Preparation and Characterization of Graphene Oxide-Modified Sapium sebiferum Oil-Based Polyurethane Composites with Improved Thermal and Mechanical Properties



Abstract
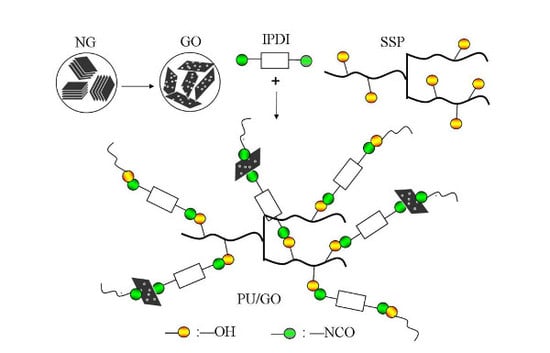
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis Methods
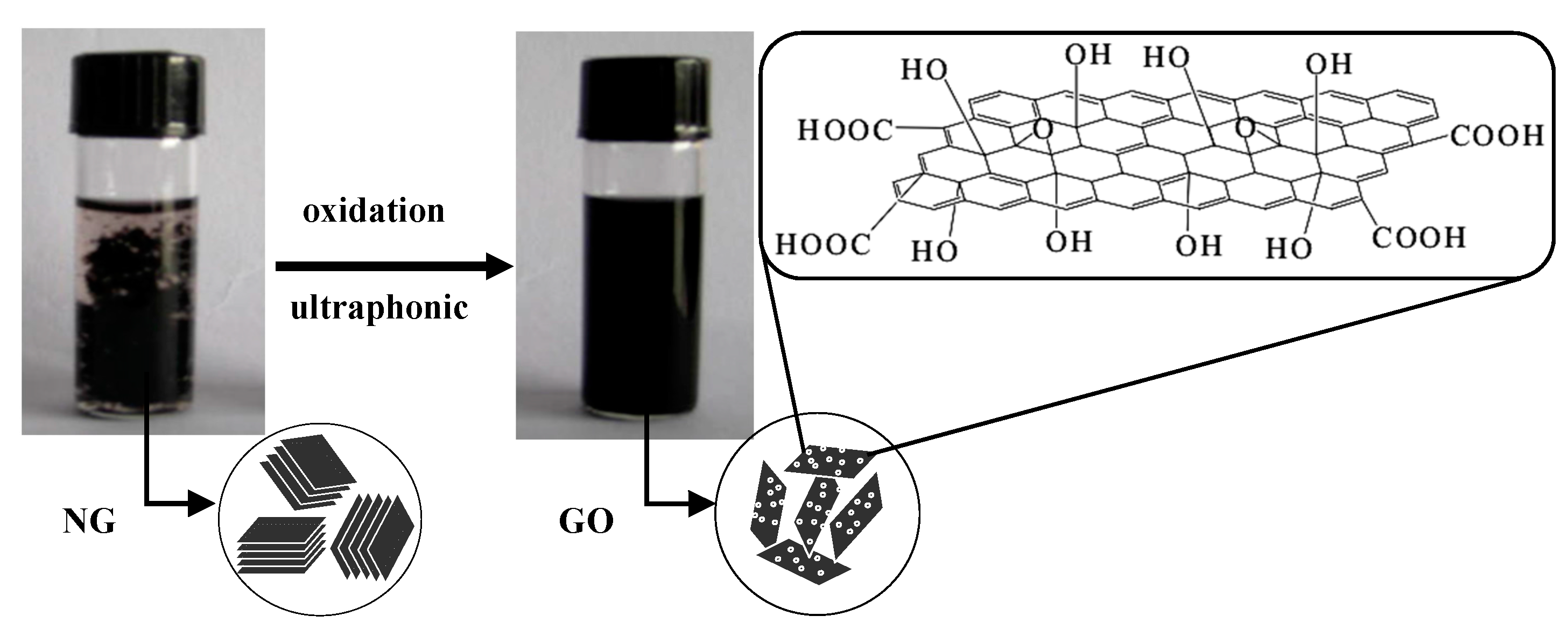
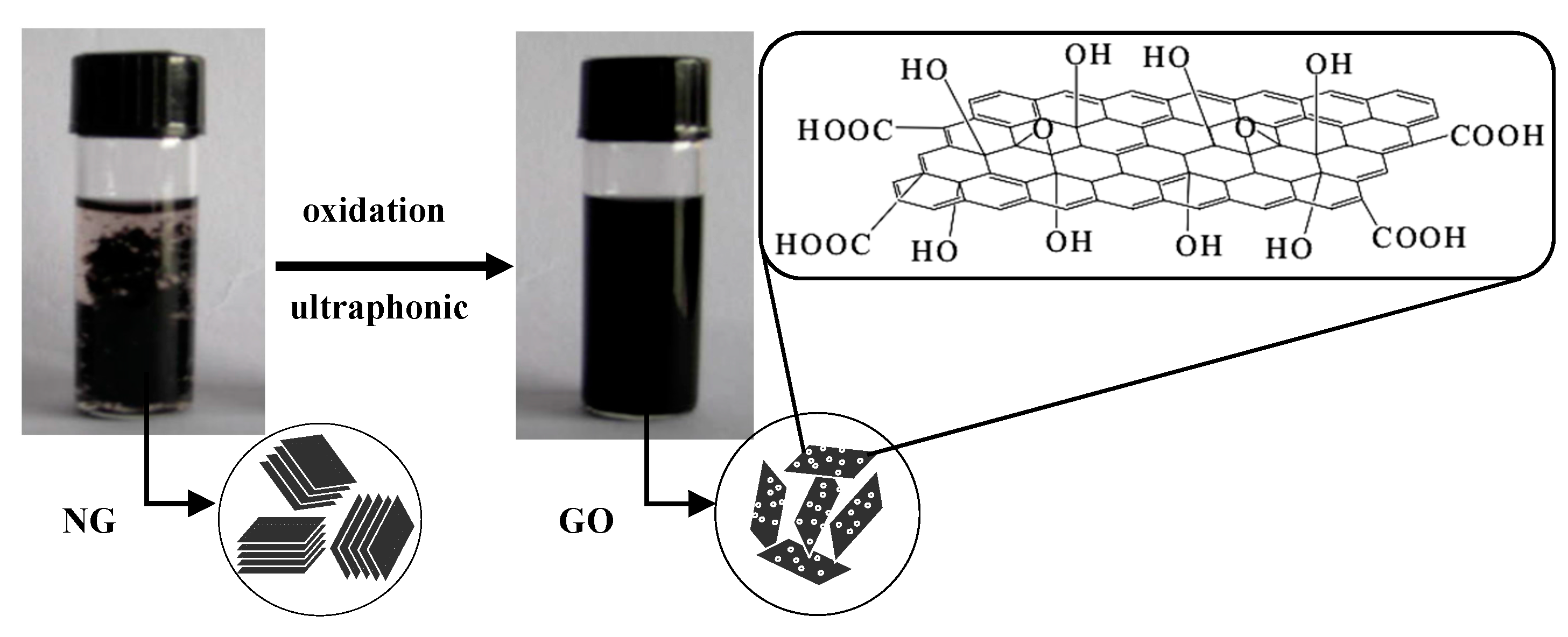
2.2.1. Preparation of GO
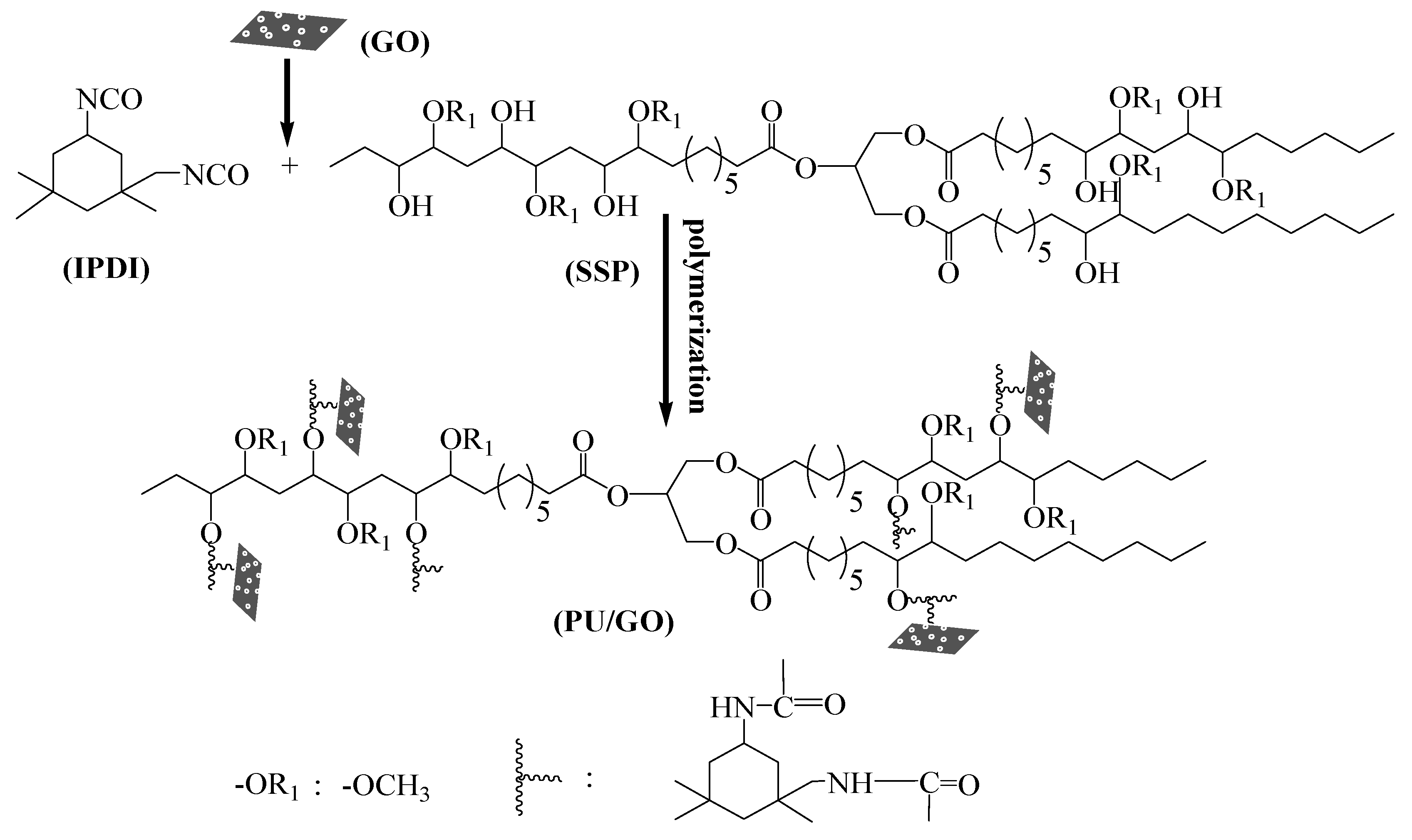
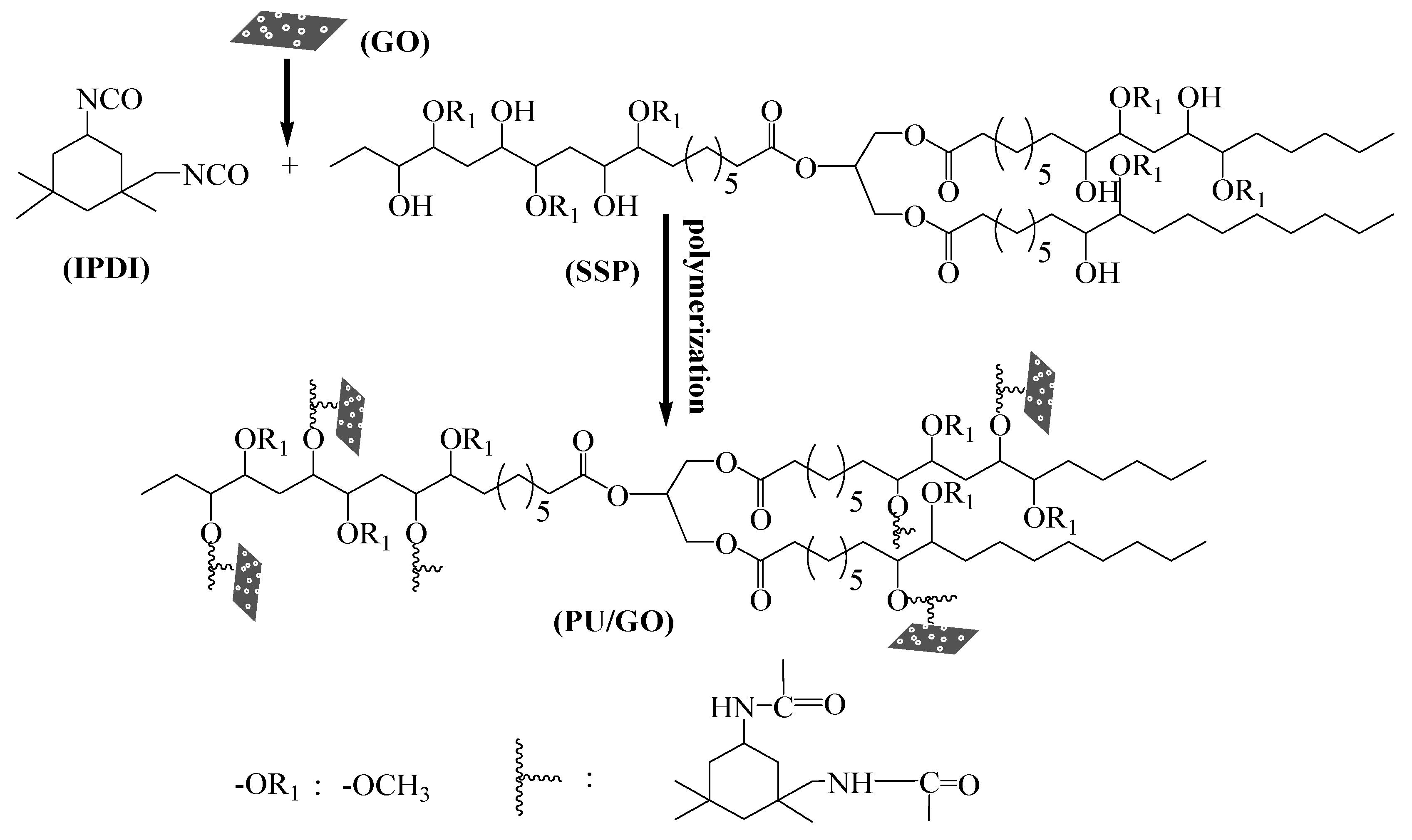
2.2.2. Synthesis of PU and PU/GO Composites
2.3. Testing and Measurement
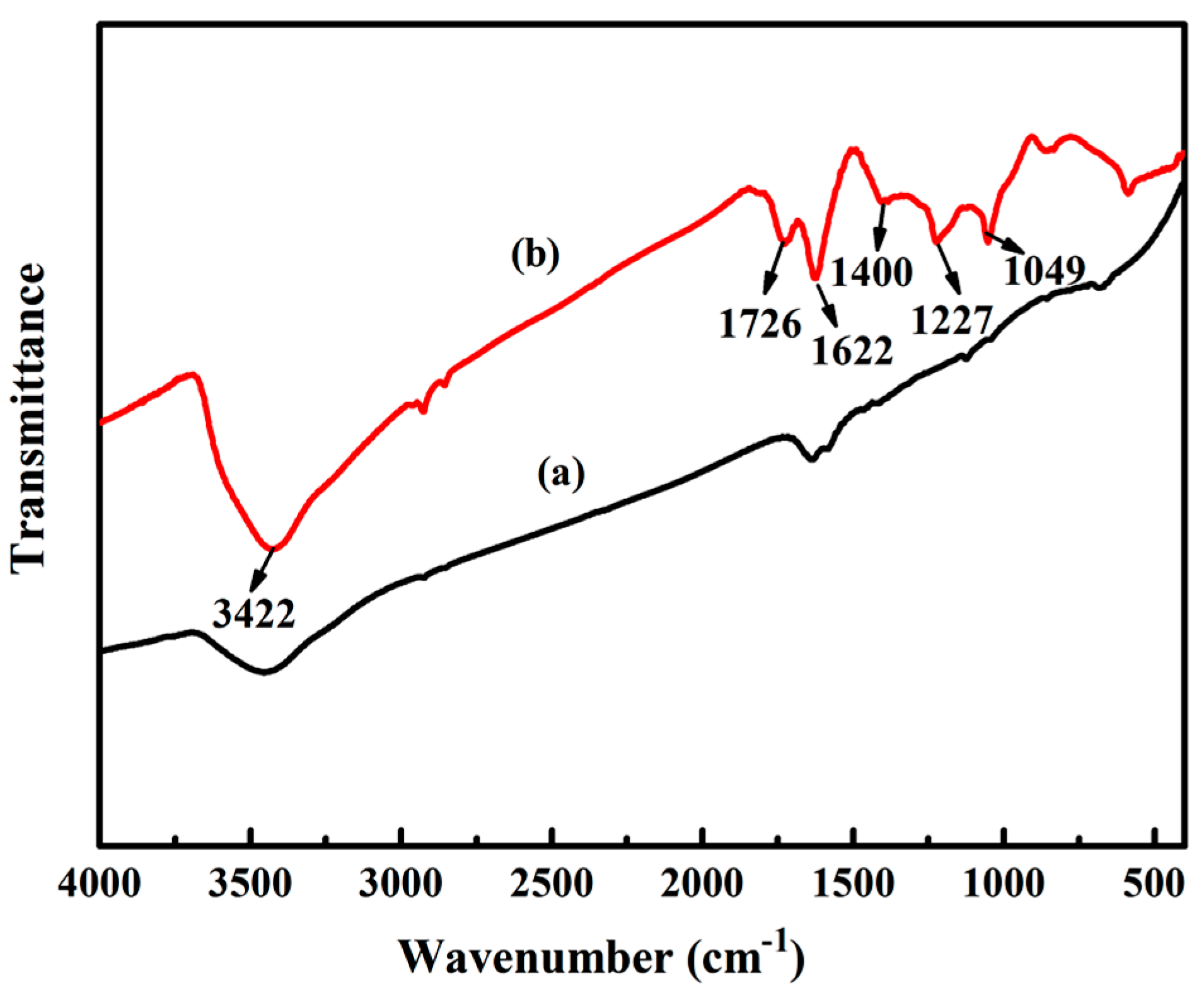
2.3.1. Fourier Transform Infrared Spectroscopy (FITR)
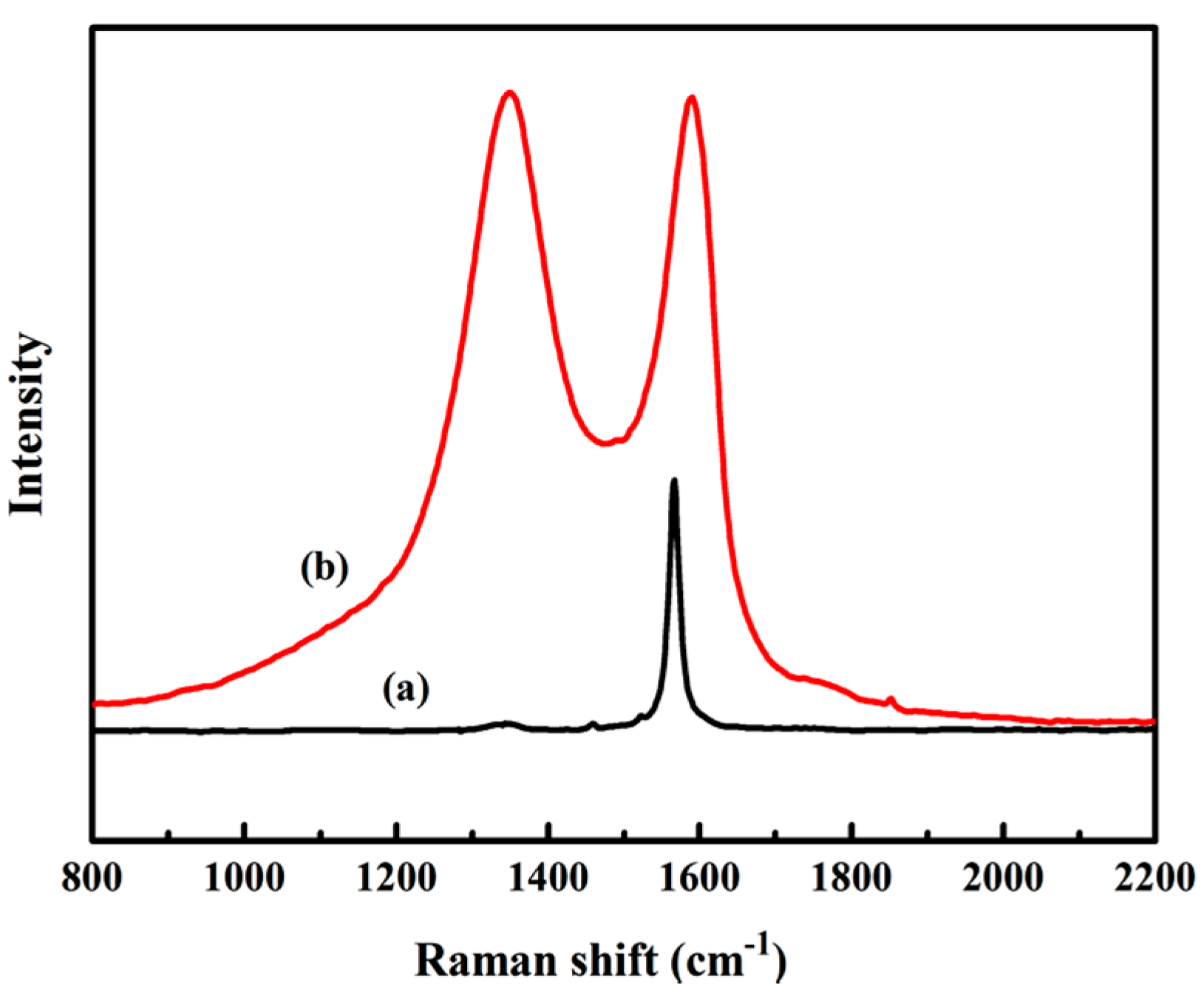
2.3.2. Raman
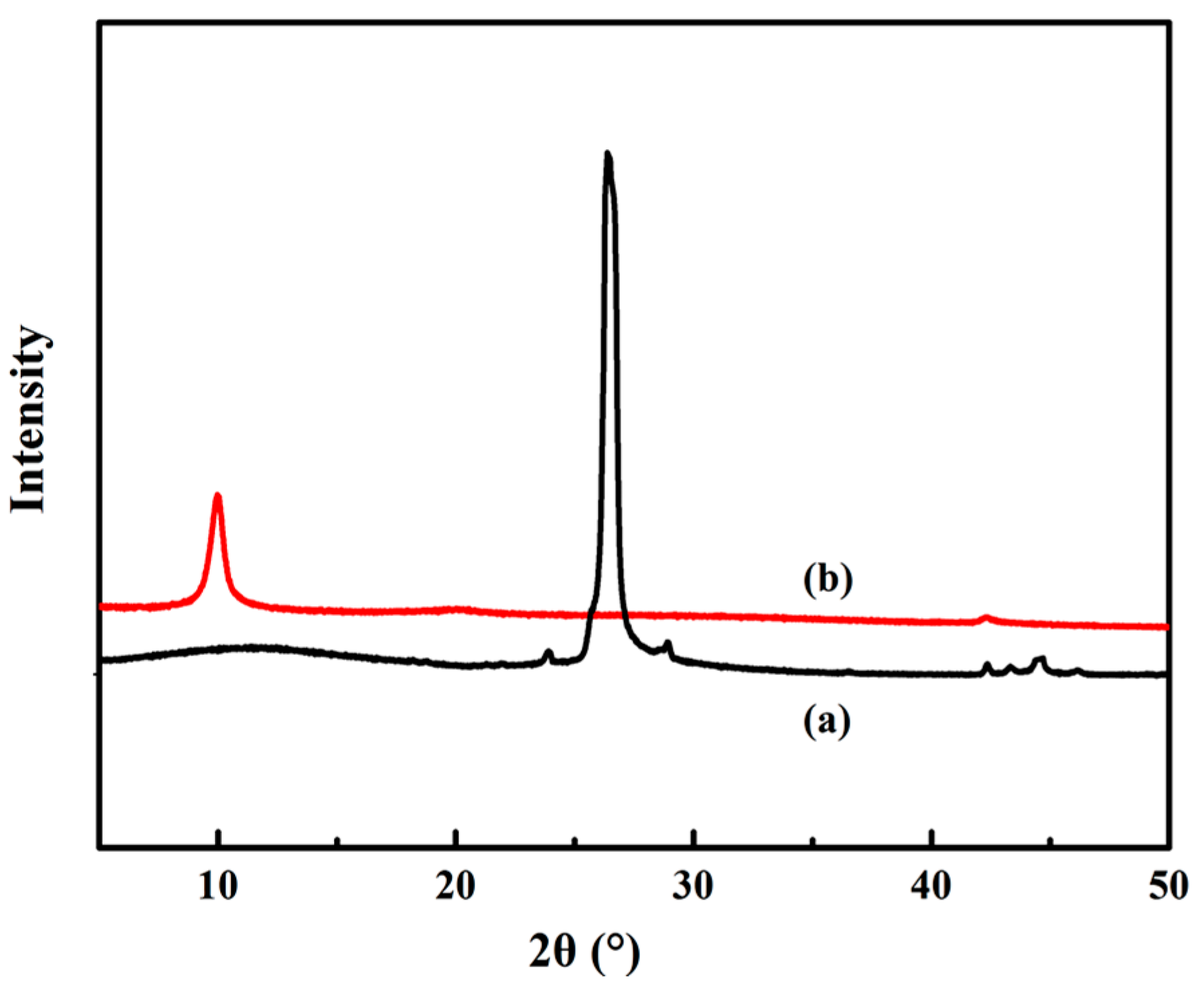
2.3.3. X-ray Diffraction (XRD)
2.3.4. Morphology Analysis
2.3.5. Thermal Stability Analysis
2.3.6. Mechanical Testing
3. Results and Discussion
3.1. Structural Characterization of GO
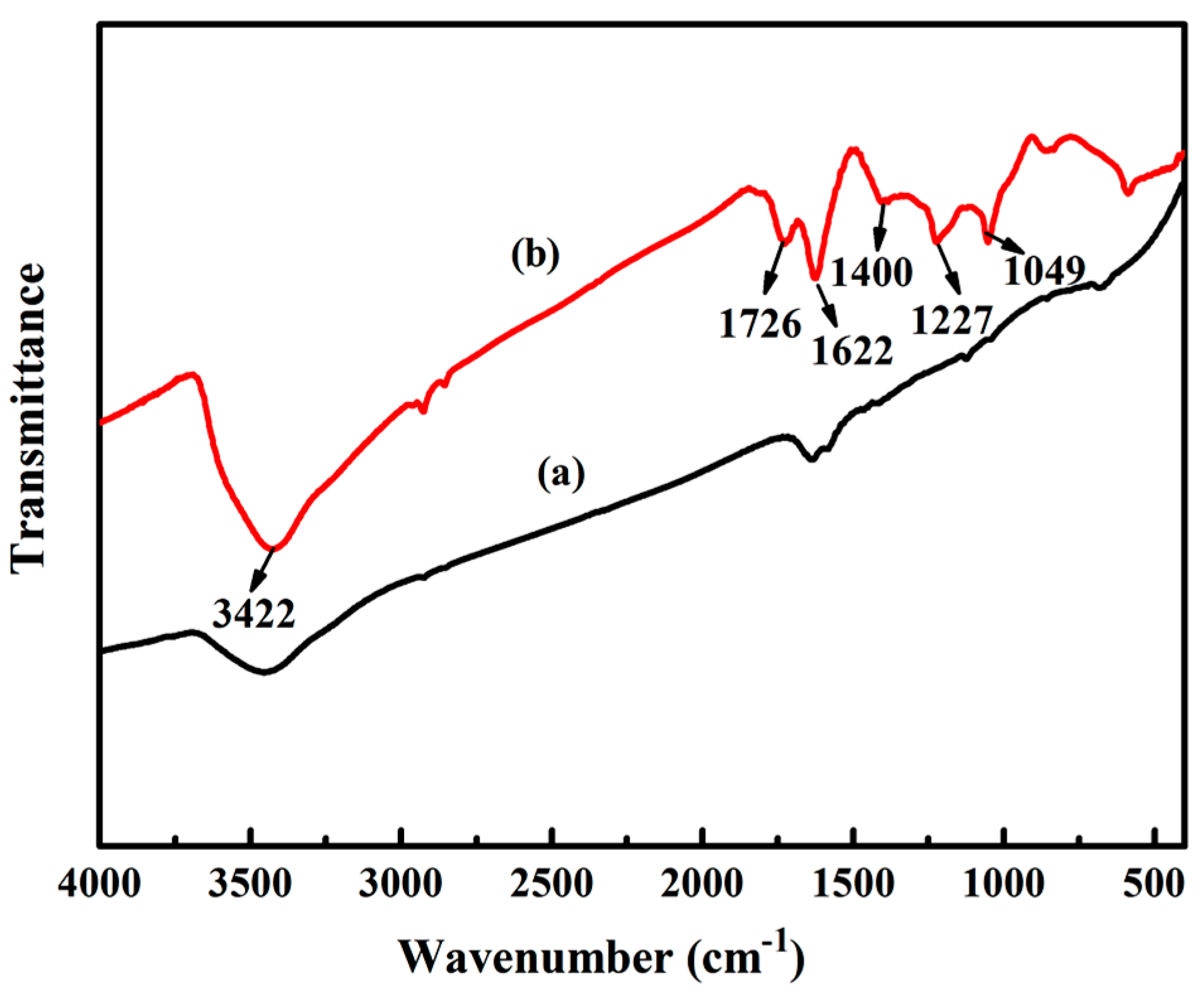
3.1.1. FTIR Analysis of GO
3.1.2. Raman Analysis of GO
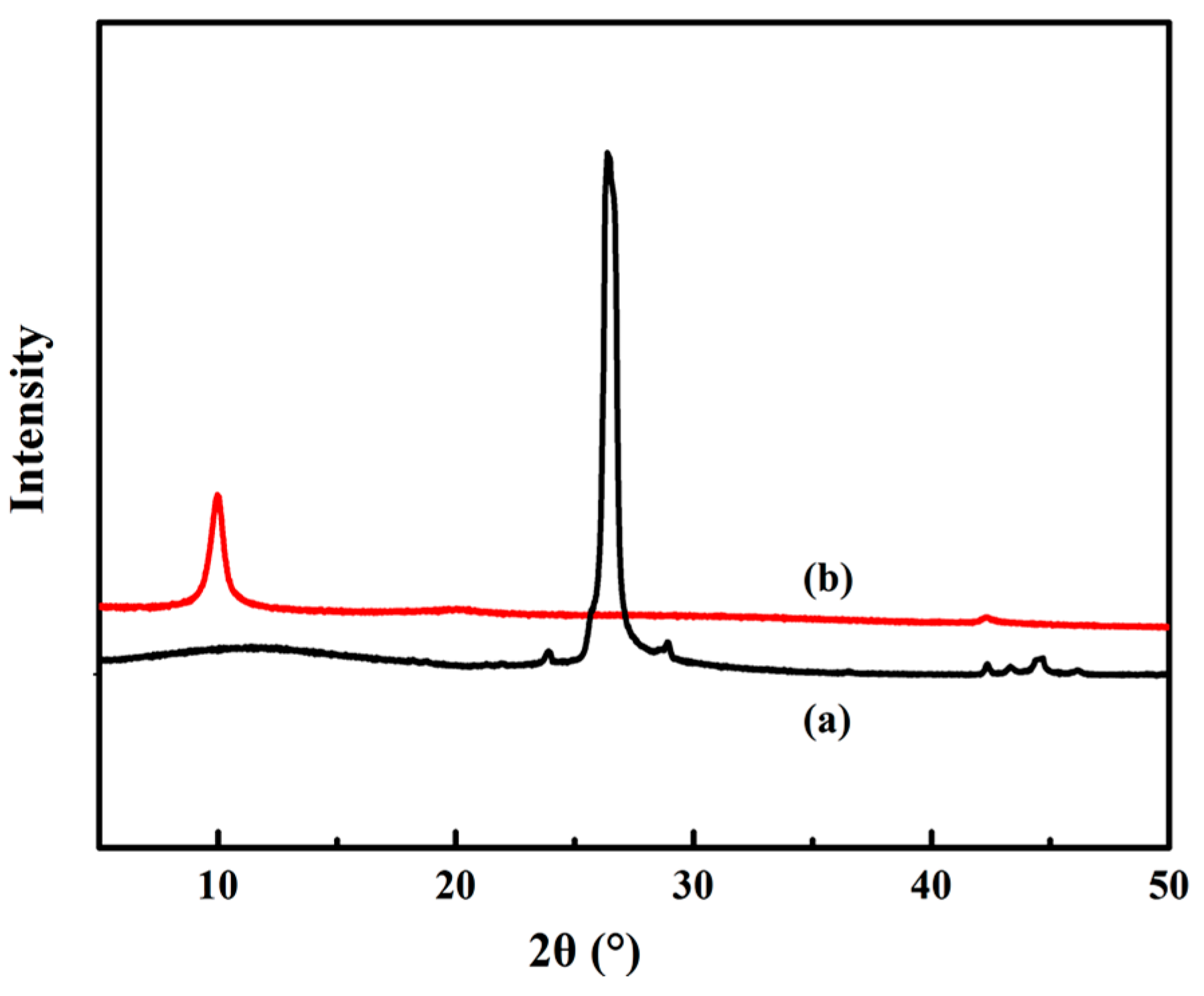
3.2. XRD Analysis of GO
3.3. TEM Analysis of GO
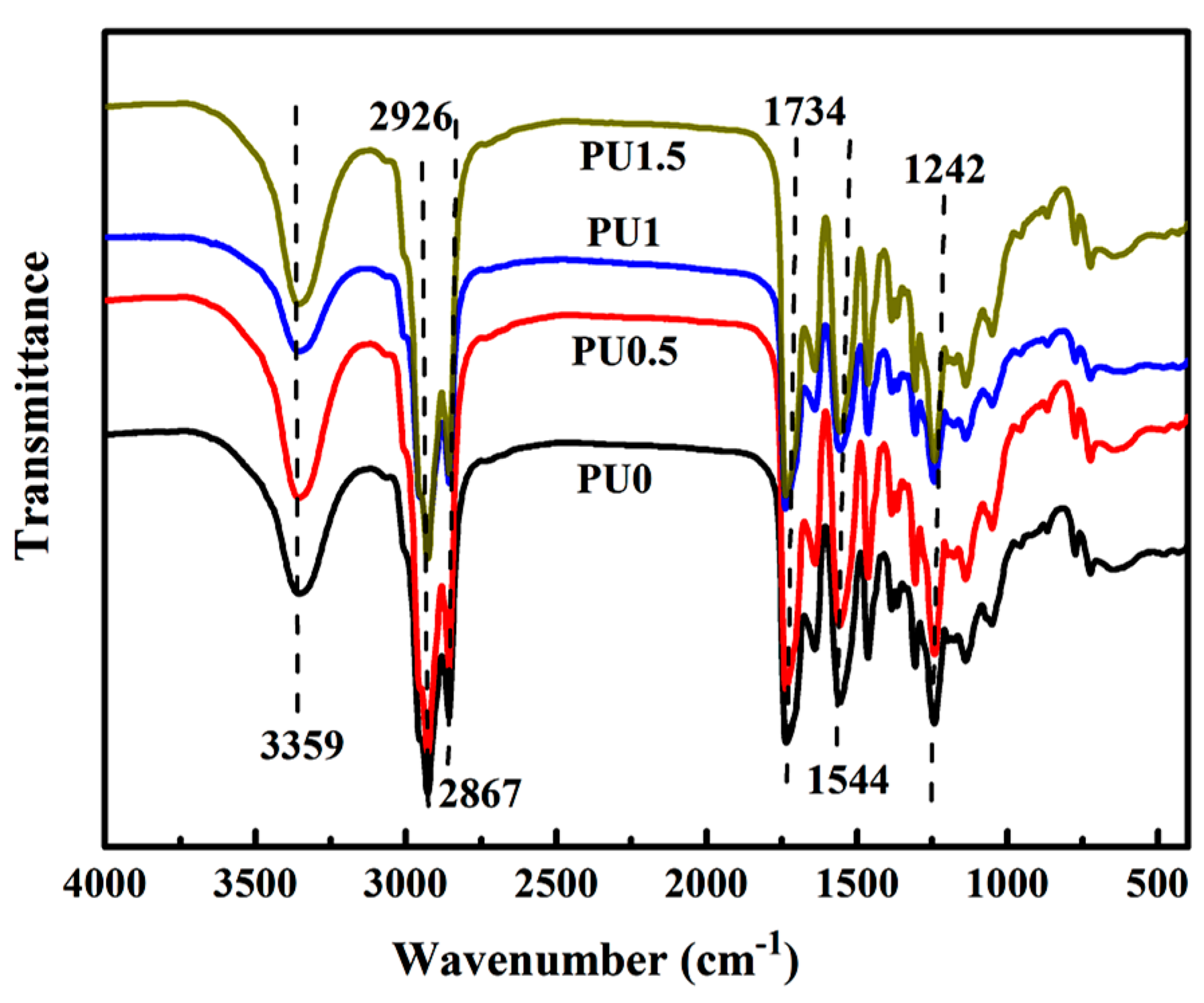
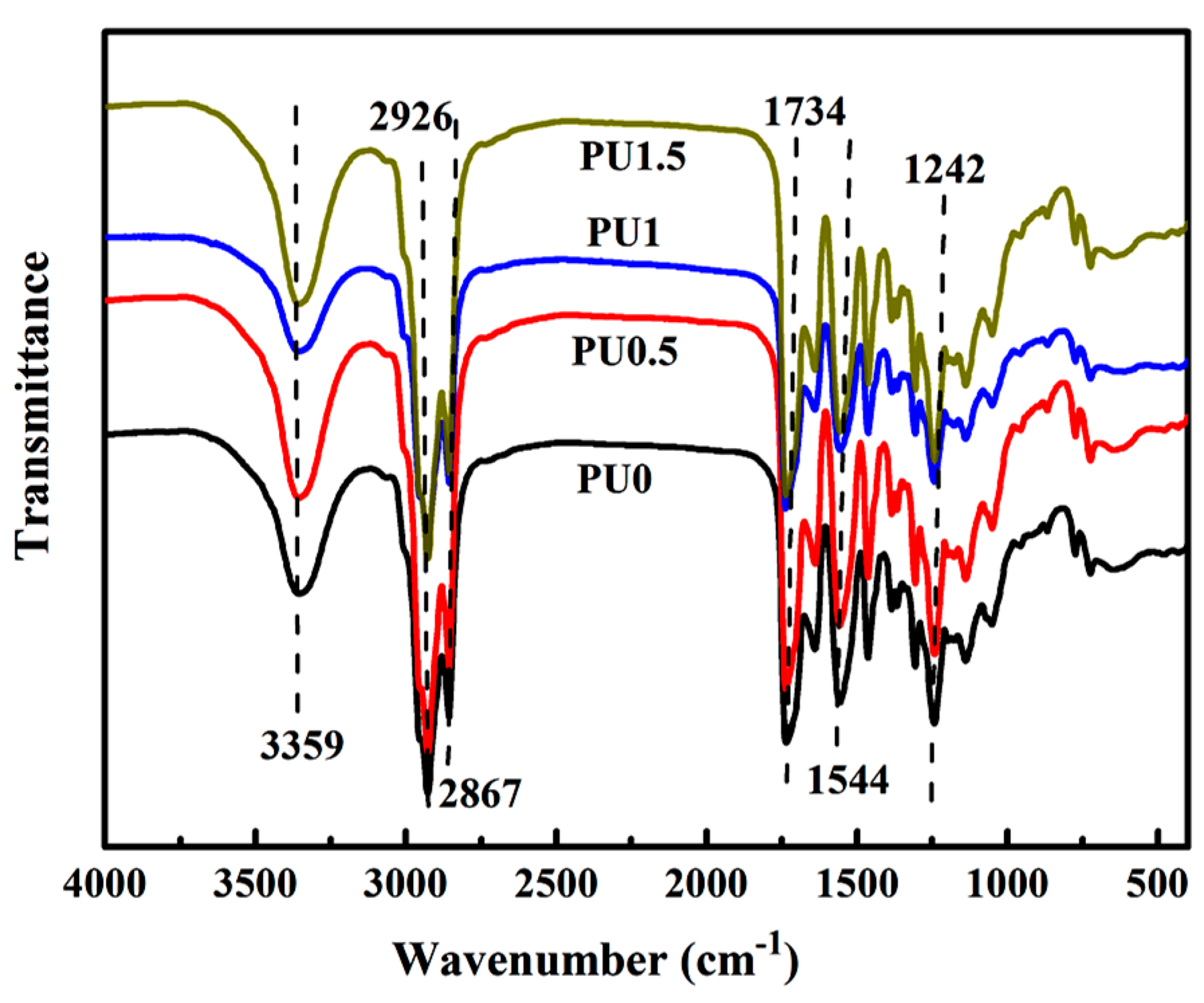
3.4. Structural Characterization of PU/GO Composites
3.5. Microstructure of Pure PU and PU/GO Composites
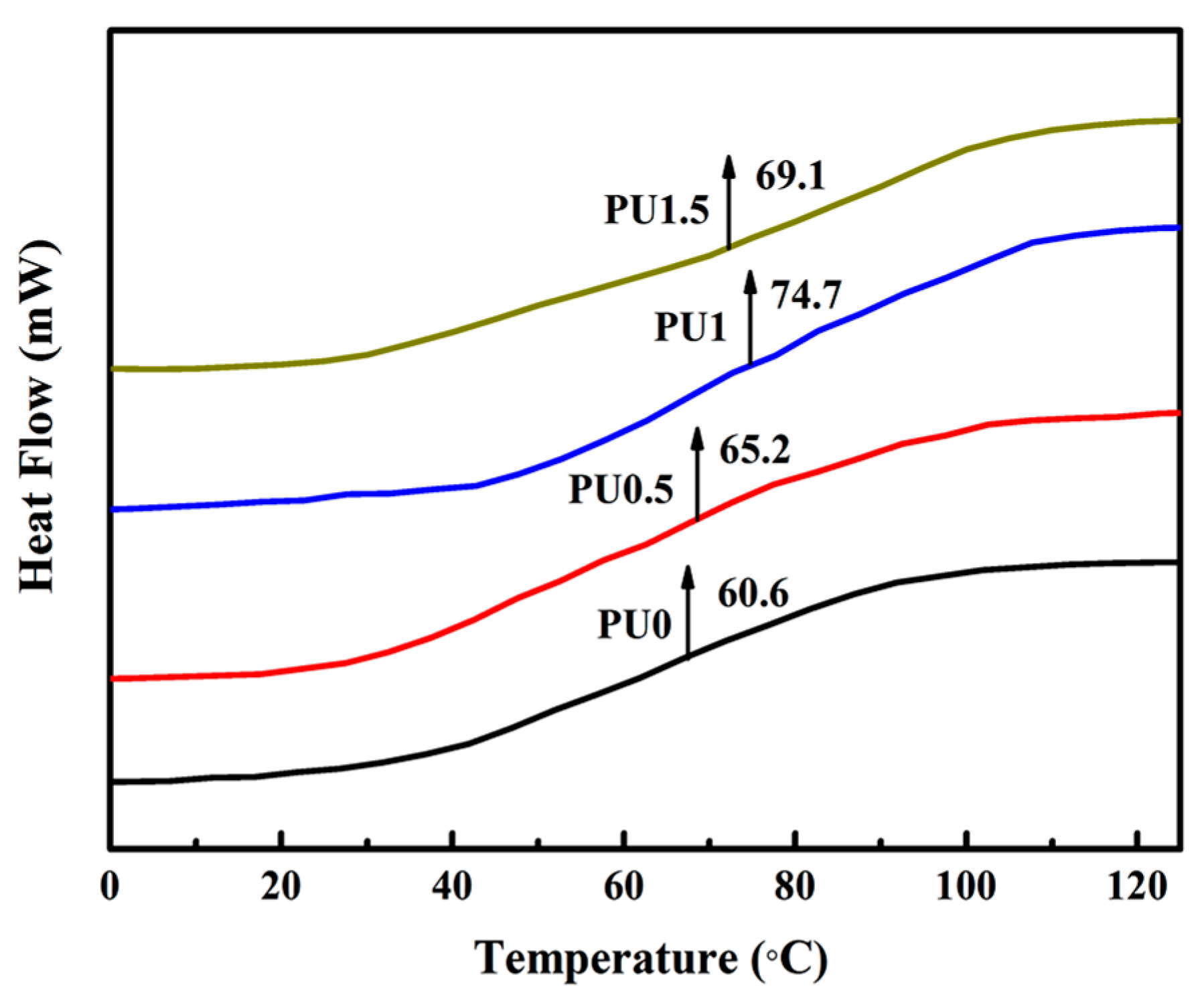
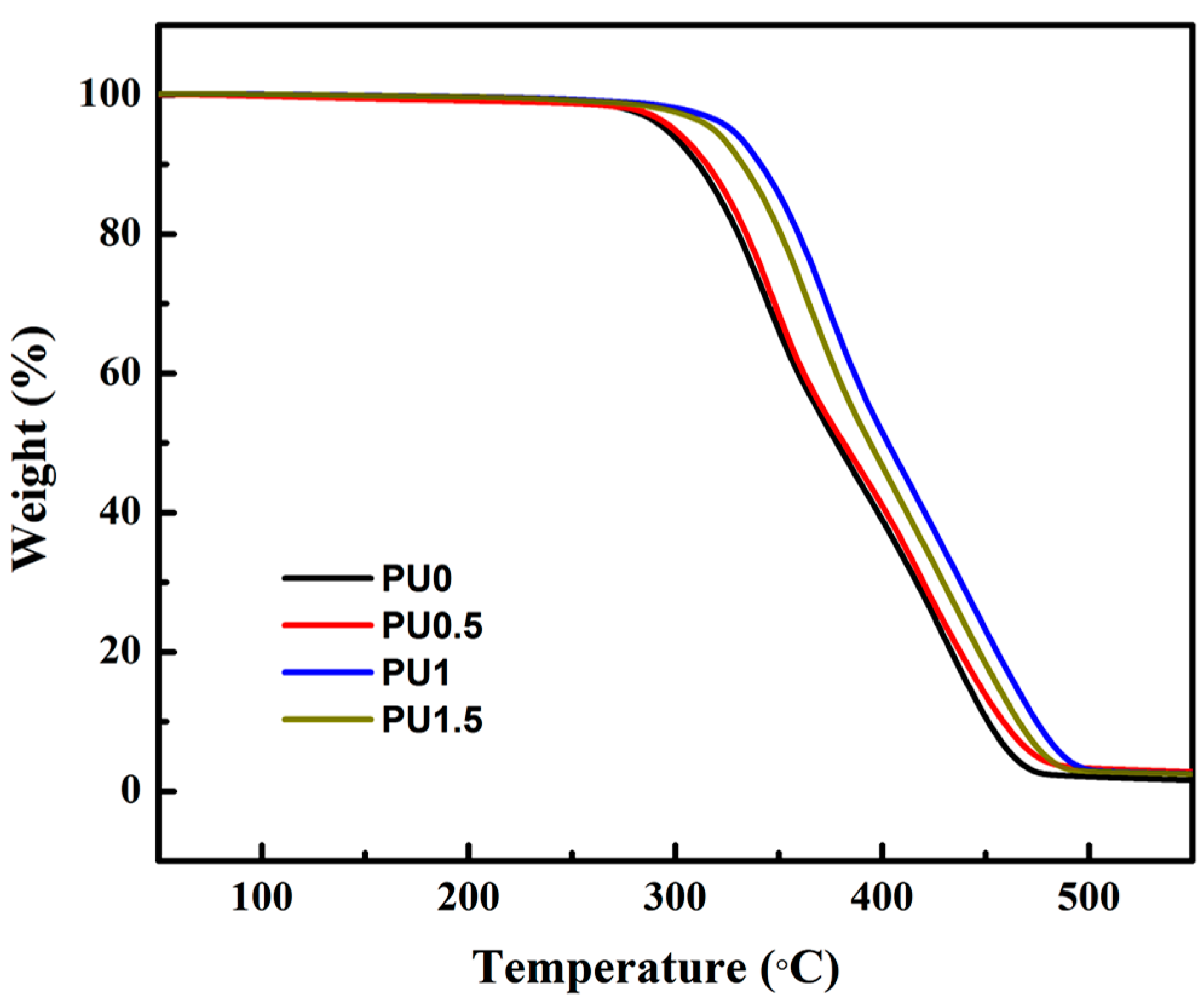
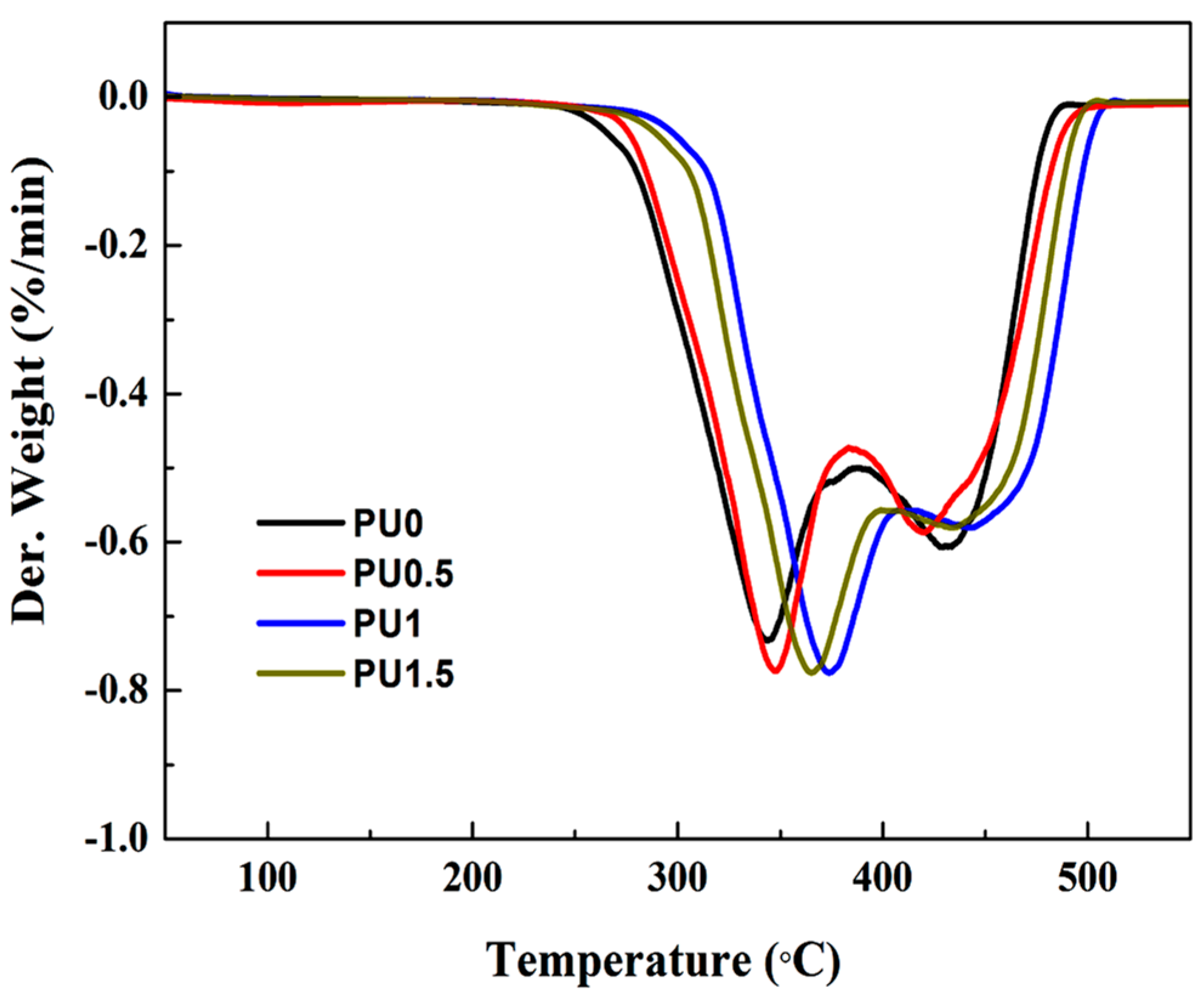
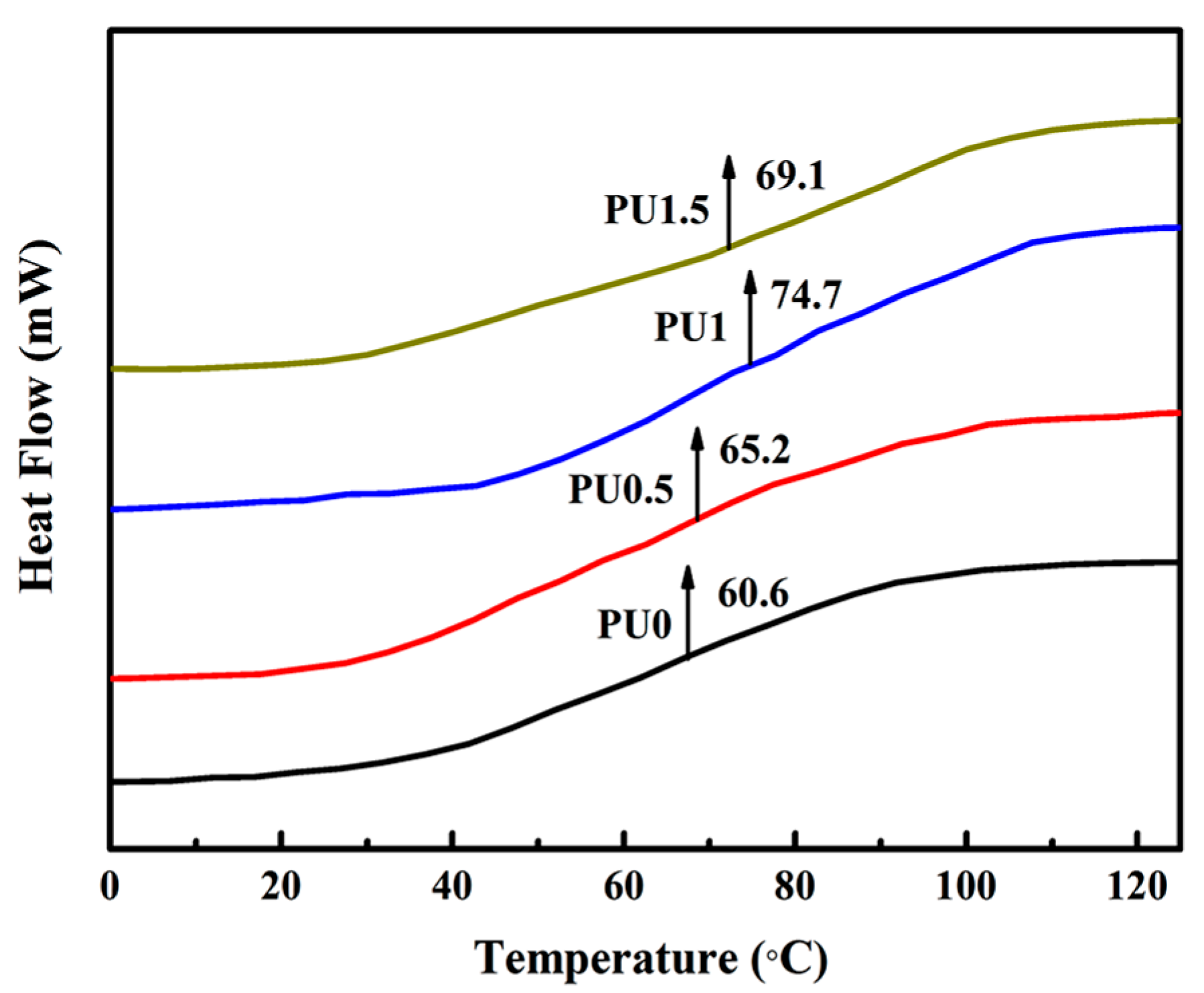
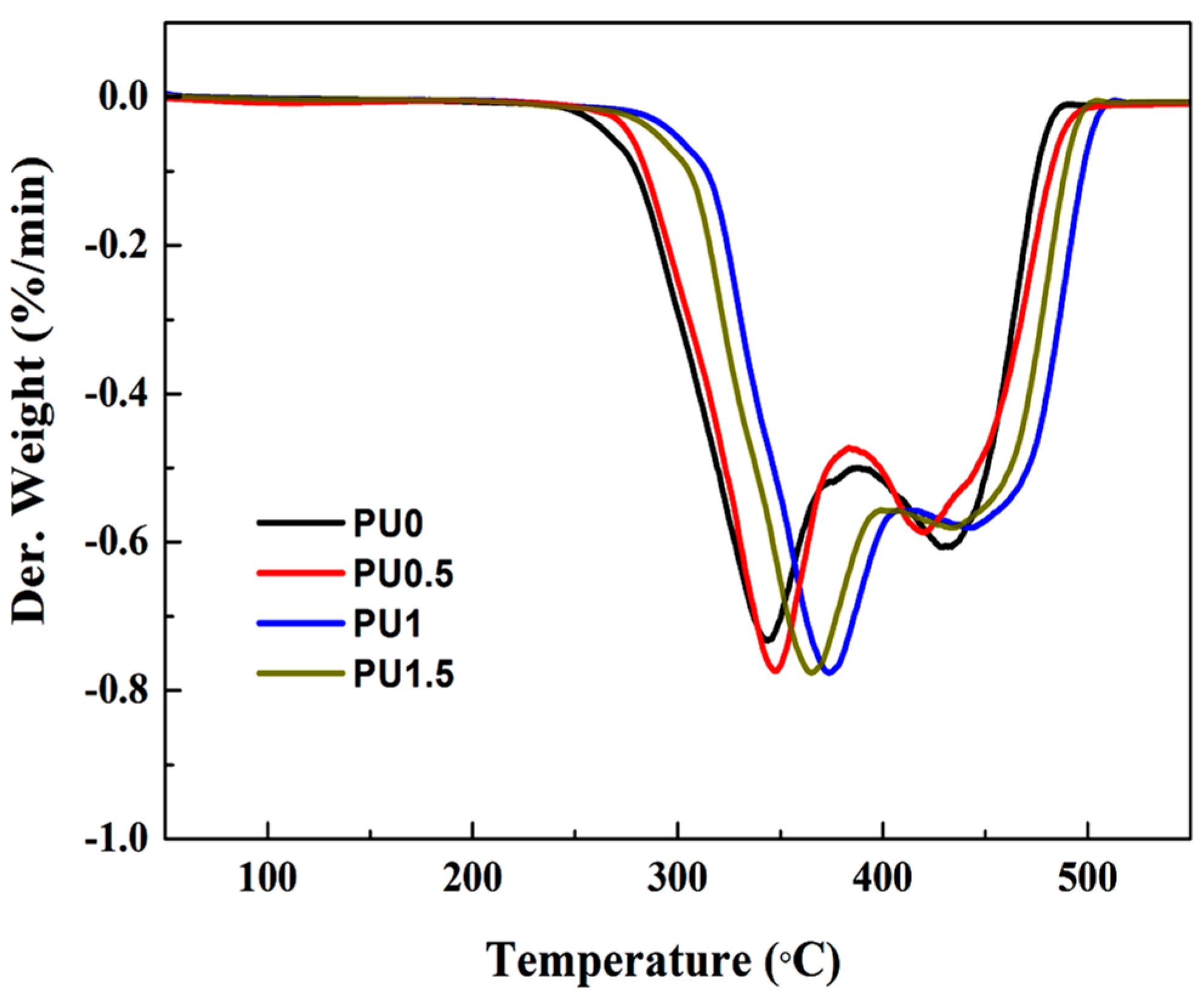
3.6. Thermal Properties of Pure PU and PU/GO Composites
3.7. Mechanical Properties of Pure PU and PU/GO Composites
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cheng, C.C.; Liao, Z.S.; Huang, J.J.; Huang, S.Y.; Fan, W.L. Incorporation of supramolecular polymer-functionalized graphene: Towards the development of bio-based high electrically conductive polymeric nanocomposites. Compos. Sci. Technol. 2017, 148, 89–96. [Google Scholar] [CrossRef]
- Leng, W.Q.; Li, J.H.; Cai, Z.Y. Synthesis and Characterization of Cellulose Nanofibril-Reinforced Polyurethane Foam. Polymers 2017, 9, 597. [Google Scholar] [CrossRef]
- Luo, W.K.; Qin, J.X.; Xiao, M.; Han, D.M.; Wang, S.J.; Meng, Y.Z. Synthesis of aliphatic carbonate macrodiols and their application as sustainable feedstock for polyurethane. ACS Omega 2017, 2, 3205–3213. [Google Scholar] [CrossRef]
- Pielichowska, K.; Bieda, J.; Szatkowski, P. Polyurethane/graphite nano-platelet composites for thermal energy storage. Renew. Energy 2016, 91, 456–465. [Google Scholar] [CrossRef]
- Kucińska-Lipka, J.; Gubanska, L.; Skwarska, A. Microporous polyurethane thin layer as a promising scaffold for tissue engineering. Polymers 2017, 9, 277. [Google Scholar] [CrossRef]
- Usman, A.; Zia, K.M.; Zuber, M.; Tabasum, S.; Rehman, S.; Zia, F. Chitin and chitosan based polyurethanes: A review of recent advances and prospective biomedical applications. Int. J. Biol. Macromol. 2016, 86, 630–645. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.Q.; Garrison, T.F.; Madbouly, S.A.; Kessler, M.R. Recent advances in vegetable oil-based polymers and their composites. Prog. Polym. Sci. 2017, 71, 91–143. [Google Scholar] [CrossRef]
- Das, S.; Pandey, P.; Mohanty, S.; Nayak, S.K. Evaluation of biodegradability of green polyurethane/nanosilica composite synthesized from transesterified castor oil and palm oil based isocyanate. Int. Biodeterior. Biodegrad. 2017, 117, 278–288. [Google Scholar] [CrossRef]
- Tenorio-Alfonso, A.; Sánchez, M.C.; Franco, J.M. Preparation, Characterization and Mechanical Properties of Bio-Based Polyurethane Adhesives from Isocyanate-Functionalized Cellulose Acetate and Castor Oil for Bonding Wood. Polymers 2017, 9, 132. [Google Scholar] [CrossRef]
- Liu, H.J.; Li, C.; Sun, X.S. Soy-oil-based waterborne polyurethane improved wet strength of soy protein adhesives on wood. Int. J. Adhes. Adhes. 2017, 73, 66–74. [Google Scholar] [CrossRef]
- Atabani, A.E.; Silitonga, A.S.; Ong, H.C.; Mahlia, T.M.I.; Masjuki, H.H.; Badruddin, I.A.; Fayaz, H. Non-edible vegetable oils: A critical evaluation of oil extraction, fatty acid compositions, biodiesel production, characteristics, engine performance and emissions production. Renew. Sustain. Energy Rev. 2013, 18, 211–245. [Google Scholar] [CrossRef]
- Li, Q.; Yan, Y.J. Production of biodiesel catalyzed by immobilized pseudomonas cepacia lipase from Sapium sebiferum oil in micro-aqueous phase. Appl. Energy 2010, 87, 3148–3154. [Google Scholar] [CrossRef]
- Jaganathan, S.K.; Mani, M.P.; Ismail, A.F.; Ayyar, M. Manufacturing and Characterization of Novel Electrospun Composite Comprising Polyurethane and Mustard Oil Scaffold with Enhanced Blood Compatibility. Polymers 2017, 9, 163. [Google Scholar] [CrossRef]
- Patel, D.K.; Singh, R.K.; Singh, S.K.; Aswal, V.K.; Rana, D.; Ray, B.; Maiti, P. Graphene as a chain extender of polyurethanes for biomedical applications. RSC Adv. 2016, 6, 58628–58640. [Google Scholar] [CrossRef]
- Jiang, S.; Li, Q.F.; Wang, J.W.; He, Z.L.; Zhao, Y.H.; Kang, M.Q. Multiscale graphene oxide-carbon fiber reinforcements for advanced polyurethane composites. Compos. A 2016, 87, 1–9. [Google Scholar] [CrossRef]
- Kaur, G.; Adhikari, R.; Cass, P.; Bown, M.; Evans, M.D.M.; Vashi, A.V.; Gunatillake, P. Graphene/polyurethane composites: Fabrication and evaluation of electrical conductivity, mechanical properties and cell viability. RSC Adv. 2015, 5, 98762–98772. [Google Scholar] [CrossRef]
- Jing, Q.F.; Liu, W.S.; Pan, Y.Z.; Silberschmidt, V.V.; Li, L.; Dong, Z.L. Chemical functionalization of graphene oxide for improving mechanical and thermal properties of polyurethane composites. Mater. Des. 2015, 85, 808–814. [Google Scholar] [CrossRef] [Green Version]
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V. Electric field effect in atomically thin carbon films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Wei, X.D.; Kysar, J.W.; James, H. Measurement of the elastic properties and intrinsic strength of monolayer graphen. Science 2008, 321, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Nair, R.R.; Blake, P.; Grigorenko, A.N.; Novoselov, K.S.; Booth, T.J.; Stauber, T.; Peres, N.M.R.; Geim, A.K. Fine structure constant defines visual transparency of graphene. Science 2008, 320, 1308. [Google Scholar] [CrossRef] [PubMed]
- Dikin, D.A.; Sasha, S.; Zimney, E.J.; Piner, R.D.; Dommett, G.H.B.; Evmenenko, G.; Nguyen, S.T.; Ruoff, R.S. Preparation and characterization of graphene oxide paper. Nature 2007, 448, 457–460. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.L.; Baji, A.; Tien, H.W.; Yang, Y.K.; Yang, S.Y.; Wu, S.Y.; Ma, C.C.; Liu, H.Y.; Mai, Y.W. Self-assembly of silver-graphene hybrid on electrospun polyurethane nanofibers as flexible transparent conductive thin films. Photochem. Photobiol. 2012, 50, 3473–3481. [Google Scholar] [CrossRef]
- Li, Y.T.; Lian, H.Q.; Hu, Y.N.; Chang, W.; Cui, X.G.; Liu, Y. Enhancement in mechanical and shape memory properties for liquid crystalline polyurethane strengthened by graphene oxide. Polymers 2016, 8, 236. [Google Scholar] [CrossRef]
- Suen, M.C.; Gu, J.H.; Lee, H.T. In situ polymerisation and characteristic properties of the waterborne grapheneoxide/poly(siloxane-urethane)s nanocomposites. Polym. Bull. 2017, 74, 4921–4942. [Google Scholar] [CrossRef]
- Al-Attabi, N.Y.; Kaur, G.; Adhikari, R. Preparation and characterization of highly conductive polyurethane composites containing graphene and gold nanoparticles. J. Mater. Sci. 2017, 52, 11774–11784. [Google Scholar] [CrossRef]
- Hummers, W.S.; Offeman, R.E. Preparation of graphitic oxide. J. Am. Chem. Soc. 1958, 80, 1339. [Google Scholar] [CrossRef]
- Marcano, D.C.; Kosynkin, D.V.; Berlin, J.M.; Sinitskii, A.; Sun, Z.Z.; Slesarev, A.; Alemany, L.B.; Lu, W.; Tour, J.M. Improved synthesis of graphene oxide. ACS Nano 2010, 4, 4806–4814. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.Y.; He, X.; Yan, Y.J. Lipase-catalyzed modification of natural Sapium sebiferum oil-based polyol for synthesis of polyurethane with improved properties. RSC Adv. 2017, 7, 1504–1512. [Google Scholar] [CrossRef]
- Wu, G.Y.; He, X.; Xu, L.; Zhang, H.J.; Yan, Y.J. Synthesis and characterization of biobased polyurethane/SiO2 nanocomposites from natural Sapium sebiferum oil. RSC Adv. 2015, 5, 27097–27106. [Google Scholar] [CrossRef]
- Wu, G.Y.; Fan, Y.L.; He, X.; Yan, Y.J. Bio-polyurethanes from Sapium sebiferum oil reinforced with carbon nanotubes: Synthesis, characterization and properties. RSC Adv. 2015, 5, 80893–80900. [Google Scholar] [CrossRef]
- Si, Y.; Samulski, E.T. Synthesis of Water Soluble Graphene. Nano Lett. 2008, 8, 1679–1682. [Google Scholar] [CrossRef] [PubMed]
- Hilder, M.; Winther-Jensen, O.; Winther-Jensen, B.; MacFarlane, D.R. Graphene/zinc nano-composites by electrochemical co-deposition. Phys. Chem. Chem. Phys. 2012, 14, 14034–14040. [Google Scholar] [CrossRef] [PubMed]
- Ramezanzadeh, B.; Ghasemi, E.; Mahdavian, M.; Changizi, E.; Mohamadzadeh Moghadam, M.H. Covalently-grafted graphene oxide nanosheets to improve barrier and corrosion protection properties of polyurethane coatings. Carbon 2015, 93, 555–573. [Google Scholar] [CrossRef]
- Kim, J.; Jeon, J.H.; Kim, H.J.; Lim, H. Durable and water-floatable ionic polymer actuator with hydrophobic and asymmetrically laser-scribed reduced graphene oxide paper electrodes. ACS Nano 2014, 8, 2986–2997. [Google Scholar] [CrossRef] [PubMed]
- Pant, H.R.; Pokharel, P.; Joshi, M.K.; Adhikari, S.; Kim, H.J.; Park, C.H.; Kim, C.S. Processing and characterization of electrospun graphene oxide/polyurethane composite nanofibers for stent coating. Chem. Eng. J. 2015, 270, 336–342. [Google Scholar] [CrossRef]
- Zhou, Y.; Liu, X.D.; Sheng, D.K.; Lin, C.H.; Ji, F.; Dong, L.; Xu, S.B.; Wu, H.H.; Yang, Y.M. Graphene oxide/polyurethane-based solid-solid phase change materials with enhanced mechanical properties. Thermochim. Acta 2017, 658, 38–64. [Google Scholar] [CrossRef]
- Zhu, Y.W.; Murali, S.; Cai, W.W.; Li, X.S.; Suk, J.W.; Potts, J.R.; Ruoff, R.S. Graphene and graphene oxide: Synthesis, properties, and applications. Adv. Mater. 2010, 22, 3906–3924. [Google Scholar] [CrossRef] [PubMed]
- Strankowski, M.; Włodarczyk, D.; Piszczyk, Ł.; Strankowska, J. Polyurethane nanocomposites containing reduced graphene oxide, FTIR, Raman, and XRD Studies. J. Spectrosc. 2016, 2016. [Google Scholar] [CrossRef]
- Tien, Y.I.; Wei, K.H. The effect of nano-sized silicate layers from montmorillonite on glass transition, dynamic mechanical, and thermal degradation properties of segmented polyurethane. J. Appl. Polym. Sci. 2002, 86, 1741–1748. [Google Scholar] [CrossRef]
- Liu, X.; Xu, K.; Liu, H.; Cai, H.L.; Su, J.X.; Fu, Z.E.; Guo, Y.; Chen, M.C. Preparation and properties of waterborne polyurethanes with natural dimer fatty acids based polyester polyol as soft segment. Prog. Org. Coat. 2011, 72, 612–620. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | IDT (°C) | Tmax | Residue at 550 °C (%) | |
|---|---|---|---|---|
| 1st Step (°C) | 2nd Step (°C) | |||
| PU0 | 294.3 | 341.8 | 429.7 | 1.8 |
| PU0.5 | 297.6 | 347.3 | 419.2 | 2.1 |
| PU1 | 326.1 | 375.2 | 442.6 | 2.2 |
| PU1.5 | 315.2 | 365.1 | 436.8 | 2.3 |
| Tensile strength (MPa) | Elongation at break (%) | Young’s modulus (MPa) | |
|---|---|---|---|
| PU0 | 12.5 ± 0.6 | 168.3 ± 12.3 | 22.3 ± 1.4 |
| PU0.5 | 19.2 ± 0.8 | 152.2 ± 12.5 | 28.4 ± 1.5 |
| PU1 | 28.3 ± 1.9 | 143.5 ± 11.4 | 45.1 ± 4.1 |
| PU1.5 | 24.7 ± 1.6 | 134.6 ± 10.8 | 35.3 ± 3.2 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, G.; Xu, X.; He, X.; Yan, Y. Preparation and Characterization of Graphene Oxide-Modified Sapium sebiferum Oil-Based Polyurethane Composites with Improved Thermal and Mechanical Properties. Polymers 2018, 10, 133. https://doi.org/10.3390/polym10020133
Wu G, Xu X, He X, Yan Y. Preparation and Characterization of Graphene Oxide-Modified Sapium sebiferum Oil-Based Polyurethane Composites with Improved Thermal and Mechanical Properties. Polymers. 2018; 10(2):133. https://doi.org/10.3390/polym10020133
Chicago/Turabian StyleWu, Guiying, Xiaoling Xu, Xin He, and Yunjun Yan. 2018. "Preparation and Characterization of Graphene Oxide-Modified Sapium sebiferum Oil-Based Polyurethane Composites with Improved Thermal and Mechanical Properties" Polymers 10, no. 2: 133. https://doi.org/10.3390/polym10020133
APA StyleWu, G., Xu, X., He, X., & Yan, Y. (2018). Preparation and Characterization of Graphene Oxide-Modified Sapium sebiferum Oil-Based Polyurethane Composites with Improved Thermal and Mechanical Properties. Polymers, 10(2), 133. https://doi.org/10.3390/polym10020133