Implantable Polymeric Drug Delivery Devices: Classification, Manufacture, Materials, and Clinical Applications





Abstract
1. Introduction
2. Implantable Polymeric Drug Delivery Device Classification
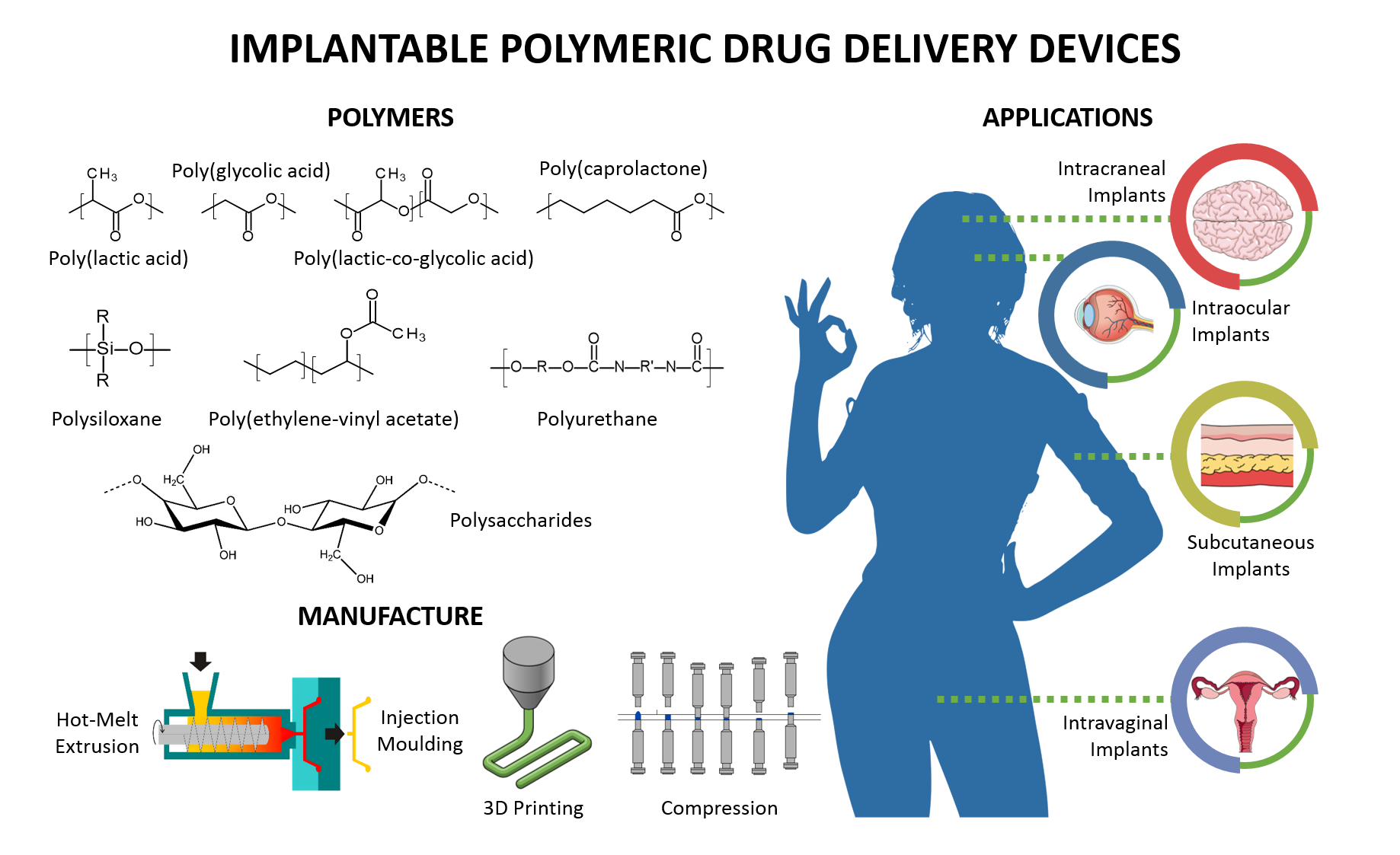
2.1. Passive Polymeric Implants
2.1.1. Non-Biodegradable Polymeric Implantable Systems
2.1.2. Biodegradable Polymeric Implants
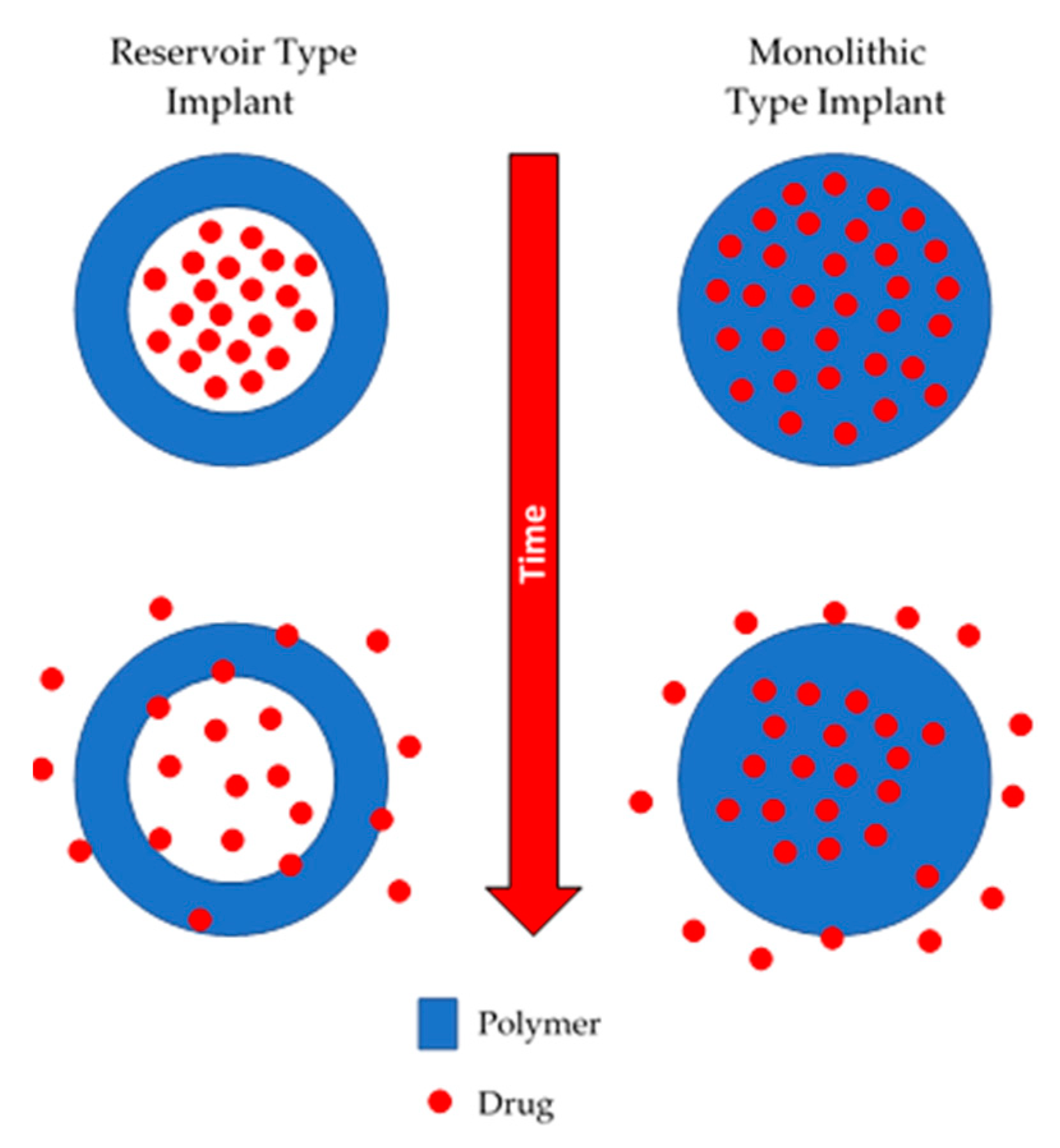
2.2. Dynamic or Active Polymeric Implants
3. Mechanism of Drug Release from Implantable Polymeric Drug Delivery Systems
3.1. Mechanism of Drug Release from Non-Biodegradable Implants
3.2. Mechanism of Drug Release from Biodegradable Implants
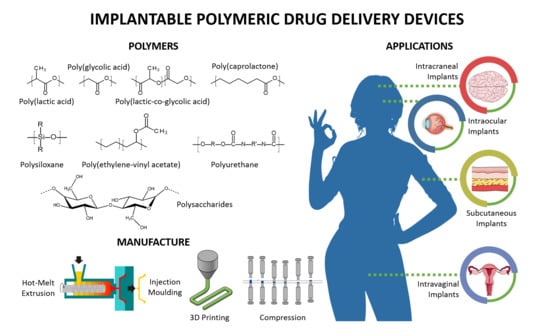
4. Polymers Used for Implantable Polymeric Drug Delivery Devices
4.1. Biodegradable Polymers
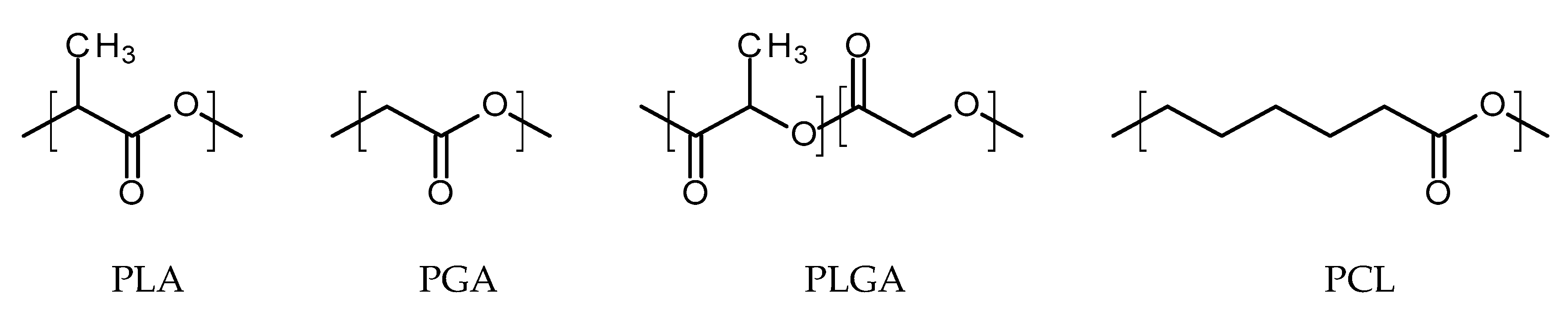
4.1.1. Thermoplastic Aliphatic Polyesters
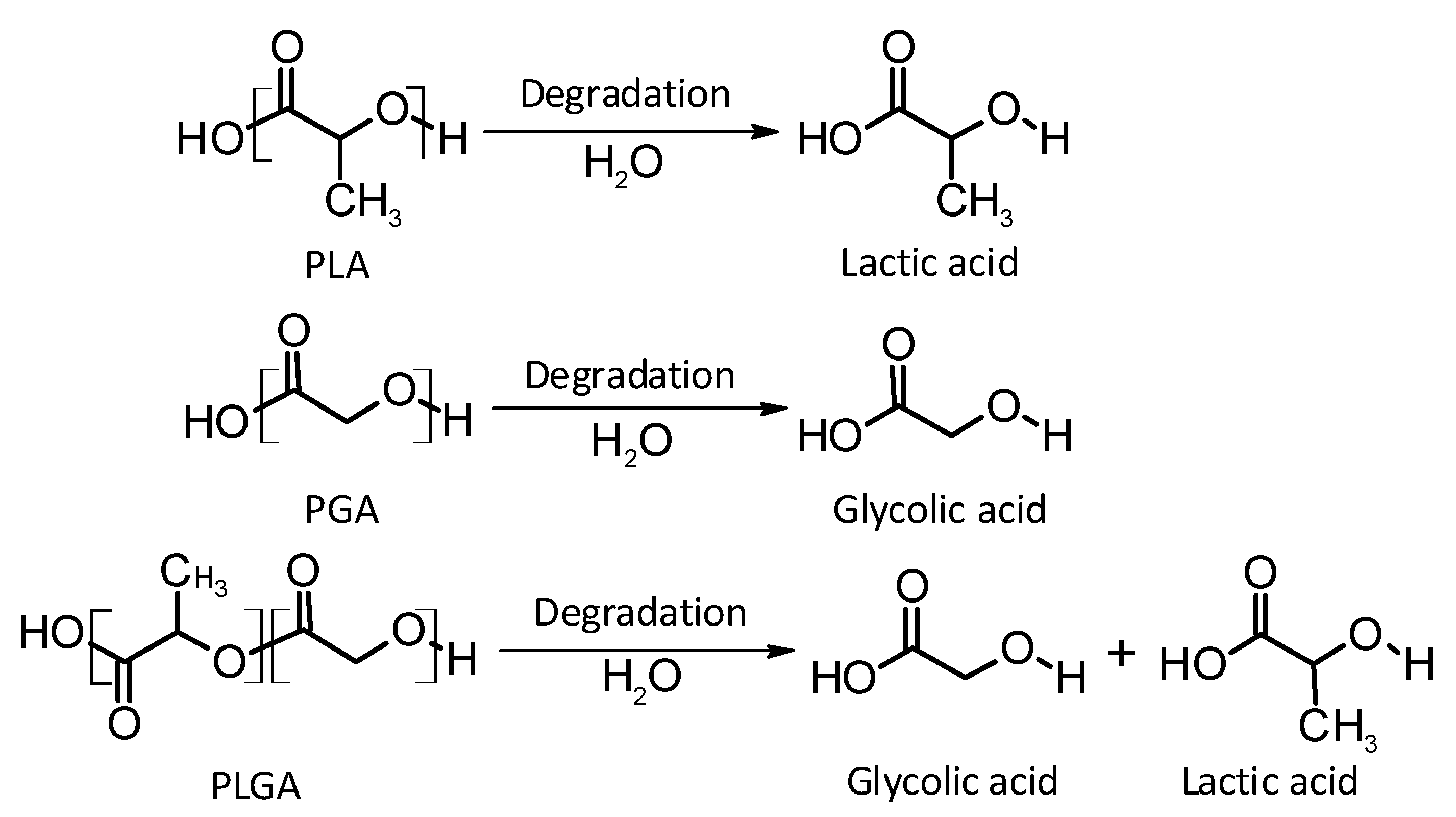
Poly(lactic acid)
Poly(glycolic acid)
Poly(lactic-co-glycolic acid)
Poly(caprolactone)
Other Biodegradable Polymers
4.2. Non-Biodegradable Polymers
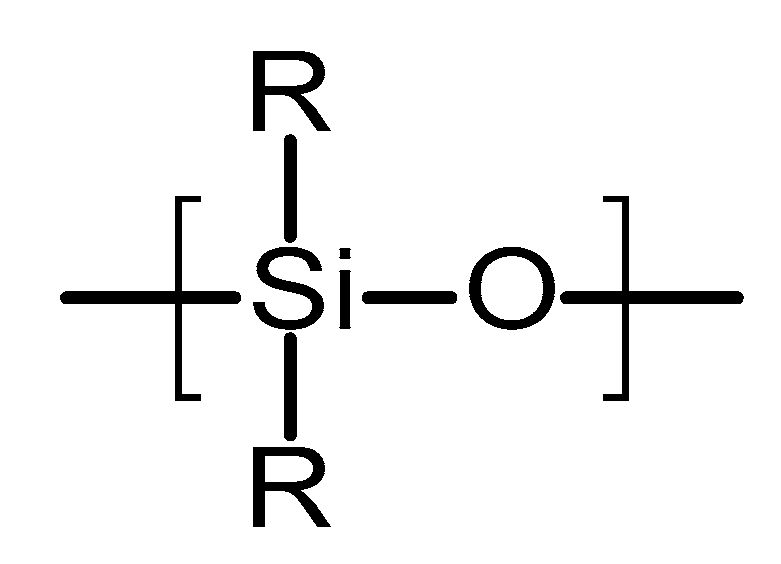
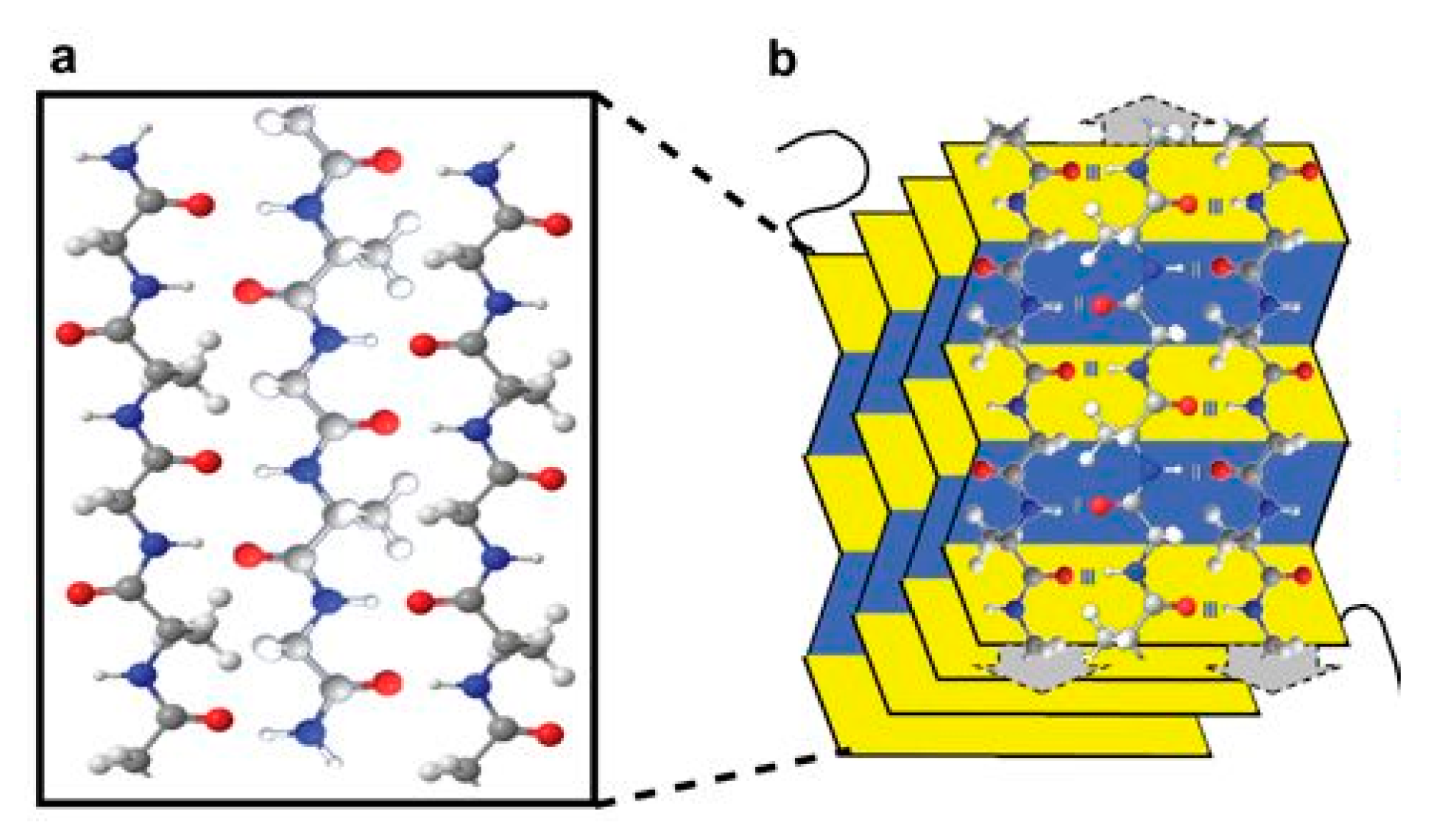
4.2.1. Poly(siloxanes)
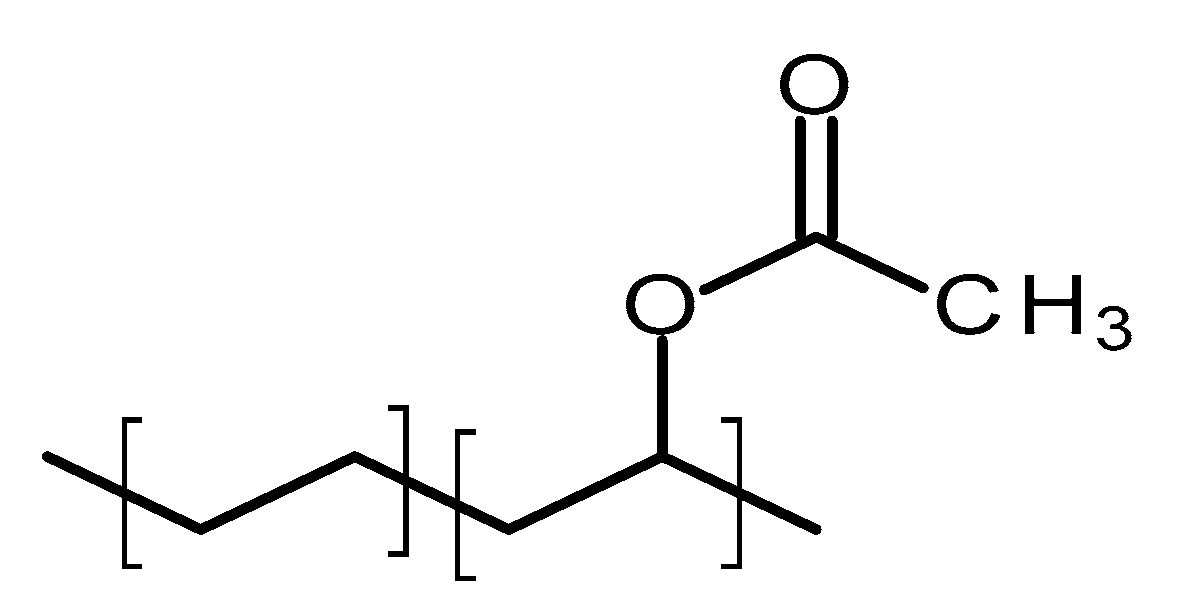
4.2.2. Poly(ethylene-vinyl acetate)
4.3. Other Polymers
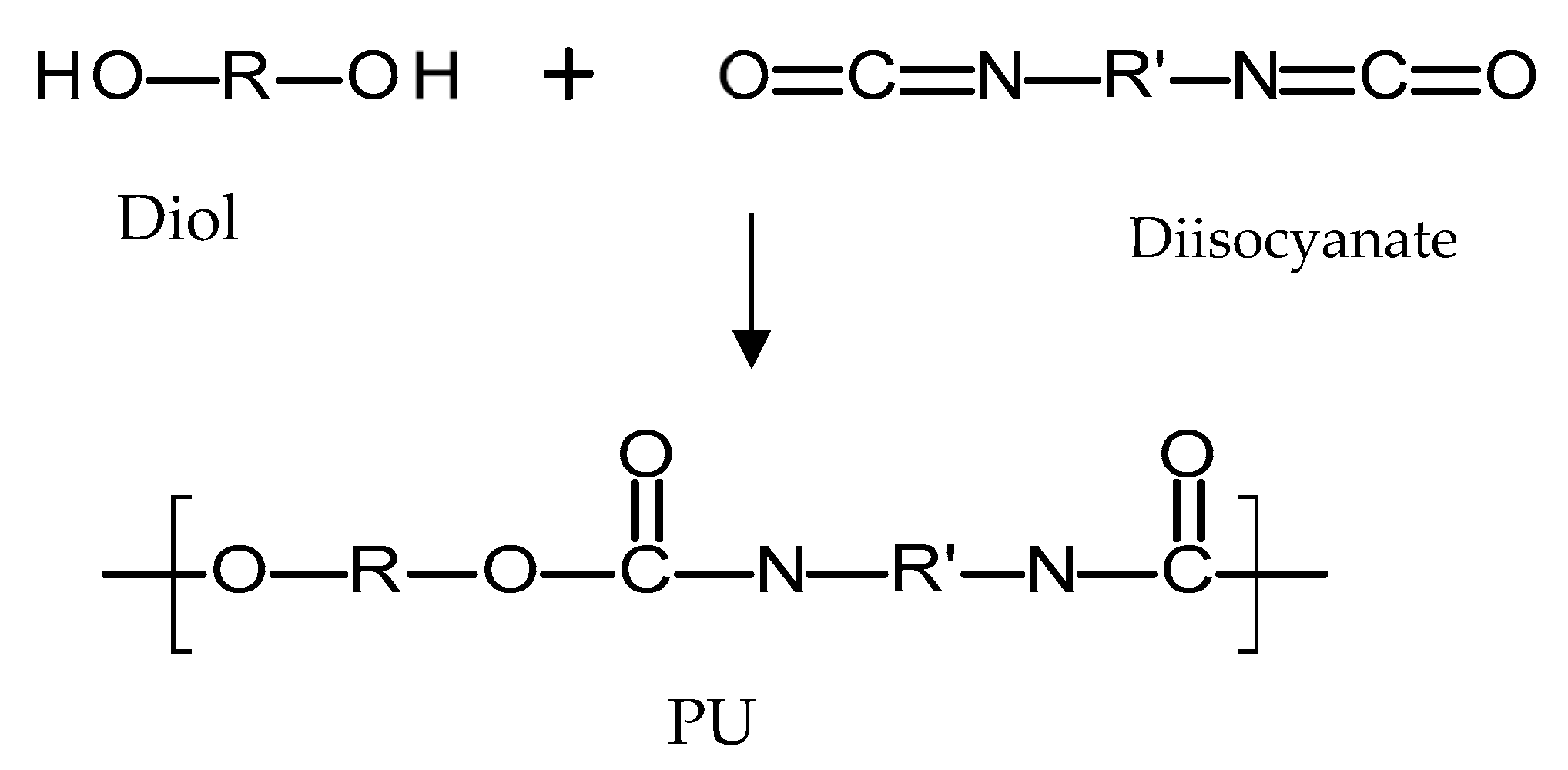
4.3.1. Poly(urethanes)
4.3.2. Natural Polymers
Cellulose
Chitosan
Silk
5. Methods of Implant Manufacture
5.1. Compression
5.2. Solvent Casting
5.3. Hot Melt Extrusion
5.4. Injection Moulding
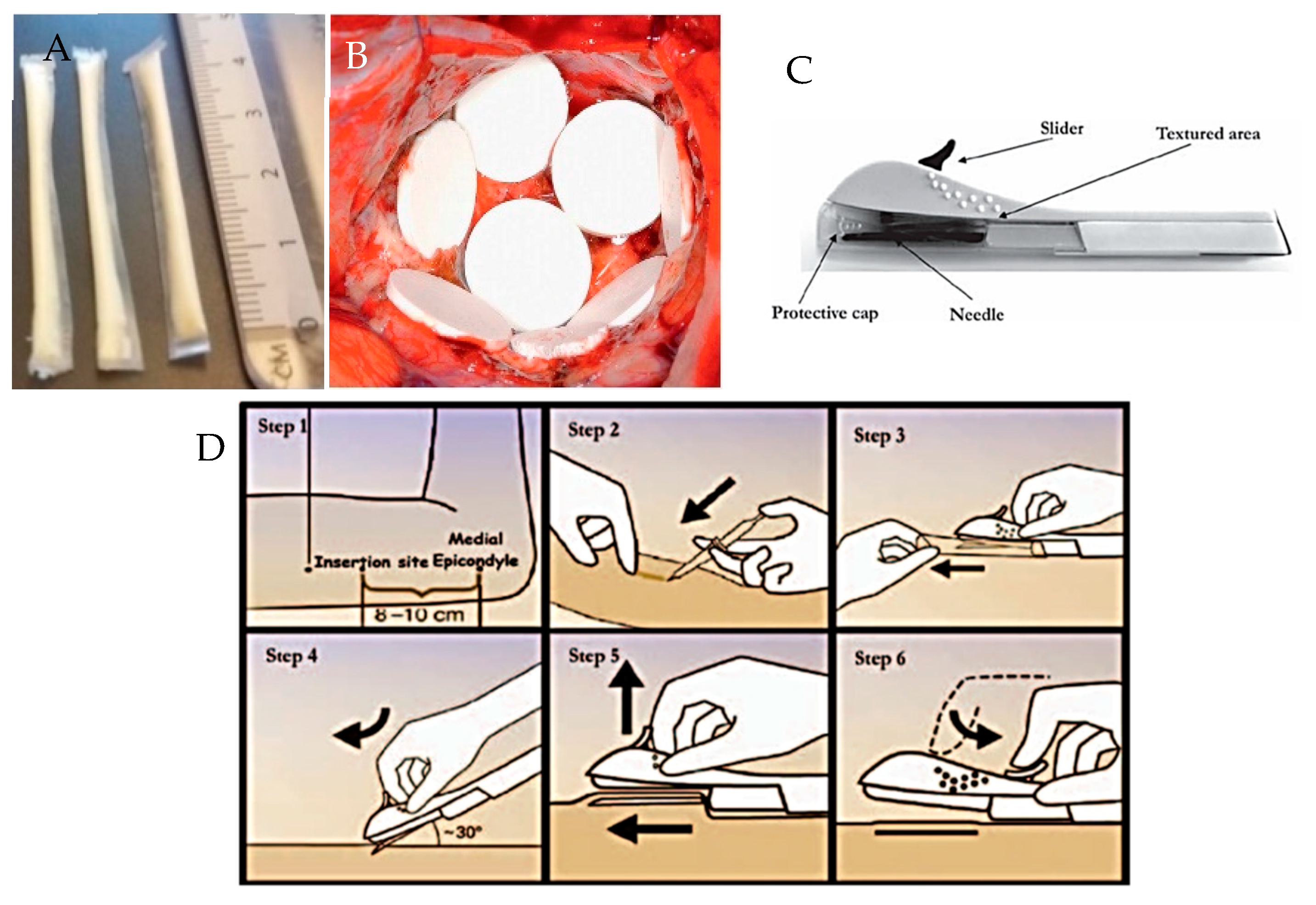
5.5. 3D Printing
6. Implantable Polymeric Device Design
7. Current Therapeutic Applications
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ravi Kumar, M.N.V.; Kumar, N. Polymeric Controlled Drug-Delivery Systems: Perspective Issues and Opportunities. Drug Dev. Ind. Pharm. 2001, 27, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Larrañeta, E.; Lutton, R.E.M.; Woolfson, A.D.; Donnelly, R.F. Microneedle arrays as transdermal and intradermal drug delivery systems: Materials science, manufacture and commercial development. Mater. Sci. Eng. R Rep. 2016, 104, 1–32. [Google Scholar] [CrossRef]
- Langer, R. New methods of drug delivery. Science 1990, 249, 1527–1533. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.W.; Gong, C.Y.; Gou, M.L.; Fu, S.Z.; Guo, Q.F.; Shi, S.; Luo, F.; Guo, G.; Qiu, L.Y.; Qian, Z.Y. Biodegradable poly(ε-caprolactone)-poly(ethylene glycol) copolymers as drug delivery system. Int. J. Pharm. 2009, 381, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Rajgor, N.; Bhaskar, V.; Patel, M. Implantable drug delivery systems: An overview. Syst. Rev. Pharm. 2011, 2, 91–95. [Google Scholar] [CrossRef]
- Dash, A.; Cudworth, G. Therapeutic applications of implantable drug delivery systems. J. Pharmacol. Toxicol. Methods 1998, 40, 1–12. [Google Scholar] [CrossRef]
- Fialho, S.L.; da Silva Cunha, A. Manufacturing Techniques of Biodegradable Implants Intended for Intraocular Application. Drug Deliv. 2005, 12, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Rabin, C.; Liang, Y.; Ehrlichman, R.S.; Budhian, A.; Metzger, K.L.; Majewski-Tiedeken, C.; Winey, K.I.; Siegel, S.J. In vitro and in vivo demonstration of risperidone implants in mice. Schizophr. Res. 2008, 98, 66–78. [Google Scholar] [CrossRef]
- Schlesinger, E.; Johengen, D.; Luecke, E.; Rothrock, G.; McGowan, I.; van der Straten, A.; Desai, T. A Tunable, Biodegradable, Thin-Film Polymer Device as a Long-Acting Implant Delivering Tenofovir Alafenamide Fumarate for HIV Pre-exposure Prophylaxis. Pharm. Res. 2016, 33, 1649–1656. [Google Scholar] [CrossRef]
- Colaris, M.J.L.; de Boer, M.; van der Hulst, R.R.; Cohen Tervaert, J.W. Two hundreds cases of ASIA syndrome following silicone implants: A comparative study of 30 years and a review of current literature. Immunol. Res. 2017, 65, 120–128. [Google Scholar] [CrossRef]
- De Witt, D.; Finley, M.; Lawin, L.; Dewitt David, M.; Finley Michael, J.L.; Laurie, R. A Blends Comprising Ethylene-Vinyl Acetate Copolymer and Poly (Alkyl (Meth) Acrylates or Poly (Aromatic (Meth) Acrylates); Implantable Medical Device; Permit Stents Releasing the Bioactive Agent over Time In Vivo; Provide Clear Coats, Durability, Biocompatibility, and Release Kinetic; Drug Delivey Device. U.S. Patent Application No. 11/099,997, 6 April 2004. [Google Scholar]
- Nunes-Pereira, J.; Ribeiro, S.; Ribeiro, C.; Gombek, C.J.; Gama, F.M.; Gomes, A.C.; Patterson, D.A.; Lanceros-Méndez, S. Poly(vinylidene fluoride) and copolymers as porous membranes for tissue engineering applications. Polym. Test. 2015, 44, 234–241. [Google Scholar] [CrossRef]
- Shastri, V.P. Non-degradable biocompatible polymers in medicine: Past, present and future. Curr. Pharm. Biotechnol. 2003, 4, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Zur, G.; Linder-Ganz, E.; Elsner, J.J.; Shani, J.; Brenner, O.; Agar, G.; Hershman, E.B.; Arnoczky, S.P.; Guilak, F.; Shterling, A. Chondroprotective effects of a polycarbonate-urethane meniscal implant: Histopathological results in a sheep model. Knee Surg. Sports Traumatol. Arthrosc. 2011, 19, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Pillai, J. Implantable drug delivery systems. In Nanostructures for the Engineering of Cells, Tissues and Organs; Elsevier: Amsterdam, The Netherlands, 2018; pp. 473–511. [Google Scholar]
- Martínez-Rus, F.; Ferreiroa, A.; Özcan, M.; Bartolomé, J.F.; Pradíes, G. Fracture resistance of crowns cemented on titanium and zirconia implant abutments: A comparison of monolithic versus manually veneered all-ceramic systems. Int. J. Oral Maxillofac. Implants 2012, 27, 1448–1455. [Google Scholar] [PubMed]
- Claes, L.; Ignatius, A. Development of new, biodegradable implants. Chirurg 2002, 73, 990–996. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Mahmoudi, M.; Lhermusier, T.; Kiramijyan, S.; Chen, F.; Torguson, R.; Suddath, W.O.; Satler, L.F.; Pichard, A.D.; Waksman, R. The influence of advancing age on implantation of drug-eluting stents. Catheter. Cardiovasc. Interv. 2016, 88, 516–521. [Google Scholar] [CrossRef] [PubMed]
- Kleiner, L.W.; Wright, J.C.; Wang, Y. Evolution of implantable and insertable drug delivery systems. J. Control. Release 2014, 181, 1–10. [Google Scholar] [CrossRef]
- Jacob, J.; Haponiuk, J.T.; Thomas, S.; Gopi, S. Biopolymer based nanomaterials in drug delivery systems: A review. Mater. Today Chem. 2018, 9, 43–55. [Google Scholar] [CrossRef]
- Herrlich, S.; Spieth, S.; Messner, S.; Zengerle, R. Osmotic micropumps for drug delivery. Adv. Drug Deliv. Rev. 2012, 64, 1617–1627. [Google Scholar] [CrossRef]
- Verma, R. Formulation aspects in the development of osmotically controlled oral drug delivery systems. J. Control. Release 2002, 79, 7–27. [Google Scholar] [CrossRef]
- Kutz, M. Biomedical Engineering and Design Handbook; McGraw-Hill: New York, NY, USA, 2009; Volume 2. [Google Scholar]
- Gulati, K.; Kogawa, M.; Prideaux, M.; Findlay, D.M.; Atkins, G.J.; Losic, D. Drug-releasing nano-engineered titanium implants: Therapeutic efficacy in 3D cell culture model, controlled release and stability. Mater. Sci. Eng. C 2016, 69, 831–840. [Google Scholar] [CrossRef] [PubMed]
- Pillai, O.; Panchagnula, R. Polymers in drug delivery. Curr. Opin. Chem. Biol. 2001, 5, 447–451. [Google Scholar] [CrossRef]
- Fu, Y.; Kao, W.J. Drug release kinetics and transport mechanisms of non-degradable and degradable polymeric delivery systems. Expert Opin. Drug Deliv. 2010, 7, 429–444. [Google Scholar] [CrossRef] [PubMed]
- Siepmann, J.; Siepmann, F. Mathematical modeling of drug delivery. Int. J. Pharm. 2008, 364, 328–343. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.B.; Hwang, K.S.; Tsay, S.Y.; Cooper, S.L. Segmental orientation studies of polyether polyurethane block copolymers with different hard segment lengths and distributions. Colloid Polym. Sci. 1985, 263, 128–140. [Google Scholar] [CrossRef]
- Ulery, B.D.; Nair, L.S.; Laurencin, C.T. Biomedical applications of biodegradable polymers. J. Polym. Sci. Part B Polym. Phys. 2011, 49, 832–864. [Google Scholar] [CrossRef] [PubMed]
- Lyu, S.; Untereker, D. Degradability of Polymers for Implantable Biomedical Devices. Int. J. Mol. Sci. 2009, 10, 4033–4065. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Mei, L.; Song, C.; Cui, X.; Wang, P. The in vivo degradation, absorption and excretion of PCL-based implant. Biomaterials 2006, 27, 1735–1740. [Google Scholar] [CrossRef]
- Odom, E.B.; Eisenberg, D.L.; Fox, I.K. Difficult removal of subdermal contraceptive implants: A multidisciplinary approach involving a peripheral nerve expert. Contraception 2017, 96, 89–95. [Google Scholar] [CrossRef]
- Nair, L.S.; Laurencin, C.T. Biodegradable polymers as biomaterials. Prog. Polym. Sci. 2007, 32, 762–798. [Google Scholar] [CrossRef]
- Ginjupalli, K.; Shavi, G.V.; Averineni, R.K.; Bhat, M.; Udupa, N.; Nagaraja Upadhya, P. Poly(α-hydroxy acid) based polymers: A review on material and degradation aspects. Polym. Degrad. Stab. 2017, 144, 520–535. [Google Scholar] [CrossRef]
- Grayson, A.C.R.; Voskerician, G.; Lynn, A.; Anderson, J.M.; Cima, M.J.; Langer, R. Differential degradation rates in vivo and in vitro of biocompatible poly(lactic acid) and poly(glycolic acid) homo- and co-polymers for a polymeric drug-delivery microchip. J. Biomater. Sci. Polym. Ed. 2004, 15, 1281–1304. [Google Scholar] [CrossRef] [PubMed]
- Middleton, J.C.; Tipton, A.J. Synthetic biodegradable polymers as orthopedic devices. Biomaterials 2000, 21, 2335–2346. [Google Scholar] [CrossRef]
- Jain, R.A. The manufacturing techniques of various drug loaded biodegradable poly(lactide-co-glycolide) (PLGA) devices. Biomaterials 2000, 21, 2475–2490. [Google Scholar] [CrossRef]
- Kumari, A.; Yadav, S.K.; Yadav, S.C. Biodegradable polymeric nanoparticles based drug delivery systems. Colloids Surf. B Biointerfaces 2010, 75, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.M.; Shive, M.S. Biodegradation and biocompatibility of PLA and PLGA microspheres. Adv. Drug Deliv. Rev. 1997, 28, 5–24. [Google Scholar] [CrossRef]
- Makadia, H.K.; Siegel, S.J. Poly Lactic-co-Glycolic Acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers 2011, 3, 1377–1397. [Google Scholar] [CrossRef] [PubMed]
- Farah, S.; Anderson, D.G.; Langer, R. Physical and mechanical properties of PLA, and their functions in widespread applications—A comprehensive review. Adv. Drug Deliv. Rev. 2016, 107, 367–392. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, T.; Kao, N. PLA Based Biopolymer Reinforced with Natural Fibre: A Review. J. Polym. Environ. 2011, 19, 714–725. [Google Scholar] [CrossRef]
- Da Silva, D.; Kaduri, M.; Poley, M.; Adir, O.; Krinsky, N.; Shainsky-Roitman, J.; Schroeder, A. Biocompatibility, biodegradation and excretion of polylactic acid (PLA) in medical implants and theranostic systems. Chem. Eng. J. 2018, 340, 9–14. [Google Scholar] [CrossRef]
- Xiao, L.; Wang, B.; Yang, G.; Gauthier, M. Poly(Lactic Acid)-Based Biomaterials: Synthesis, Modification and Applications. In Biomedical Science, Engineering and Technology; InTech: London, UK, 2012. [Google Scholar]
- Avérous, L.; Pollet, E. (Eds.) Environmental Silicate Nano-Biocomposites; Green Energy and Technology; Springer: London, UK, 2012; ISBN 978-1-4471-4101-3. [Google Scholar]
- Bao, L.; Dorgan, J.R.; Knauss, D.; Hait, S.; Oliveira, N.S.; Maruccho, I.M. Gas permeation properties of poly(lactic acid) revisited. J. Membr. Sci. 2006, 285, 166–172. [Google Scholar] [CrossRef]
- Maurus, P.B.; Kaeding, C.C. Bioabsorbable implant material review. Oper. Tech. Sports Med. 2004, 12, 158–160. [Google Scholar] [CrossRef]
- Xinteng, Z.; Weisan, P.; Ruhua, Z.; Feng, Z. Preparation and evaluation of poly (d, l-lactic acid) (PLA) or d, l-lactide/glycolide copolymer (PLGA) microspheres with estradiol. Pharmazie 2002, 57, 695–697. [Google Scholar] [PubMed]
- Jamshidian, M.; Tehrany, E.A.; Imran, M.; Jacquot, M.; Desobry, S. Poly-Lactic Acid: Production, Applications, Nanocomposites, and Release Studies. Compr. Rev. Food Sci. Food Saf. 2010, 9, 552–571. [Google Scholar] [CrossRef]
- Fukushima, K.; Hirata, M.; Kimura, Y. Synthesis and Characterization of Stereoblock Poly(lactic acid)s with Nonequivalent D/L Sequence Ratios. Macromolecules 2007, 40, 3049–3055. [Google Scholar] [CrossRef]
- Mooney, D.J.; Baldwin, D.F.; Suh, N.P.; Vacanti, J.P.; Langer, R. Novel approach to fabricate porous sponges of poly(d,l-lactic-co-glycolic acid) without the use of organic solvents. Biomaterials 1996, 17, 1417–1422. [Google Scholar] [CrossRef]
- Grizzi, I.; Garreau, H.; Li, S.; Vert, M. Hydrolytic degradation of devices based on poly(dl-lactic acid) size-dependence. Biomaterials 1995, 16, 305–311. [Google Scholar] [CrossRef]
- Pitt, C.G.; Gratzl, M.M.; Kimmel, G.L.; Surles, J.; Schindler, A. Aliphatic polyesters II. The degradation of poly (dl-lactide), poly (epsilon-caprolactone), and their copolymers in vivo. Biomaterials 1981, 2, 215–220. [Google Scholar] [CrossRef]
- Bini, R.A.; Silva, M.F.; Varanda, L.C.; da Silva, M.A.; Dreiss, C.A. Soft nanocomposites of gelatin and poly(3-hydroxybutyrate) nanoparticles for dual drug release. Colloids Surf. B Biointerfaces 2017, 157, 191–198. [Google Scholar] [CrossRef]
- Goldbart, R.; Traitel, T.; Lapidot, S.A.; Kost, J. Enzymatically controlled responsive drug delivery systems. Polym. Adv. Technol. 2002, 13, 1006–1018. [Google Scholar] [CrossRef]
- Athanasiou, K.; Agrawal, C.; Barber, F.; Burkhart, S. Orthopaedic applications for PLA-PGA biodegradable polymers. Arthrosc. J. Arthrosc. Relat. Surg. 1998, 14, 726–737. [Google Scholar] [CrossRef]
- Castilla-Cortázar, I.; Más-Estellés, J.; Meseguer-Dueñas, J.M.; Escobar Ivirico, J.L.; Marí, B.; Vidaurre, A. Hydrolytic and enzymatic degradation of a poly(ε-caprolactone) network. Polym. Degrad. Stab. 2012, 97, 1241–1248. [Google Scholar] [CrossRef]
- Mondal, D.; Griffith, M.; Venkatraman, S.S. Polycaprolactone-based biomaterials for tissue engineering and drug delivery: Current scenario and challenges. Int. J. Polym. Mater. Polym. Biomater. 2016, 65, 255–265. [Google Scholar] [CrossRef]
- Woodruff, M.A.; Hutmacher, D.W. The return of a forgotten polymer—Polycaprolactone in the 21st century. Prog. Polym. Sci. 2010, 35, 1217–1256. [Google Scholar] [CrossRef]
- Jenkins, M.J.; Harrison, K.L. The effect of molecular weight on the crystallization kinetics of polycaprolactone. Polym. Adv. Technol. 2006, 17, 474–478. [Google Scholar] [CrossRef]
- Escobar Ivirico, J.L.; Salmerón Sánchez, M.; Sabater i Serra, R.; Meseguer Dueñas, J.M.; Gómez Ribelles, J.L.; Monleón Pradas, M. Structure and Properties of Poly(ε-caprolactone) Networks with Modulated Water Uptake. Macromol. Chem. Phys. 2006, 207, 2195–2205. [Google Scholar] [CrossRef]
- I Serra, R.S.; Escobar Ivirico, J.L.; Meseguer Dueñas, J.M.; Andrio Balado, A.; Gómez Ribelles, J.L.; Salmerón Sánchez, M. Dielectric relaxation spectrum of poly (ε-caprolactone) networks hydrophilized by copolymerization with 2-hydroxyethyl acrylate. Eur. Phys. J. E 2007, 22, 293–302. [Google Scholar] [CrossRef]
- Luckachan, G.E.; Pillai, C.K.S. Biodegradable Polymers—A Review on Recent Trends and Emerging Perspectives. J. Polym. Environ. 2011, 19, 637–676. [Google Scholar] [CrossRef]
- Uhrich, K.E.; Cannizzaro, S.M.; Langer, R.S.; Shakesheff, K.M. Polymeric Systems for Controlled Drug Release. Chem. Rev. 1999, 99, 3181–3198. [Google Scholar] [CrossRef]
- Doppalapudi, S.; Jain, A.; Khan, W.; Domb, A.J. Biodegradable polymers-an overview. Polym. Adv. Technol. 2014, 25, 427–435. [Google Scholar] [CrossRef]
- Goonoo, N.; Jeetah, R.; Bhaw-Luximon, A.; Jhurry, D. Polydioxanone-based bio-materials for tissue engineering and drug/gene delivery applications. Eur. J. Pharm. Biopharm. 2015, 97, 371–391. [Google Scholar] [CrossRef] [PubMed]
- Rahimi, A.; Mashak, A. Review on rubbers in medicine: Natural, silicone and polyurethane rubbers. Plast. Rubber Compos. 2013, 42, 223–230. [Google Scholar] [CrossRef]
- Woolfson, A.D.; Malcolm, R.K.; Gorman, S.P.; Jones, D.S.; Brown, A.F.; McCullagh, S.D. Self-lubricating silicone elastomer biomaterials. J. Mater. Chem. 2003, 13, 2465–2470. [Google Scholar] [CrossRef]
- Mashak, A.; Taghizadeh, S.M. In vitro progesterone release from γ-irradiated cross-linked polydimethylsiloxane. Radiat. Phys. Chem. 2006, 75, 229–235. [Google Scholar] [CrossRef]
- Schneider, C.; Langer, R.; Loveday, D.; Hair, D. Applications of ethylene vinyl acetate copolymers (EVA) in drug delivery systems. J. Control. Release 2017, 262, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Guillet, A.A.C. Determination of Primary Relaxation Temperatures and Melting Points of Ethylene Vinyl Acetate Copolymers. J. Therm. Anal. Calorim. 2000, 61, 681–685. [Google Scholar]
- Joseph, J.; Patel, R.M.; Wenham, A.; Smith, J.R. Biomedical applications of polyurethane materials and coatings. Trans. IMF 2018, 96, 121–129. [Google Scholar] [CrossRef]
- Sharmin, E.; Zafar, F. Polyurethane: An Introduction. In Polyurethane; InTech: London, UK, 2012. [Google Scholar]
- Guo, Y.; Zhang, R.; Xiao, Q.; Guo, H.; Wang, Z.; Li, X.; Chen, J.; Zhu, J. Asynchronous fracture of hierarchical microstructures in hard domain of thermoplastic polyurethane elastomer: Effect of chain extender. Polymer 2018, 138, 242–254. [Google Scholar] [CrossRef]
- Jana, R.N.; Bhunia, H. Accelerated Hygrothermal and UV Aging of Thermoplastic Polyurethanes. High Perform. Polym. 2010, 22, 3–15. [Google Scholar] [CrossRef]
- Nakkabi, A.; Sadiki, M.; Fahim, M.; Ittobane, N.; Ibnsouda Koraichi, S.; Barkai, H.; El Abed, S. Biodegradation of poly(ester urethane)s by Bacillus subtilis. Int. J. Environ. Res. 2015, 9, 157–162. [Google Scholar]
- Barrioni, B.R.; de Carvalho, S.M.; Oréfice, R.L.; de Oliveira, A.A.R.; de Magalhães Pereira, M. Synthesis and characterization of biodegradable polyurethane films based on HDI with hydrolyzable crosslinked bonds and a homogeneous structure for biomedical applications. Mater. Sci. Eng. C 2015, 52, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Rebelo, R.; Fernandes, M.; Fangueiro, R. Biopolymers in Medical Implants: A Brief Review. Procedia Eng. 2017, 200, 236–243. [Google Scholar] [CrossRef]
- Klemm, D.; Heublein, B.; Fink, H.-P.; Bohn, A. Cellulose: Fascinating Biopolymer and Sustainable Raw Material. Angew. Chem. Int. Ed. 2005, 44, 3358–3393. [Google Scholar] [CrossRef] [PubMed]
- Godinho, M.; Almeida, P.; Figueirinhas, J. From Cellulosic Based Liquid Crystalline Sheared Solutions to 1D and 2D Soft Materials. Materials 2014, 7, 4601–4627. [Google Scholar] [CrossRef] [PubMed]
- Cherian, B.M.; Leão, A.L.; de Souza, S.F.; de Olyveira, G.M.; Costa, L.M.M.; Brandão, C.V.S.; Narine, S.S. Bacterial Nanocellulose for Medical Implants. In Advances in Natural Polymers; Springer: Berlin/Heidelberg, Germany, 2013; pp. 337–359. [Google Scholar]
- Lin, N.; Dufresne, A. Nanocellulose in biomedicine: Current status and future prospect. Eur. Polym. J. 2014, 59, 302–325. [Google Scholar] [CrossRef]
- Abeer, M.M.; Mohd Amin, M.C.I.; Martin, C. A review of bacterial cellulose-based drug delivery systems: Their biochemistry, current approaches and future prospects. J. Pharm. Pharmacol. 2014, 66, 1047–1061. [Google Scholar] [CrossRef] [PubMed]
- Modulevsky, D.J.; Cuerrier, C.M.; Pelling, A.E. Biocompatibility of Subcutaneously Implanted Plant-Derived Cellulose Biomaterials. PLoS ONE 2016, 11, e0157894. [Google Scholar] [CrossRef]
- Jorfi, M.; Foster, E.J. Recent advances in nanocellulose for biomedical applications. J. Appl. Polym. Sci. 2015, 132. [Google Scholar] [CrossRef]
- Rebelo, R.; Vila, N.; Rana, S.; Fangueiro, R. Poly Lactic Acid Fibre Based Biodegradable Stents and Their Functionalization Techniques. In Natural Fibres: Advances in Science and Technology towards Industrial Applications; Springer: Dordrecht, The Netherlands, 2016; pp. 331–342. [Google Scholar]
- Dash, M.; Chiellini, F.; Ottenbrite, R.M.; Chiellini, E. Chitosan—A versatile semi-synthetic polymer in biomedical applications. Prog. Polym. Sci. 2011, 36, 981–1014. [Google Scholar] [CrossRef]
- Hu, L.; Sun, Y.; Wu, Y. Advances in chitosan-based drug delivery vehicles. Nanoscale 2013, 5, 3103–3111. [Google Scholar] [CrossRef]
- Bernkop-Schnürch, A.; Dünnhaupt, S. Chitosan-based drug delivery systems. Eur. J. Pharm. Biopharm. 2012, 81, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Younes, I.; Rinaudo, M. Chitin and Chitosan Preparation from Marine Sources. Structure, Properties and Applications. Mar. Drugs 2015, 13, 1133–1174. [Google Scholar] [CrossRef] [PubMed]
- Yucel, T.; Lovett, M.L.; Kaplan, D.L. Silk-based biomaterials for sustained drug delivery. J. Control. Release 2014, 190, 381–397. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wenk, E.; Matsumoto, A.; Meinel, L.; Li, C.; Kaplan, D.L. Silk microspheres for encapsulation and controlled release. J. Control. Release 2007, 117, 360–370. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yucel, T.; Lu, Q.; Hu, X.; Kaplan, D.L. Silk nanospheres and microspheres from silk/pva blend films for drug delivery. Biomaterials 2010, 31, 1025–1035. [Google Scholar] [CrossRef] [PubMed]
- Leisk, G.G.; Lo, T.J.; Yucel, T.; Lu, Q.; Kaplan, D.L. Electrogelation for Protein Adhesives. Adv. Mater. 2010, 22, 711–715. [Google Scholar] [CrossRef] [PubMed]
- Yucel, T.; Kojic, N.; Leisk, G.G.; Lo, T.J.; Kaplan, D.L. Non-equilibrium silk fibroin adhesives. J. Struct. Biol. 2010, 170, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Fini, M.; Motta, A.; Torricelli, P.; Giavaresi, G.; Nicoli Aldini, N.; Tschon, M.; Giardino, R.; Migliaresi, C. The healing of confined critical size cancellous defects in the presence of silk fibroin hydrogel. Biomaterials 2005, 26, 3527–3536. [Google Scholar] [CrossRef]
- Kim, U.-J.; Park, J.; Li, C.; Jin, H.-J.; Valluzzi, R.; Kaplan, D.L. Structure and Properties of Silk Hydrogels. Biomacromolecules 2004, 5, 786–792. [Google Scholar] [CrossRef]
- Wang, Y.; Rudym, D.D.; Walsh, A.; Abrahamsen, L.; Kim, H.-J.; Kim, H.S.; Kirker-Head, C.; Kaplan, D.L. In vivo degradation of three-dimensional silk fibroin scaffolds. Biomaterials 2008, 29, 3415–3428. [Google Scholar] [CrossRef]
- Guziewicz, N.A.; Massetti, A.J.; Perez-Ramirez, B.J.; Kaplan, D.L. Mechanisms of monoclonal antibody stabilization and release from silk biomaterials. Biomaterials 2013, 34, 7766–7775. [Google Scholar] [CrossRef] [PubMed]
- Cebe, P.; Hu, X.; Kaplan, D.L.; Zhuravlev, E.; Wurm, A.; Arbeiter, D.; Schick, C. Beating the Heat—Fast Scanning Melts Silk Beta Sheet Crystals. Sci. Rep. 2013, 3, 1130. [Google Scholar] [CrossRef] [PubMed]
- Elzoghby, A.O.; Samy, W.M.; Elgindy, N.A. Protein-based nanocarriers as promising drug and gene delivery systems. J. Control. Release 2012, 161, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Jivraj, M.; Martini, L.G.; Thomson, C.M. An overview of the different excipients useful for the direct compression of tablets. Pharm. Sci. Technol. Today 2000, 3, 58–63. [Google Scholar] [CrossRef]
- Santoveña, A.; García, J.T.; Oliva, A.; Llabrés, M.; Fariña, J.B. A mathematical model for interpreting in vitro rhGH release from laminar implants. Int. J. Pharm. 2006, 309, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Dorta, M.J.; Santoveña, A.; Llabrés, M.; Fariña, J.B. Potential applications of PLGA film-implants in modulating in vitro drugs release. Int. J. Pharm. 2002, 248, 149–156. [Google Scholar] [CrossRef]
- Umeki, N.; Sato, T.; Harada, M.; Takeda, J.; Saito, S.; Iwao, Y.; Itai, S. Preparation and evaluation of biodegradable films containing the potent osteogenic compound BFB0261 for localized delivery. Int. J. Pharm. 2011, 404, 10–18. [Google Scholar] [CrossRef]
- Breitenbach, J. Melt extrusion: From process to drug delivery technology. Eur. J. Pharm. Biopharm. 2002, 54, 107–117. [Google Scholar] [CrossRef]
- Repka, M.A.; Gerding, T.G.; Repka, S.L.; McGinity, J.W. Influence of Plasticizers and Drugs on the Physical-Mechanical Properties of Hydroxypropylcellulose Films Prepared by Hot Melt Extrusion. Drug Dev. Ind. Pharm. 1999, 25, 625–633. [Google Scholar] [CrossRef]
- Rothen-Weinhold, A.; Besseghir, K.; Vuaridel, E.; Sublet, E.; Oudry, N.; Kubel, F.; Gurny, R. Injection-molding versus extrusion as manufacturing technique for the preparation of biodegradable implants. Eur. J. Pharm. Biopharm. 1999, 48, 113–121. [Google Scholar] [CrossRef]
- Shi, Y.; Kramer, G.; Schröder, A.; Kirkpatrick, C.J.; Seekamp, A.; Schmidt, H.; Fuchs, S. Early endothelial progenitor cells as a source of myeloid cells to improve the pre-vascularisation of bone constructs. Eur. Cells Mater. 2014, 27, 64–80. [Google Scholar] [CrossRef]
- Norman, J.; Madurawe, R.D.; Moore, C.M.V.; Khan, M.A.; Khairuzzaman, A. A new chapter in pharmaceutical manufacturing: 3D-printed drug products. Adv. Drug Deliv. Rev. 2017, 108, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Wan, L. The levonorgestrel two-rod implant for long-acting contraception 10 years of clinical experience. Obstet. Gynecol. 2003, 102, 24–26. [Google Scholar] [CrossRef] [PubMed]
- Larrañeta, E.; Stewart, S.; Ervine, M.; Al-Kasasbeh, R.; Donnelly, R. Hydrogels for Hydrophobic Drug Delivery. Classification, Synthesis and Applications. J. Funct. Biomater. 2018, 9, 13. [Google Scholar] [CrossRef] [PubMed]
- Gunawardana, M.; Remedios-Chan, M.; Miller, C.S.; Fanter, R.; Yang, F.; Marzinke, M.A.; Hendrix, C.W.; Beliveau, M.; Moss, J.A.; Smith, T.J.; et al. Pharmacokinetics of Long-Acting Tenofovir Alafenamide (GS-7340) Subdermal Implant for HIV Prophylaxis. Antimicrob. Agents Chemother. 2015, 59, 3913–3919. [Google Scholar] [CrossRef] [PubMed]
- Wolinsky, J.B.; Colson, Y.L.; Grinstaff, M.W. Local drug delivery strategies for cancer treatment: Gels, nanoparticles, polymeric films, rods, and wafers. J. Control. Release 2012, 159, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Palomba, S.; Falbo, A.; Di Cello, A.; Materazzo, C.; Zullo, F. Nexplanon: the new implant for long-term contraception. A comprehensive descriptive review. Gynecol. Endocrinol. 2012, 28, 710–721. [Google Scholar] [CrossRef]
- Rademacher, K.H.; Vahdat, H.L.; Dorflinger, L.; Owen, D.H.; Steiner, M.J. Global Introduction of a Low-Cost Contraceptive Implant. In Critical Issues in Reproductive Health; Springer: Dordrecht, The Netherlands, 2014; pp. 285–306. [Google Scholar]
- Mansour, D.; Inki, P.; Gemzell-Danielsson, K. Efficacy of contraceptive methods: A review of the literature. Eur. J. Contracept. Reprod. Health Care 2010, 15, S19–S31. [Google Scholar] [CrossRef]
- Affandi, B.; Santoso, S.S.I.; Hadisaputra, W.; Moeloek, F.A.; Prihartono, J.; Lubis, F.; Samil, R.S. Five-year experience with Norplant®. Contraception 1987, 36, 417–428. [Google Scholar] [CrossRef]
- Brache, V. WHO Symposium WHO. Background and study methodology of a multicentre randomized clinical trial of two implantable contraceptives for women: Jadelle and Implanon. Eur. J. Contracept. Reprod. Health Care 2014, 19, 1. [Google Scholar]
- Baum, M.M.; Butkyavichene, I.; Gilman, J.; Kennedy, S.; Kopin, E.; Malone, A.M.; Nguyen, C.; Smith, T.J.; Friend, D.R.; Clark, M.R.; et al. An Intravaginal Ring for the Simultaneous Delivery of Multiple Drugs. J. Pharm. Sci. 2012, 101, 2833–2843. [Google Scholar] [CrossRef] [PubMed]
- Friend, D.R. Advances in vaginal drug delivery. Drug Deliv. Transl. Res. 2011, 1, 183–184. [Google Scholar] [CrossRef] [PubMed]
- Mulders, T. Use of the novel combined contraceptive vaginal ring NuvaRing for ovulation inhibition. Fertil. Steril. 2001, 75, 865–870. [Google Scholar] [CrossRef]
- Uhm, S.; Pope, R.; Schmidt, A.; Bazella, C.; Perriera, L. Home or office etonogestrel implant insertion after pregnancy: A randomized trial. Contraception 2016, 94, 567–571. [Google Scholar] [CrossRef] [PubMed]
- Mansour, D. Nexplanon®: What Implanon® did next. J. Fam. Plan. Reprod. Health Care 2010, 36, 187–189. [Google Scholar] [CrossRef] [PubMed]
- De Souza, R.; Zahedi, P.; Allen, C.J.; Piquette-Miller, M. Polymeric drug delivery systems for localized cancer chemotherapy. Drug Deliv. 2010, 17, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Goldspiel, B.R.; Kohler, D.R. Goserelin Acetate Implant: A Depot Luteinizing Hormone-Releasing Hormone Analog for Advanced Prostate Cancer. DICP 1991, 25, 796–804. [Google Scholar] [CrossRef] [PubMed]
- MHRA. Prolonged-Release Suspension for Injection in Pre-Filled Syringe (Leuprorelin Acetate) UKPAR Prolonged-Release Suspension for Injection in Pre-Filled Syringe (Leuprorelin Acetate) Lay Summary; MHRA: London, UK, 2015; pp. 1–41.
- Westphal, M.; Hilt, D.C.; Bortey, E.; Delavault, P.; Olivares, R.; Warnke, P.C.; Whittle, I.R.; Jääskeläinen, J.; Ram, Z. A phase 3 trial of local chemotherapy with biodegradable carmustine (BCNU) wafers (Gliadel wafers) in patients with primary malignant glioma. Neuro-Oncology 2003, 5, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Elstad, N.L.; Fowers, K.D. OncoGel (ReGel/paclitaxel)—Clinical applications for a novel paclitaxel delivery system. Adv. Drug Deliv. Rev. 2009, 61, 785–794. [Google Scholar] [CrossRef] [PubMed]
- Schlegel, P. A review of the pharmacokinetic and pharmacological properties of a once-yearly administered histrelin acetate implant in the treatment of prostate cancer. BJU Int. 2009, 103, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Shore, N. Introducing Vantas: The First Once-Yearly Luteinising Hormone-Releasing Hormone Agonist. Eur. Urol. Suppl. 2010, 9, 701–705. [Google Scholar] [CrossRef]
- Daneshmand, S.; Pohar, K.S.; Steinberg, G.D.; Aron, M.; Cutie, C. Effect of GemRIS (gemcitabine-releasing intravesical system, TAR-200) on antitumor activity in muscle-invasive bladder cancer (MIBC). J. Clin. Oncol. 2017, 35, e16000. [Google Scholar] [CrossRef]
- Waite, D.; Wang, Y.; Jones, D.; Stitt, A.; Raj Singh, T.R. Posterior drug delivery via periocular route: Challenges and opportunities. Ther. Deliv. 2017, 8, 685–699. [Google Scholar] [CrossRef] [PubMed]
- Manickavasagam, D.; Oyewumi, M.O. Critical Assessment of Implantable Drug Delivery Devices in Glaucoma Management. J. Drug Deliv. 2013, 2013, 895013. [Google Scholar] [CrossRef] [PubMed]
- Gooch, N.; Molokhia, S.A.; Condie, R.; Burr, R.M.; Archer, B.; Ambati, B.K.; Wirostko, B. Ocular Drug Delivery for Glaucoma Management. Pharmaceutics 2012, 4, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Macoul, K.L.; Pavan-Langston, D. Pilocarpine Ocusert System for Sustained Control of Ocular Hypertension. Arch. Ophthalmol. 1975, 93, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Haghjou, N.; Soheilian, M.; Abdekhodaie, M.J. Sustained release intraocular drug delivery devices for treatment of uveitis. J. Ophthalmic Vis. Res. 2011, 6, 317–329. [Google Scholar]
- Wong, I.B.; Teoh, S.C.; Yeoh, A.E.; Lingam, G. Sustained-release ganciclovir implant as prophylaxis for cytomegalovirus retinitis in a child undergoing bone marrow transplantation. Eye 2013, 27, 890–891. [Google Scholar] [CrossRef] [PubMed]
- Bobo, W.V.; Shelton, R.C. Risperidone long-acting injectable (Risperdal Consta®) for maintenance treatment in patients with bipolar disorder. Expert Rev. Neurother. 2010, 10, 1637–1658. [Google Scholar] [CrossRef]
- Siegel, S. Surgically Implantable Long-term Antipsychotic Delivery Systems for the Treatment of Schizophrenia. Neuropsychopharmacology 2002, 26, 817–823. [Google Scholar] [CrossRef]
- Grossman, S.A.; Roberts, N. Analgesic applications for a subcutaneous implant that continuously releases hydromorphone. Eur. J. Pain Suppl. 2011, 5, 439–442. [Google Scholar] [CrossRef]
- Lee, S.H.; Choy, Y. Bin Implantable Devices for Sustained, Intravesical Drug Delivery. Int. Neurourol. J. 2016, 20, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Nickel, J.C.; Jain, P.; Shore, N.; Anderson, J.; Giesing, D.; Lee, H.; Kim, G.; Daniel, K.; White, S.; Larrivee-Elkins, C.; et al. Continuous Intravesical Lidocaine Treatment for Interstitial Cystitis/Bladder Pain Syndrome: Safety and Efficacy of a New Drug Delivery Device. Sci. Transl. Med. 2012, 4, 143ra100. [Google Scholar] [CrossRef] [PubMed]
- Itzoe, M.; Guarnieri, M. New developments in managing opioid addiction: Impact of a subdermal buprenorphine implant. Drug Des. Dev. Ther. 2017, 11, 1429–1437. [Google Scholar] [CrossRef] [PubMed]
- Gangadharam, P.R.; Ashtekar, D.R.; Farhi, D.C.; Wise, D.L. Sustained release of isoniazid in vivo from a single implant of a biodegradable polymer. Tubercle 1991, 72, 115–122. [Google Scholar] [CrossRef]
- Gangadharam, P.R.; Geeta, N.; Hsu, Y.Y.; Wise, D.L. Chemotherapy of tuberculosis in mice using single implants of isoniazid and pyrazinamide. Int. J. Tuberc. Lung Dis. 1999, 3, 515–520. [Google Scholar] [PubMed]
- Dammerman, R.; Kim, S.; Adera, M.; Schwarz, A. Pharmacokinetics and Safety of Risperidone Subcutaneous Implants in Stable Patients With Schizophrenia. Clin. Pharmacol. Drug Dev. 2018, 7, 298–310. [Google Scholar] [CrossRef]
- Schwarz, A.; Thoroughman, S.; Winstead, D.; Decker, S.; Varughese, J. Development of a subcutaneous implant using polyurethane as a semi-permeable membrane for the controlled release of risperidone. In Proceedings of the Annual Meeting of the Controlled Release Society 2012, Quebec City, QC, Canada, 15–18 July 2012. [Google Scholar]
- Anselmo, A.C.; Mitragotri, S. An overview of clinical and commercial impact of drug delivery systems. J. Control. Release 2014, 190, 15–28. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Product Name | Implant Type | Material | Drug Delivered | Indication | References |
|---|---|---|---|---|---|
| Norplant® | Sub-cutaneous | Silicone | Levonorgestrel | Contraception | [118,119] |
| Jadelle® | |||||
| Estring® | Intra-vaginal | Silicone | Estradiol | Menopausal symptoms | [120] |
| Nuvaring® | Intra-vaginal | PEVA | Etonogestrel, Ethinyl estradiol | Contraception | [121,122] |
| Implanon® | Sub-cutaneous | PEVA | Etonogestrel | Contraception | [123,124] |
| Nexplanon® |
| Product Name | Implant Type | Material | Drug Delivered | Indication | References |
|---|---|---|---|---|---|
| Zoladex® | Sub-cutaneous | PLGA | Goserelin | Prostate cancer | [126] |
| Prostap®SR | Sub-cutaneous | PLGA | Leuprolide | Prostate cancer | [127] |
| Gliadel Wafers® | Intra-tumoral | Silicone | Carmustine (BCNU) | Primary malignant glioma | [114,128] |
| Oncogel® | Intra-tumoral | PLGA-PEG-PLGA | Paclitaxel | Oesophageal cancer | [129] |
| Vantas® | Sub-cutaneous | Methacrylate based hydrogel | Histrelin | Prostate Cancer | [130,131] |
| GemRIS® | Intra-vesical | ND | Gemcitabine | Non-muscle invasive Bladder Cancer | [132] |
| Product Name | Implant Type | Material | Drug Delivered | Indication | Reference |
|---|---|---|---|---|---|
| Ocusert® | Intra-ocular | PEVA | Pilocarpine, Alginic acid | Open angle glaucoma | [136] |
| Retisert® | Intra-ocular | Microcrystalline cellulose, PVA, Magnesium stearate | Fluocinolone | Non-infectious uveitis | [137] |
| Vitrasert® | Intra-ocular | PVA, PEVA | Ganciclovir | CMV retinitis in AIDS patients | [138] |
| Therapeutic Indication | Product Name | Implant Type | Material | Drug Delivered | Indication | References |
|---|---|---|---|---|---|---|
| Pain | ND (Axxia Pharmaceuticals) | Sub-cutaneous | PU, PEG/PPG/PTMEG | Hydromorphine | Chronic neuropathic pain | [141] |
| LiRIS® | Intra-vesical | Silicone | Lidocaine | Interstitial cystitis/bladder pain syndrome | [142,143] | |
| Probuphine® | Sub-cutaneous | PEVA | Buprenorphine | Opioid abuse | [144] | |
| Infectious Diseases | ND | ND | PLGA | Isoniazid | TB | [145] |
| ND | ND | PLGA | Isoniazid, Pyrazinamide | TB | [146] | |
| Central Nervous System disorders | Med-Launch | Sub-cutaneous | PLGA | Risperidone | Schizophrenia | [8,147] |
| ND | Sub-cutaneous | PU | Risperidone | Schizophrenia | [148] | |
| Risperdal consta® | Intra-muscular | PLGA | Risperidone | Schizophrenia | [149] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stewart, S.A.; Domínguez-Robles, J.; Donnelly, R.F.; Larrañeta, E. Implantable Polymeric Drug Delivery Devices: Classification, Manufacture, Materials, and Clinical Applications. Polymers 2018, 10, 1379. https://doi.org/10.3390/polym10121379
Stewart SA, Domínguez-Robles J, Donnelly RF, Larrañeta E. Implantable Polymeric Drug Delivery Devices: Classification, Manufacture, Materials, and Clinical Applications. Polymers. 2018; 10(12):1379. https://doi.org/10.3390/polym10121379
Chicago/Turabian StyleStewart, Sarah A., Juan Domínguez-Robles, Ryan F. Donnelly, and Eneko Larrañeta. 2018. "Implantable Polymeric Drug Delivery Devices: Classification, Manufacture, Materials, and Clinical Applications" Polymers 10, no. 12: 1379. https://doi.org/10.3390/polym10121379
APA StyleStewart, S. A., Domínguez-Robles, J., Donnelly, R. F., & Larrañeta, E. (2018). Implantable Polymeric Drug Delivery Devices: Classification, Manufacture, Materials, and Clinical Applications. Polymers, 10(12), 1379. https://doi.org/10.3390/polym10121379