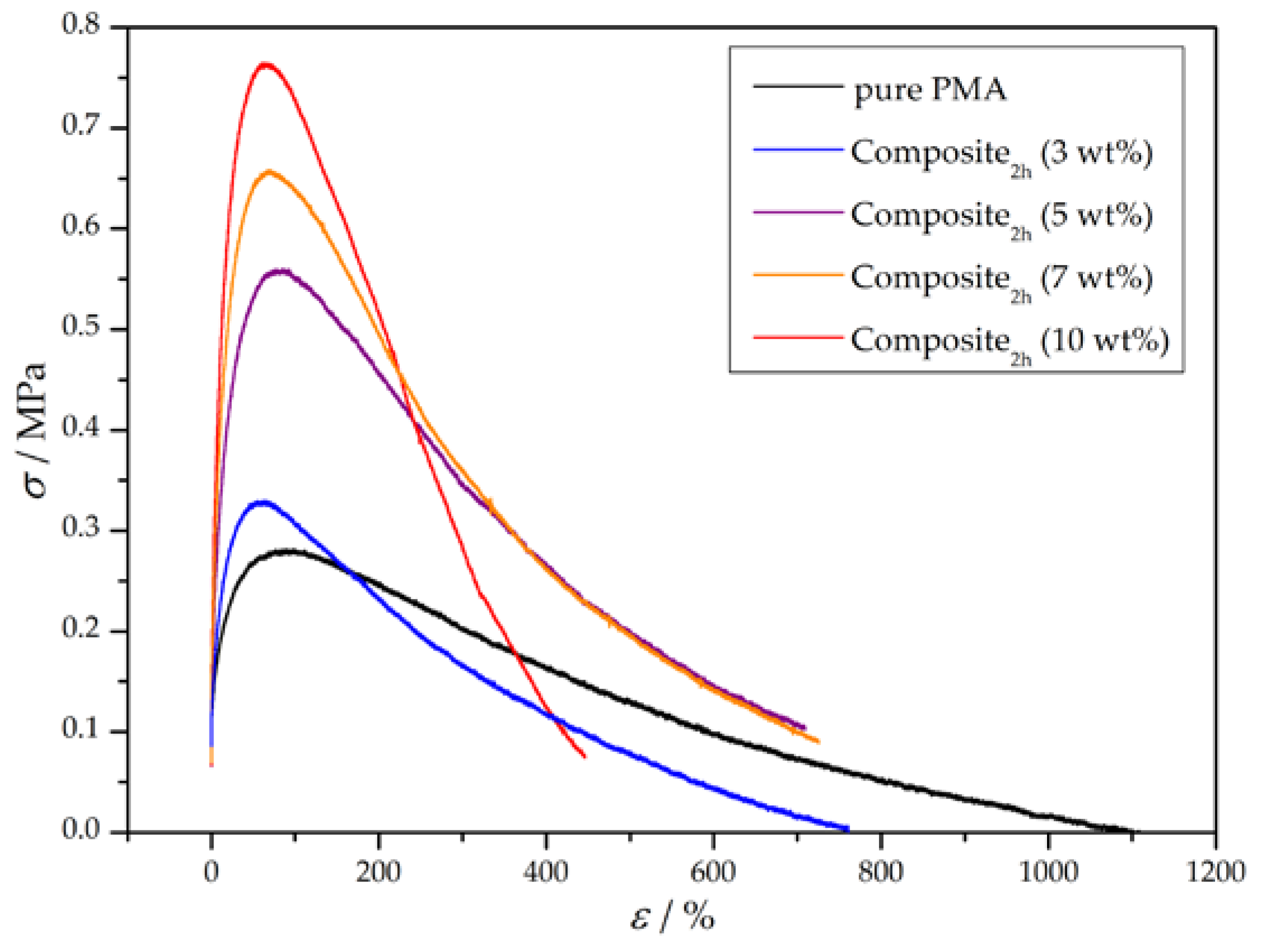
3.3. Preparation and Properties of Wood-Reinforced Thermoplastics
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kumar, V.; Tyagi, L.; Sinha, S. Wood flour-reinforced plastic composites: A review. Rev. Chem. Eng. 2011, 27, 253–264. [Google Scholar] [CrossRef]
- Kim, J.K.; Kaushik, P. Recent Advances in the Processing of Wood–Plastic Composites; Springer: Heidelberg/Berlin, Germany, 2010; ISBN 9783642148767. [Google Scholar]
- Li, Y. Wood-Polymer Composites. In Advances in Composite Materials—Analysis of Natural and Man—Made Materials; Tesinova, P., Ed.; InTech: Rijeka, Yugoslavia, 2011; pp. 229–284. ISBN 978-953-307-449-8. [Google Scholar]
- El-Haggar, S.M.; Kamel, M.A. Wood Plastic Composites. In Advances in Composite Materials—Analysis of Natural and Man—Made Materials; Tesinova, P., Ed.; Intech: Rijeka, Yugoslavia, 2011; pp. 325–344. ISBN 978-953-307-449-8. [Google Scholar]
- McDonough, W.; Braungart, M. Cradle to Cradle: Remaking the Way We Make Things, 1st ed.; North Point Press: New York, NY, USA, 2002; ISBN 978-0865475878. [Google Scholar]
- Forest Products Laboratory. Wood Handbook: Wood as an Engineering Material; Ross, R.J., Ed.; CreateSpace Independent Publishing Platform: Madison, WI, USA, 2010; ISBN 1892529025. [Google Scholar]
- Bledzki, A.K.; Reihmane, S.; Gassan, J. Thermoplastics reinforced with wood fillers: A literature review. Polym. Plast. Technol. Eng. 1998, 37, 451–468. [Google Scholar] [CrossRef]
- Bledzki, A.K.; Gassan, J. Composites reinforced with cellulose based fibres. Prog. Polym. Sci. 1999, 24, 221–274. [Google Scholar] [CrossRef]
- Müller, A.H.E.; Matyjaszewski, K. Controlled and Living Polymerizations, 1st ed.; Matyjaszewski, K., Müller, A.H.E., Eds.; WILEY-VCH Verlag: Weinheim, Germany, 2009; ISBN 9783527629091. [Google Scholar]
- Matiyashevskiy, K.; Thomas, P.D. Handbook of Radical Polymerization, 1st ed.; Matyjaszewski, K., Davis, T.P., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2002; ISBN 047139274X. [Google Scholar]
- Matyjaszewski, K.; Xia, J. Atom Transfer Radical Polymerization. Chem. Rev. 2001, 101, 2921–2990. [Google Scholar] [CrossRef] [PubMed]
- Khabibullin, A.; Mastan, E.; Matyjaszewski, K.; Zhu, S. Surface-Initiated Atom Transfer Radical Polymerization. In Controlled Radical Polymerization at and from Solid Surfaces; Vana, P., Ed.; Springer International Publishing: Cham, Switzerland, 2016; pp. 29–76. ISBN 978-3-319-22138-0. [Google Scholar]
- Matyjaszewski, K.; Hongchen, D.; Jakubowski, W.; Pietrasik, J.; Kusumo, A. Grafting from surfaces for “everyone”: ARGET ATRP in the presence of air. Langmuir 2007, 23, 4528–4531. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Wang, X.; Zhou, F. A versatile macro-initiator with dual functional anchoring groups for surface-initiated atom transfer radical polymerization on various substrates. Polym. Chem. 2012, 3, 2129–2137. [Google Scholar] [CrossRef]
- Xing, T.; Hu, W.; Li, S.; Chen, G. Preparation, structure and properties of multi-functional silk via ATRP method. Appl. Surf. Sci. 2012, 258, 3208–3213. [Google Scholar] [CrossRef]
- Lindqvist, J.; Malmström, E. Surface modification of natural substrates by atom transfer radical polymerization. J. Appl. Polym. Sci. 2006, 100, 4155–4162. [Google Scholar] [CrossRef]
- Morsi, S.M.; Pakzad, A.; Amin, A.; Yassar, R.S.; Heiden, P.A. Chemical and nanomechanical analysis of rice husk modified by ATRP-grafted oligomer. J. Colloid Interface Sci. 2011, 360, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.; Al-Khouja, A.; Washburn, N.R. Lignin-Based Graft Copolymers via ATRP and Click Chemistry. In Green Polymer Chemistry: Biocatalysis and Materials II; Cheng, H.N., Gross, R.H., Smith, P.B., Eds.; American Chemical Society: Washington, DC, USA, 2013; pp. 373–391. ISBN 9780841228955. [Google Scholar]
- Liu, X.; Yin, H.; Zhang, Z.; Diao, B.; Li, J. Functionalization of lignin through ATRP grafting of poly(2-dimethylaminoethyl methacrylate) for gene delivery. Colloids Surf. B Biointerfaces 2015, 125, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Hansson, S.; Östmark, E.; Carlmark, A.; Malmström, E. ARGET ATRP for versatile grafting of cellulose using various monomers. ACS Appl. Mater. Interfaces 2009, 1, 2651–2659. [Google Scholar] [CrossRef] [PubMed]
- Carlmark, A.; Malmström, E. Atom Transfer Radical Polymerization from Cellulose Fibers at Ambient Temperature. J. Am. Chem. Soc. 2002, 124, 900–901. [Google Scholar] [CrossRef] [PubMed]
- Carlmark, A.; Larsson, E.; Malmström, E. Grafting of cellulose by ring-opening polymerisation—A review. Eur. Polym. J. 2012, 48, 1646–1659. [Google Scholar] [CrossRef]
- Meng, T.; Gao, X.; Zhang, J.; Yuan, J.; Zhang, Y.; He, J. Graft copolymers prepared by atom transfer radical polymerization (ATRP) from cellulose. Polymer 2009, 50, 447–454. [Google Scholar] [CrossRef]
- Castelvetro, V.; Geppi, M.; Giaiacopi, S.; Mollica, G. Cotton fibers encapsulated with homo- and block copolymers: Synthesis by the atom transfer radical polymerization grafting-from technique and solid-state NMR dynamic investigations. Biomacromolecules 2007, 8, 498–508. [Google Scholar] [CrossRef] [PubMed]
- Nyström, D.; Lindqvist, J.; Östmark, E.; Antoni, P.; Carlmark, A.; Hult, A.; Malmström, E. Superhydrophobic and self-cleaning bio-fiber surfaces via ATRP and subsequent postfunctionalization. ACS Appl. Mater. Interfaces 2009, 1, 816–823. [Google Scholar] [CrossRef] [PubMed]
- Worthley, C.H.; Constantopoulos, K.T.; Ginic-Markovic, M.; Pillar, R.J.; Matisons, J.G.; Clarke, S. Surface modification of commercial cellulose acetate membranes using surface-initiated polymerization of 2-hydroxyethyl methacrylate to improve membrane surface biofouling resistance. J. Membr. Sci. 2011, 385–386, 30–39. [Google Scholar] [CrossRef]
- Xiao, M.; Li, S.; Chanklin, W.; Zheng, A.; Xiao, H. Surface-initiated atom transfer radical polymerization of butyl acrylate on cellulose microfibrils. Carbohydr. Polym. 2011, 83, 512–519. [Google Scholar] [CrossRef]
- Plackett, D.; Jankova, K.; Egsgaard, H.; Hvilsted, S. Modification of jute fibers with polystyrene via atom transfer radical polymerization. Biomacromolecules 2005, 6, 2474–2484. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.M.; Ji, J.F.; Tang, Y.J.; Zhao, Y. Wood Pulp Fibers Grafted with Polyacrylamide through Atom Transfer Radical Polymerization. In Advances in Chemical Engineering: ICCMME 2011; advanced materials research; Trans Tech Publications: Zurich, Switzerland, 2012; Volume 396, pp. 1458–1461. [Google Scholar]
- Zampano, G.; Bertoldo, M.; Bronco, S. Poly(ethyl acrylate) surface-initiated ATRP grafting from wood pulp cellulose fibers. Carbohydr. Polym. 2009, 75, 22–31. [Google Scholar] [CrossRef]
- Fu, Y.; Li, G.; Yu, H.; Liu, Y. Hydrophobic modification of wood via surface-initiated ARGET ATRP of MMA. Appl. Surf. Sci. 2012, 258, 2529–2533. [Google Scholar] [CrossRef]
- Cabane, E.; Keplinger, T.; Künniger, T.; Merk, V.; Burgert, I. Functional lignocellulosic materials prepared by ATRP from a wood scaffold. Nat. Publ. Gr. 2016, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Fu, Y.; Li, G.; Liu, Y. Antimicrobial surfaces of quaternized poly[(2-dimethyl amino)ethyl methacrylate] grafted on wood via ARGET ATRP. Holzforschung 2013, 67, 455–461. [Google Scholar] [CrossRef]
- Yu, F.; Yang, W.; Song, J.; Wu, Q.; Chen, L. Investigation on hydrophobic modification of bamboo flour surface by means of atom transfer radical polymerization method. Wood Sci. Technol. 2014, 48, 289–299. [Google Scholar] [CrossRef]
- Babu, K.; Dhamodharan, R. Grafting of poly(methyl methacrylate) brushes from magnetite nanoparticles using a phosphonic acid based initiator by Ambient Temperature Atom Transfer Radical Polymerization (ATATRP). Nanoscale Res. Lett. 2008, 3, 109–117. [Google Scholar] [CrossRef]
- Jakubowski, W.; Min, K.; Matyjaszewski, K. Activators Regenerated by Electron Transfer for Atom Transfer Radical Polymerization of Styrene. Macromolecules 2006, 39, 39–45. [Google Scholar] [CrossRef]
- Matyjaszewski, K.; Gao, H.; Min, K. Use of Ascorbic Acid as Reducing Agent for Synthesis of Well-Defined Polymers by ARGET ATRP. Macromolecules 2007, 40, 1789–1791. [Google Scholar] [CrossRef]
- Schwellenbach, J.; Kosiol, P.; Sölter, B.; Taft, F.; Villain, L.; Strube, J. Controlling the polymer-nanolayer architecture on anion-exchange membrane adsorbers via surface-initiated atom transfer radical polymerization. React. Funct. Polym. 2016, 106, 32–42. [Google Scholar] [CrossRef]
- Hansson, S.; Antoni, P.; Bergenudd, H.; Malmström, E. Selective cleavage of polymer grafts from solid surfaces: Assessment of initiator content and polymer characteristics. Polym. Chem. 2011, 2, 556. [Google Scholar] [CrossRef]
- Grubisic, Z.; Rempp, P.; Benoit, H. A universal calibration for gel permeation chromatography. J. Polym. Sci. Part B Polym. Lett. 1967, 5, 753–759. [Google Scholar] [CrossRef]
- Buback, M.; Kurz, C.H.; Schmaltz, C. Pressure dependence of propagation rate coefficients in free- radical homopolymerizations of methyl acrylate and dodecyl acrylate. Macromol. Chem. Phys. 1998, 199, 1721–1727. [Google Scholar] [CrossRef]
- Bombalski, L.; Min, K.; Dong, H.; Tang, C.; Matyjaszewski, K. Preparation of well-defined hybrid materials by ATRP in miniemulsion. Macromolecules 2007, 40, 7429–7432. [Google Scholar] [CrossRef]
- Kwak, Y.; Matyjaszewski, K. ARGET ATRP of methyl methacrylate in the presence of nitrogen-based ligands as reducing agents. Polym. Int. 2009, 58, 242–247. [Google Scholar] [CrossRef]
- Matyjaszewski, K.; Jakubowski, W.; Spanswick, J. Polymerization Process with Catalyst Reactivation. U.S. Patent 20090312505 A1, 1 March 2007. [Google Scholar]
- Haken, J.K.; Werner, R.L. Infrared Spectrum of Polymethyl Acrylate. Br. Polym. J. 2007, 3, 263–265. [Google Scholar] [CrossRef]
- Schacher, F.; Müllner, M.; Schmalz, H.; Müller, A.H.E. New Block Copolymers with Poly(N,N-dimethylaminoethyl methacrylate) as a Double Stimuli-Responsive Block. Macromol. Chem. Phys. 2009, 210, 256–262. [Google Scholar] [CrossRef]
- Gašparovič, L.; Koreňová, Z.; Jelemenský, L. Kinetic study of wood chips decomposition by TGA. Chem. Pap. 2010, 64, 174–181. [Google Scholar] [CrossRef]
- Zhou, Q.; Greffe, L.; Baumann, M.J.; Malmström, E.; Teeri, T.T.; Brumer, H. Use of xyloglucan as a molecular anchor for the elaboration of polymers from cellulose surfaces: A general route for the design of biocomposites. Macromolecules 2005, 38, 3547–3549. [Google Scholar] [CrossRef]
- Barsbay, M.; Güven, O.; Stenzel, M.H.; Davis, T.P.; Barner-Kowollik, C.; Barner, L. Verification of controlled grafting of styrene from cellulose via radiation-induced RAFT polymerization. Macromolecules 2007, 40, 7140–7147. [Google Scholar] [CrossRef]
- Lee, S.B.; Koepsel, R.R.; Morley, S.W.; Matyjaszewski, K.; Sun, Y.; Russell, A.J. Permanent, nonleaching antibacterial surfaces, 1. Synthesis by atom transfer radical polymerization. Biomacromolecules 2004, 5, 877–882. [Google Scholar] [CrossRef] [PubMed]
- Zoppe, J.O.; Habibi, Y.; Rojas, O.J.; Venditti, R.A.; Johansson, L.; Efimenko, K.; Monika, O.; Osterberg, M.; Laine, J. Poly(N-isopropylacrylamide) brushes grafted from cellulose nanocrystals via surface-initiated single-electron transfer living radical polymerization. Biomacromolecules 2010, 11, 2683–2691. [Google Scholar] [CrossRef] [PubMed]
- Kamada, J.; Koynov, K.; Corten, C.; Juhari, A.; Yoon, J.A.; Urban, M.W.; Balazs, A.C.; Matyjaszewski, K. Redox responsive behavior of thiol/disulfide-functionalized star polymers synthesized via atom transfer radical polymerization. Macromolecules 2010, 43, 4133–4139. [Google Scholar] [CrossRef]
- Tsarevsky, N.V.; Matyjaszewski, K. Reversible redox cleavage/coupling of polystyrene with disulfide or thiol groups prepared by atom transfer radical polymerization. Macromolecules 2002, 35, 9009–9014. [Google Scholar] [CrossRef]
- Larsson, E.; Pendergraph, S.A.; Kaldéus, T.; Malmström, E.; Carlmark, A. Cellulose grafting by photoinduced controlled radical polymerisation. Polym. Chem. 2014, 23–25. [Google Scholar] [CrossRef]
- Hendrich, M.; Lewerdomski, L.; Vana, P. Biomimetic Triblock and Multiblock Copolymers Containing l-Phenylalanine Moieties Showing Healing and Enhanced Mechanical Properties. J. Polym. Sci. Part. A 2015, 1–11. [Google Scholar] [CrossRef]

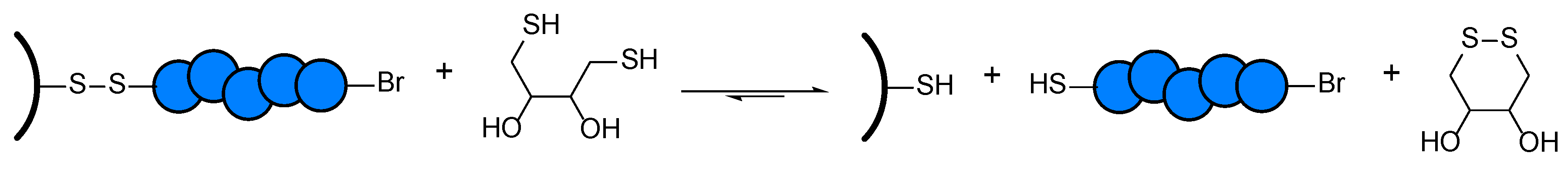

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
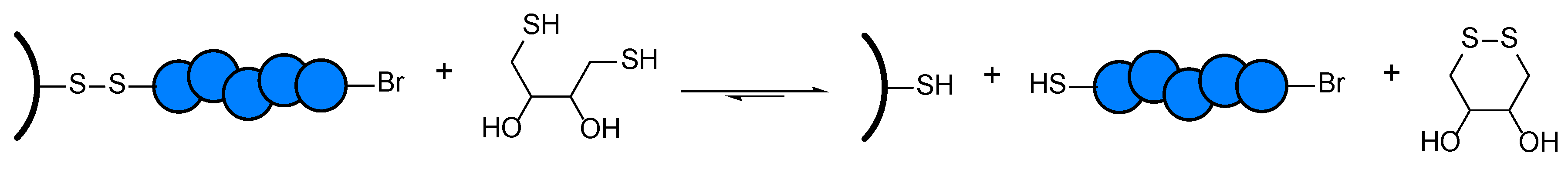{kind=link}
| Wood Stage | PMA Stage | ||||
|---|---|---|---|---|---|
| Ton/°C | Tmax/°C | ML/% | Tmax/°C | ML/% | |
| Wood | 200 | 360 |  | - | - |
| WoodBr | 160 | 360 | - |  | |
| Wood-PMA0.75h | 160 | 360 | 410 | ||
| Wood-PMA2h | 170 | 365 | 410 | ||
| Wood-PMA5h | 190 | 370 | 410 | ||
| Wood-PMA9.5h | 190 | - | 400 | ||
| PMA | 220 | - | - | 400 | |
| w450°C/% | wpolymer/% | |
|---|---|---|
| Wood-PMA0.75h | 24 | 0 |
| Wood-PMA2h | 16 | 33 |
| Wood-PMA5h | 13 | 46 |
| Wood-PMA9.5h | 4 | 83 |
| ATRP initiator | Base | Br wt % (EDX) |
|---|---|---|
| BIBB | TEA | 1.7 ± 0.7 |
| Disulfide | Py | 2.0 ± 0.6 |
| t/h | Mn,max/103 g·mol−1 | Đ |
|---|---|---|
| 2 | 290 | 1.84 |
| 5 | 410 | 1.88 |
| Sample | E/MPa | σy/MPa | UT/MPa |
|---|---|---|---|
| pure PMA | 8 ± 3 | 0.27 ± 0.06 | 126 ± 31 |
| Compositeunf | 7 ± 3 | 0.30 ± 0.17 | 50 ± 17 |
| Composite1h | 11 ± 1 | 0.40 ± 0.03 | 106 ± 18 |
| Composite2h | 20 ± 2 | 0.58 ± 0.02 | 235 ± 16 |
| Composite5h | 26 ± 1 | 0.73 ± 0.02 | 297 ± 5 |
| Composite7h | 29 ± 2 | 0.74 ± 0.01 | 398 ± 15 |
| Composite9.5h | 36 ± 2 | 0.79 ± 0.02 | 299 ± 68 |
| Sample | E/MPa | σy/MPa | UT/MPa |
|---|---|---|---|
| pure PMA | 8 ± 3 | 0.27 ± 0.06 | 126 ± 31 |
| Composite2h (3 wt %) | 8 ± 1 | 0.32 ± 0.01 | 97 ± 13 |
| Composite2h (5 wt %) | 20 ± 2 | 0.58 ± 0.02 | 235 ± 16 |
| Composite2h (7 wt %) | 22 ± 3 | 0.65 ± 0.01 | 228 ± 34 |
| Composite2h (10 wt %) | 22 ± 8 | 0.74 ± 0.14 | 166 ± 49 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaßel, M.; Gerke, J.; Ley, A.; Vana, P. Surface Modification of Wood Flour via ARGET ATRP and Its Application as Filler in Thermoplastics. Polymers 2018, 10, 354. https://doi.org/10.3390/polym10040354
Kaßel M, Gerke J, Ley A, Vana P. Surface Modification of Wood Flour via ARGET ATRP and Its Application as Filler in Thermoplastics. Polymers. 2018; 10(4):354. https://doi.org/10.3390/polym10040354
Chicago/Turabian StyleKaßel, Martin, Julia Gerke, Adrian Ley, and Philipp Vana. 2018. "Surface Modification of Wood Flour via ARGET ATRP and Its Application as Filler in Thermoplastics" Polymers 10, no. 4: 354. https://doi.org/10.3390/polym10040354
APA StyleKaßel, M., Gerke, J., Ley, A., & Vana, P. (2018). Surface Modification of Wood Flour via ARGET ATRP and Its Application as Filler in Thermoplastics. Polymers, 10(4), 354. https://doi.org/10.3390/polym10040354