Tying Together Multiscale Calculations for Charge Transport in P3HT: Structural Descriptors, Morphology, and Tie-Chains


Abstract
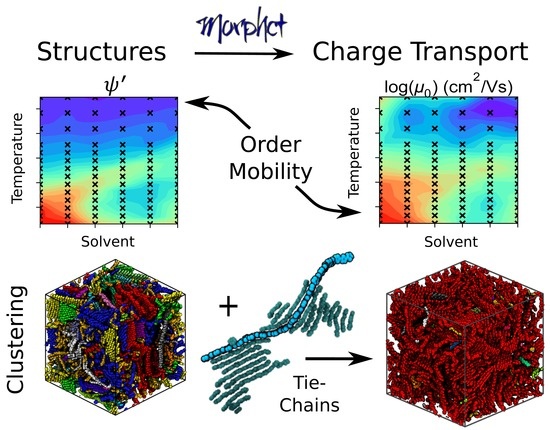
1. Introduction
2. Methods
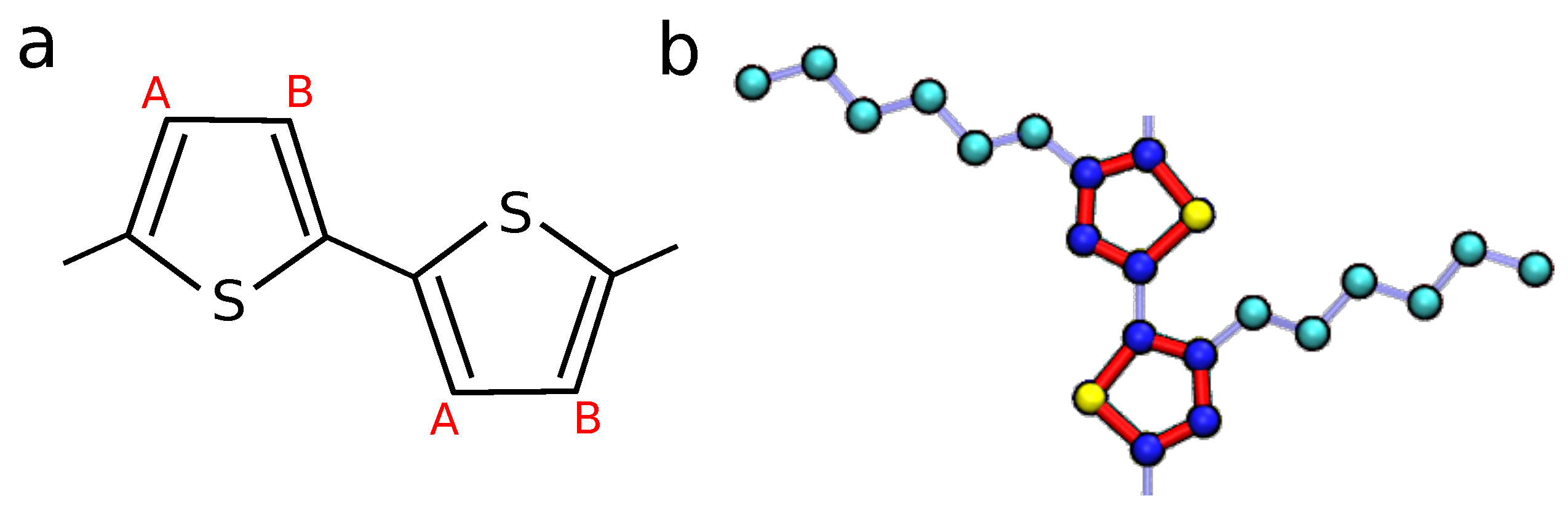
2.1. Molecular Dynamics Simulations
2.2. Kinetic Monte Carlo Simulations
3. Results and Discussion
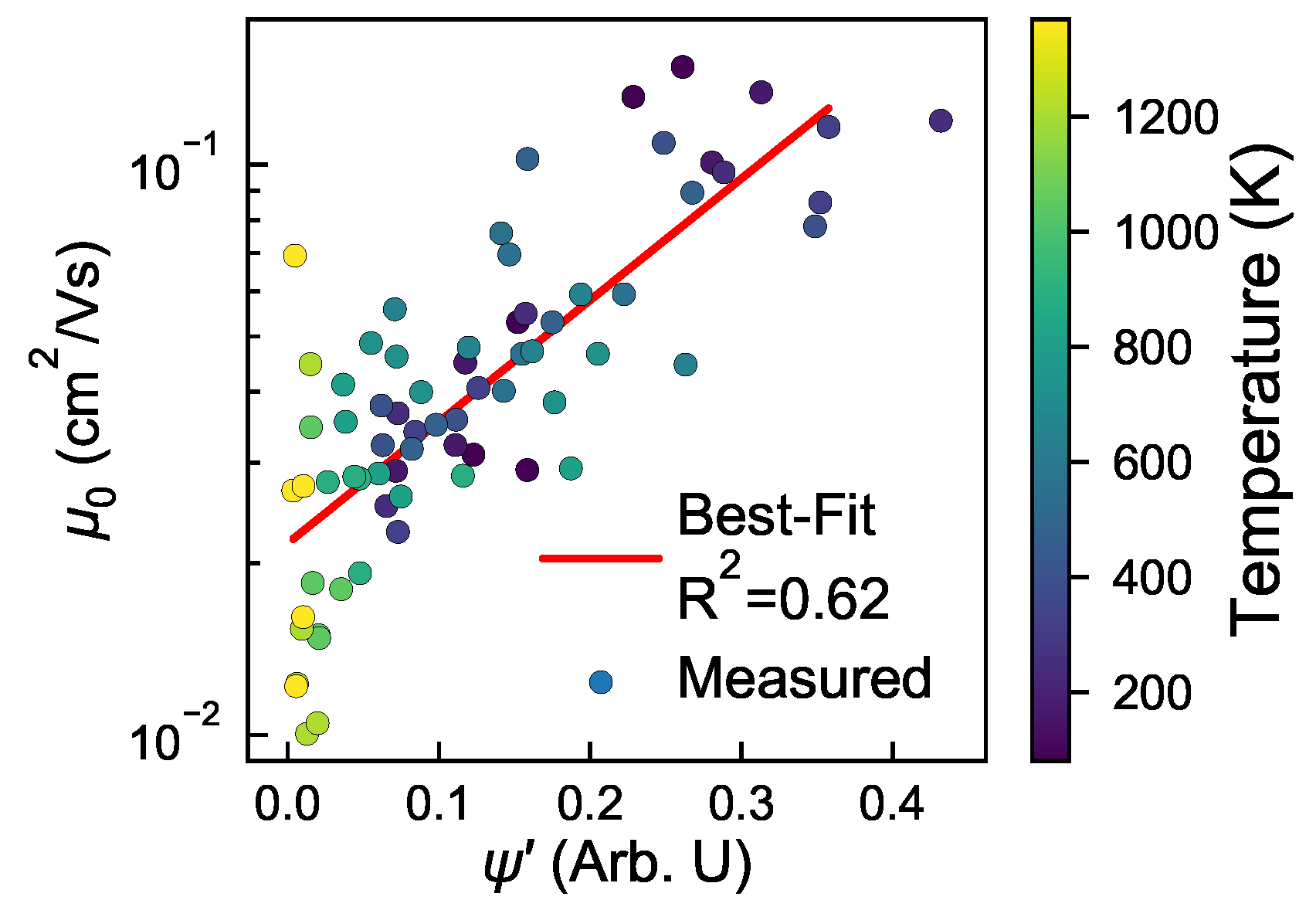
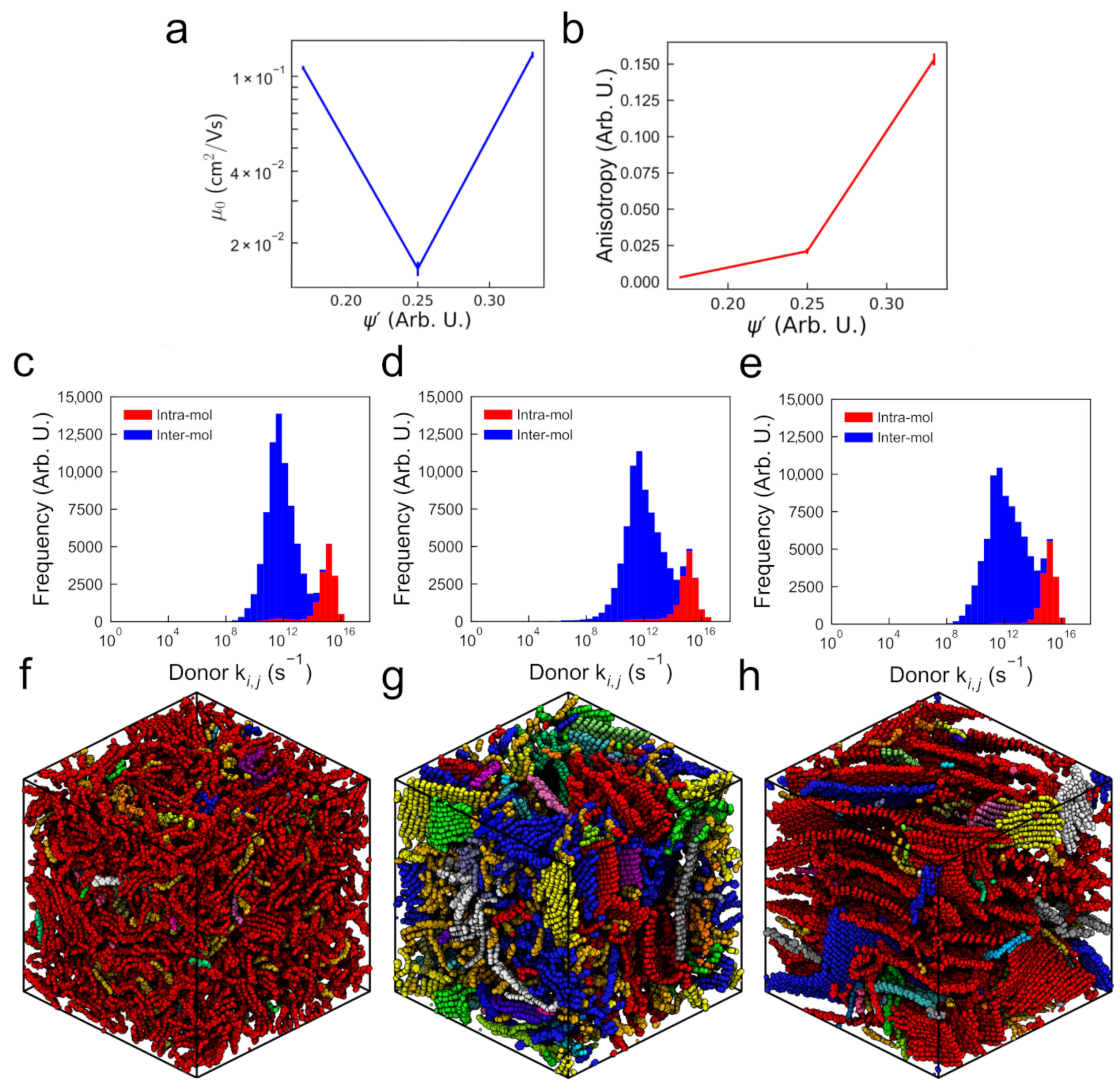
3.1. Structure and Mobility in “Small” Morphologies
3.2. Structure and Mobility in “Large” and Polydisperse Cases
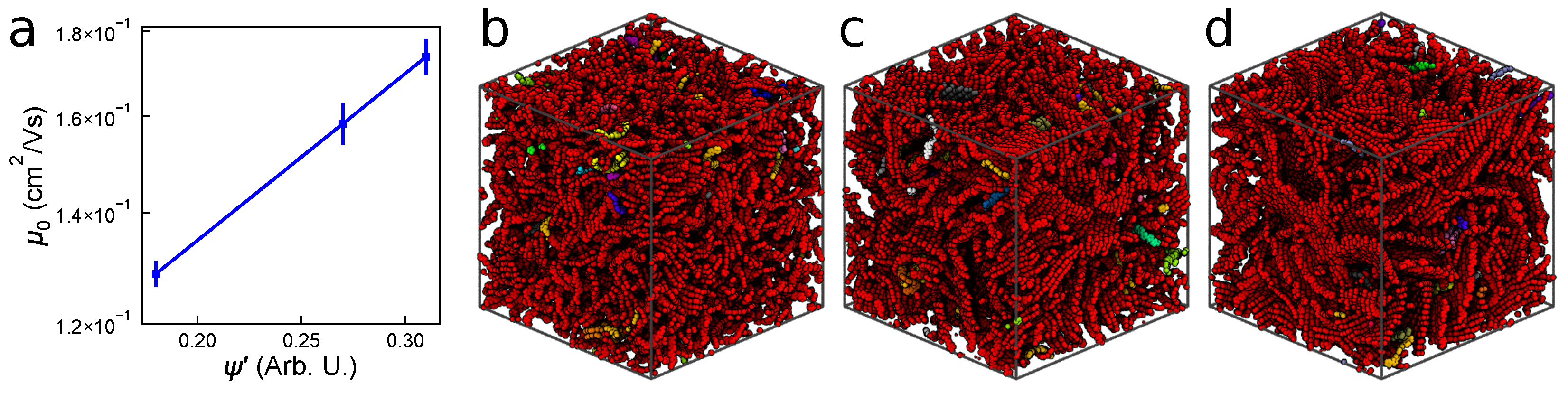
3.2.1. Mobility and Carrier Behavior
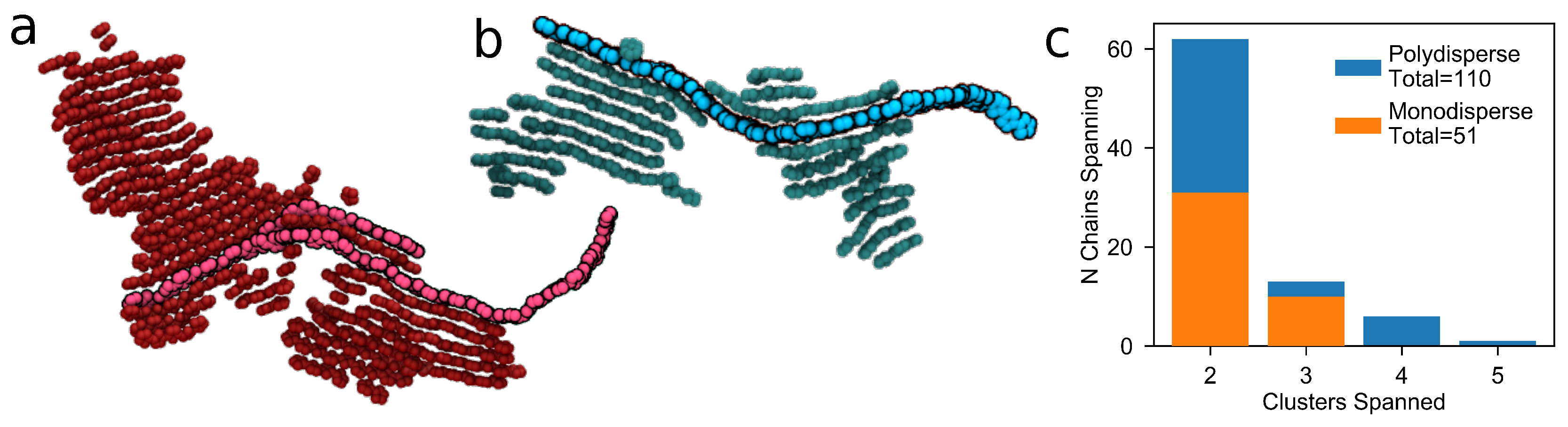
3.2.2. Tie Chains in Polydisperse Systems
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 15mer | P3HT chain containing 15 monomers |
| 50mer | P3HT chain containing 50 monomers |
| solvent quality | |
| KMC | Kinetic Monte Carlo |
| MD | Molecular Dynamics |
| Zero-field Mobility | |
| OPLS | Optimized Performance for Liquid Simulations |
| OPV | Organic Photovoltaic |
| P3HT | Poly(3-hexylthiophene) |
| order parameter | |
| modified order parameter | |
| density | |
| standard deviation | |
| T | Temperature |
| VRH | Variable Range Hopping |
| QCC | Quantum Chemical Calculations |
References
- Shaheen, S.E.; Ginley, D.S.; Jabbour, G.E. Organic-Based Photovoltaics: Toward Low-Cost Power Generation. MRS Bull. 2005, 30, 10–19. [Google Scholar] [CrossRef]
- Coakley, K.M.; McGehee, M.D. Conjugated polymer photovoltaic cells. Chem. Mater. 2004, 16, 4533–4542. [Google Scholar] [CrossRef]
- Espinosa, N.; Hösel, M.; Angmo, D.; Krebs, F.C. Solar Cells with One-Day Energy Payback for the Factories of the Future. Energy Environ. Sci. 2012, 5, 5117–5132. [Google Scholar] [CrossRef]
- Meng, L.; Zhang, Y.; Wan, X.; Li, C.; Zhang, X.; Wang, Y.; Ke, X.; Xiao, Z.; Ding, L.; Xia, R.; et al. Organic and solution-processed tandem solar cells with 17.3% efficiency. Science 2018, 361, 1094–1098. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Loos, J. Toward high-performance polymer solar cells: The importance of morphology control. Macromolecules 2007, 40, 1353–1362. [Google Scholar] [CrossRef]
- Chen, W.; Nikiforov, M.P.; Darling, S.B. Morphology characterization in organic and hybrid solar cells. Energy Environ. Sci. 2012, 5, 8045–8074. [Google Scholar] [CrossRef]
- Dang, M.T.; Wantz, G.; Bejbouji, H.; Urien, M.; Dautel, O.J.; Vignau, L.; Hirsch, L. Polymeric Solar Cells Based on P3HT:PCBM: Role of the Casting Solvent. Sol. Energy Mater. Sol. Cells 2011, 95, 3408–3418. [Google Scholar] [CrossRef]
- Surin, M.; Leclère, P.; Lazzaroni, R.; Yuen, J.D.; Wang, G.; Moses, D.; Heeger, A.J.; Cho, S.; Lee, K. Relationship between the microscopic morphology and the charge transport properties in poly(3-hexylthiophene) field-effect transistors. J. Appl. Phys. 2006, 100, 033712. [Google Scholar] [CrossRef]
- Verploegen, E.; Mondal, R.; Bettinger, C.J.; Sok, S.; Toney, M.F.; Bao, Z. Effects of Thermal Annealing Upon the Morphology of Polymer-Fullerene Blends. Adv. Funct. Mater. 2010, 20, 3519–3529. [Google Scholar] [CrossRef]
- Park, J.H.; Kim, J.S.; Lee, J.H.; Lee, W.H.; Cho, K. Effect of Annealing Solvent Solubility on the Performance of Poly(3-hexylthiophene)/Methanofullerene Solar Cells. J. Phys. Chem. C 2009, 113, 17579–17584. [Google Scholar] [CrossRef]
- Bertho, S.; Janssen, G.; Cleij, T.J.; Conings, B.; Moons, W.; Gadisa, A.; D’Haen, J.; Goovaerts, E.; Lutsen, L.; Manca, J.; et al. Effect of temperature on the morphological and photovoltaic stability of bulk heterojunction polymer:fullerene solar cells. Sol. Energy Mater. Sol. Cells 2008, 92, 753–760. [Google Scholar] [CrossRef]
- Miller, E.; Jones, M.; Henry, M.; Chery, P.; Miller, K.; Jankowski, E. Optimization and Validation of Efficient Models for Predicting Polythiophene Self-Assembly. Polymers 2018, 10, 1305. [Google Scholar] [CrossRef]
- Weis, M.; Lin, J.; Taguchi, D.; Manaka, T.; Iwamoto, M. Analysis of transient currents in organic field effect transistor: The time-of-flight method. J. Phys. Chem. C 2009, 113, 18459–18461. [Google Scholar] [CrossRef]
- Mazzio, K.A.; Luscombe, C.K. The future of organic photovoltaics. Chem. Soc. Rev. 2015, 44, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Dang, M.T.; Hirsch, L.; Wantz, G. P3HT:PCBM, best seller in polymer photovoltaic research. Adv. Mater. 2011, 23, 3597–3602. [Google Scholar] [CrossRef] [PubMed]
- Sirringhaus, H.; Brown, P.J.; Friend, R.H.; Nielsen, M.M.; Bechgaard, K.; Langeveld-Voss, B.M.W.; Spiering, A.J.H.; Janssen, R.A.J.; Meijer, E.W.; Herwig, P.; et al. Two-Dimensional Charge Transport in Self-Organized, High-Mobility Conjugated Polymers. Nature 1999, 401, 685–688. [Google Scholar] [CrossRef]
- Chang, J.F.; Sun, B.; Breiby, D.W.; Nielsen, M.M.; Sölling, T.I.; Giles, M.; McCulloch, I.; Sirringhaus, H. Enhanced Mobility of poly(3-hexylthiophene) transistors by spin-coating from high-boiling-point solvents. Chem. Mater. 2004, 16, 4772–4776. [Google Scholar] [CrossRef]
- Pandey, S.S.; Takashima, W.; Nagamatsu, S.; Endo, T.; Rikukawa, M.; Kaneto, K. Regioregularity vs. Regiorandomness: Effect on Photocarrier Transport in Poly(3-hexylthiophene). Jpn. J. Appl. Phys. 2000, 39, L94–L97. [Google Scholar] [CrossRef]
- Kim, Y.; Cook, S.; Tuladhar, S.M.; Choulis, S.A.; Nelson, J.; Durrant, J.R.; Bradley, D.D.C.; Giles, M.; McCulloch, I.; Ha, C.S.S.; et al. A Strong Regioregularity Effect in Self-Organizing Conjugated Polymer Films and High-Efficiency Polythiophene:Fullerene Solar Cells. Nat. Mater. 2006, 5, 197–203. [Google Scholar] [CrossRef]
- Ballantyne, A.M.; Chen, L.; Dane, J.; Hammant, T.; Braun, F.M.; Heeney, M.; Duffy, W.; McCulloch, I.; Bradley, D.D.C.; Nelson, J. The Effect of Poly(3-hexylthiophene) Molecular Weight on Charge Transport and the Performance of Polymer:Fullerene Solar Cells. Adv. Funct. Mater. 2008, 18, 2373–2380. [Google Scholar] [CrossRef]
- Mauer, R.; Kastler, M.; Laquai, F. The Impact of Polymer Regioregularity on Charge Transport and Efficiency of P3HT:PCBM Photovoltaic Devices. Adv. Funct. Mater. 2010, 20, 2085–2092. [Google Scholar] [CrossRef]
- Barker, J.A.; Ramsdale, C.M.; Greenham, N.C. Modeling the Current-Voltage Characteristics of Bilayer Polymer Photovoltaic Devices. Phys. Rev. B 2003, 67, 075205. [Google Scholar] [CrossRef]
- Koster, L.J.A.; Smits, E.C.P.; Mihailetchi, V.D.; Blom, P.W.M. Device Model for the Operation of Polymer/Fullerene Bulk Heterojunction Solar Cells. Phys. Rev. B 2005, 72, 085205. [Google Scholar] [CrossRef]
- Einax, M.; Dierl, M.; Nitzan, A. Heterojunction Organic Photovoltaic Cells as Molecular Heat Engines: A Simple Model for the Performance Analysis. J. Phys. Chem. C 2011, 115, 21396–21401. [Google Scholar] [CrossRef]
- Rolland, N.; Franco-Gonzalez, J.F.; Volpi, R.; Linares, M.; Zozoulenko, I.V. Understanding Morphology- Mobility Dependence in PEDOT:Tos. Phys. Rev. Mater. 2018, 2, 045605. [Google Scholar] [CrossRef]
- Cheung, D.L.; McMahon, D.P.; Troisi, A. Computational Study of the Structure and Charge-Transfer Parameters in Low-Molecular-Mass P3HT. J. Phys. Chem. B 2009, 113, 9393–9401. [Google Scholar] [CrossRef] [PubMed]
- Groves, C.; Kimber, R.G.E.; Walker, A.B. Simulation of Loss Mechanisms in Organic Solar Cells: A Description of the Mesoscopic Monte Carlo Technique and an Evaluation of the First Reaction Method. J. Chem. Phys. 2010, 133, 144110. [Google Scholar] [CrossRef]
- Jones, M.L.; Huang, D.M.; Chakrabarti, B.; Groves, C. Relating Molecular Morphology to Charge Mobility in Semicrystalline Conjugated Polymers. J. Phys. Chem. C 2016, 120, 4240–4250. [Google Scholar] [CrossRef]
- Jones, M.L.; Jankowski, E. Computationally connecting organic photovoltaic performance to atomistic arrangements and bulk morphology. Mol. Simul. 2017, 43, 756–773. [Google Scholar] [CrossRef]
- Jones, M.L.; Dyer, R.; Clarke, N.; Groves, C. Are Hot Charge Transfer States the Primary Cause of Efficient Free-Charge Generation in Polymer:Fullerene Organic Photovoltaic Devices? A Kinetic Monte Carlo Study. Phys. Chem. Chem. Phys. 2014, 16, 20310–20320. [Google Scholar] [CrossRef]
- Van, E.; Jones, M.; Jankowski, E.; Wodo, O. Using graphs to quantify energetic and structural order in semicrystalline oligothiophene thin films. Mol. Syst. Des. Eng. 2018, 1, 273–277. [Google Scholar] [CrossRef]
- Wang, L.; Li, Q.; Shuai, Z.; Chen, L.; Shi, Q. Multiscale study of charge mobility of organic semiconductor with dynamic disorders. Phys. Chem. Chem. Phys. 2010, 12, 3309–3314. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.K.; Huang, C.I. A Theoretical Study of the Charge Transfer Behavior of the Highly Regioregular Poly-3-hexylthiophene in the Ordered State. J. Phys. Chem. B 2008, 112, 14857–14862. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.L. matty-jones/MorphCT: MorphCT v3.0. Zenodo 2018. [Google Scholar] [CrossRef]
- Huang, D.M.; Faller, R.; Do, K.; Moulé, A.J. Coarse-Grained Computer Simulations of Polymer/Fullerene Bulk Heterojunctions for Organic Photovoltaic Applications. J. Chem. Theory Comput. 2010, 6, 526–537. [Google Scholar] [CrossRef] [PubMed]
- Bhatta, R.S.; Yimer, Y.Y.; Perry, D.S.; Tsige, M. Improved Force Field for Molecular Modeling of Poly(3-hexylthiophene). J. Phys. Chem. B 2013, 117, 10035–10045. [Google Scholar] [CrossRef] [PubMed]
- McMahon, D.P.; Cheung, D.L.; Goris, L.; Dacuña, J.; Salleo, A.; Troisi, A. Relation between Microstructure and Charge Transport in Polymers of Different Regioregularity. J. Phys. Chem. C 2011, 115, 19386–19393. [Google Scholar] [CrossRef]
- Murthy, D.H.K.; Gao, M.; Vermeulen, M.J.W.; Siebbeles, L.D.A.; Savenije, T.J. Mechanism of Mobile Charge Carrier Generation in Blends of Conjugated Polymers and Fullerenes: Significance of Charge Delocalization and Excess Free Energy. J. Phys. Chem. C 2012, 116, 9214–9220. [Google Scholar] [CrossRef]
- Deng, W.Q.; Goddard, W.A. Predictions of Hole Mobilities in Oligoacene Organic Semiconductors from Quantum Mechanical Calculations. J. Phys. Chem. B 2004, 108, 8614–8621. [Google Scholar] [CrossRef]
- Brédas, J.L.; Beljonne, D.; Coropceanu, V.; Cornil, J. Charge-Transfer and Energy-Transfer Processes in π-Conjugated Oligomers and Polymers: A Molecular Picture. Chem. Rev. 2004, 104, 4971–5004. [Google Scholar] [CrossRef]
- Marcus, R.A. Chemical and Electrochemical Electron-Transfer Theory. Annu. Rev. Phys. Chem. 1964, 15, 155–196. [Google Scholar] [CrossRef]
- Mott, N. Conduction in glasses containing transition metal ions. J. Non-Cryst. Solids 1968, 1, 1–17. [Google Scholar] [CrossRef]
- Tsigankov, D.N.; Efros, A.L. Variable Range Hopping in Two-Dimensional Systems of Interacting Electrons. Phys. Rev. Lett. 2002, 88, 176602. [Google Scholar] [CrossRef] [PubMed]
- Fornari, R.P.; Troisi, A. Theory of charge hopping along a disordered polymer chain. Phys. Chem. Chem. Phys. 2014, 16, 9997–10007. [Google Scholar] [CrossRef] [PubMed]
- Johansson, E.; Larsson, S. Electronic Structure and Mechanism for Conductivity in Thiophene Oligomers and Regioregular Polymer. Synth. Met. 2004, 144, 183–191. [Google Scholar] [CrossRef]
- Coropceanu, V.; Cornil, J.; da Silva Filho, D.A.; Olivier, Y.; Silbey, R.; Brédas, J.L. Charge Transport in Organic Semiconductors. Chem. Rev. 2007, 107, 926–952. [Google Scholar] [CrossRef]
- Carbone, P.; Troisi, A. Charge Diffusion in Semiconducting Polymers: Analytical Relation between Polymer Rigidity and Time Scales for Intrachain and Interchain Hopping. J. Phys. Chem. Lett. 2014, 5, 2637–2641. [Google Scholar] [CrossRef]
- Novikov, S.V. Diffusion of a Particle in the Gaussian Random-Energy Landscape: Einstein Relation and Analytical Properties of Average Velocity and Diffusivity as Functions of Driving Force. Phys. Rev. E 2018, 98, 012128. [Google Scholar] [CrossRef]
- Noriega, R.; Rivnay, J.; Vandewal, K.; Koch, F.P.V.; Stingelin, N.; Smith, P.; Toney, M.F.; Salleo, A. A general relationship between disorder, aggregation and charge transport in conjugated polymers. Nat. Mater. 2013, 12, 1038–1044. [Google Scholar] [CrossRef]
- Franco-Gonzalez, J.F.; Rolland, N.; Zozoulenko, I.V. Substrate-Dependent Morphology and Its Effect on Electrical Mobility of Doped Poly(3,4-ethylenedioxythiophene) (PEDOT) Thin Films. ACS Appl. Mater. Interfaces 2018, 10, 29115–29126. [Google Scholar] [CrossRef]
- Brinkmann, M.; Wittmann, J.C. Orientation of regioregular poly(3-hexylthiophene) by directional solidification: A simple method to reveal the semicrystalline structure of a conjugated polymer. Adv. Mater. 2006, 18, 860–863. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhao, X.; Roders, M.; Qu, G.; Diao, Y.; Ayzner, A.L.; Mei, J. Complementary Semiconducting Polymer Blends for Efficient Charge Transport. Chem. Mater. 2015, 27, 7164–7170. [Google Scholar] [CrossRef]
- Crossland, E.J.W.; Tremel, K.; Fischer, F.; Rahimi, K.; Reiter, G.; Steiner, U.; Ludwigs, S. Anisotropic Charge Transport in Spherulitic Poly(3-hexylthiophene) Films. Adv. Mater. 2012, 24, 839–844. [Google Scholar] [CrossRef] [PubMed]
- Towns, J.; Cockerill, T.; Dahan, M.; Foster, I.; Gaither, K.; Grimshaw, A.; Hazlewood, V.; Lathrop, S.; Lifka, D.; Peterson, G.D.; et al. XSEDE: Accelerating Scientific Discovery. Comput. Sci. Eng. 2014, 16, 62–74. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
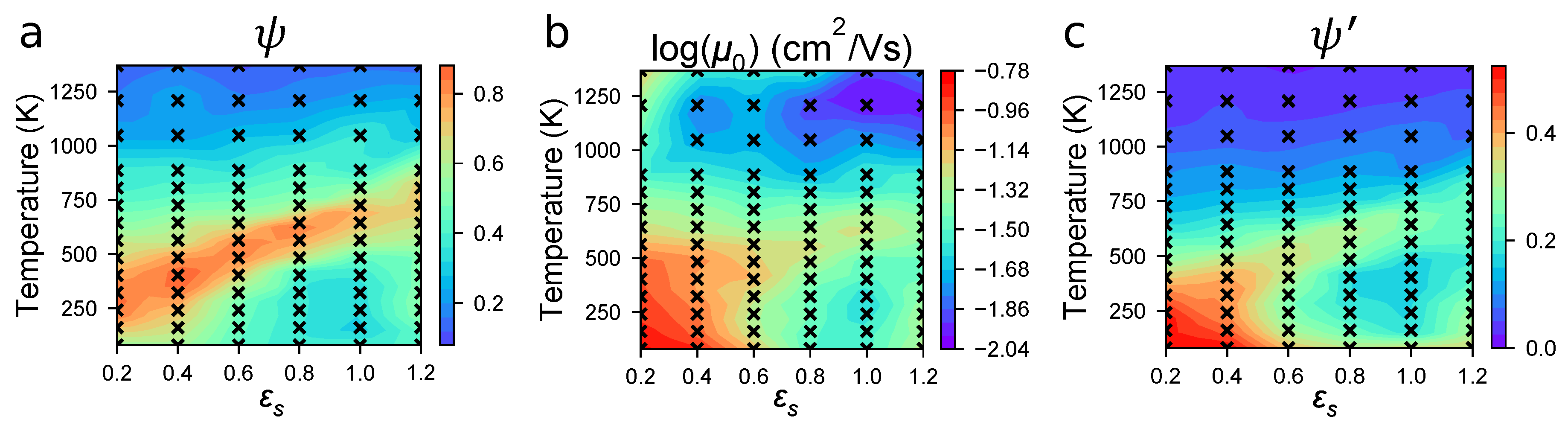{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Property | Amorphous | Semi-Crystalline | Crystalline |
|---|---|---|---|
| Mobility (cm/Vs) | |||
| Anisotropy (Arb. U.) | |||
| Intra-molecular rate (s) | |||
| Inter-molecular rate (s) | |||
| std (eV) | |||
| Total clusters (Arb. U.) | |||
| Large (>6) clusters (Arb. U.) | |||
| Largest cluster size (Arb. U.) |
| Property | Amorphous | Semi-Crystalline | Crystalline |
|---|---|---|---|
| Mobility (cm/Vs) | |||
| Anisotropy (Arb. U.) | |||
| Intra-molecular rate (s) | |||
| Inter-molecular rate (s) | |||
| std (eV) | |||
| Total clusters (Arb. U.) | |||
| Large (>6) clusters (Arb. U.) | |||
| Largest cluster size (Arb. U.) | 11,200 ± 260 | 13,200 ± 200 | 13,500 ± 100 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miller, E.D.; Jones, M.L.; Jankowski, E. Tying Together Multiscale Calculations for Charge Transport in P3HT: Structural Descriptors, Morphology, and Tie-Chains. Polymers 2018, 10, 1358. https://doi.org/10.3390/polym10121358
Miller ED, Jones ML, Jankowski E. Tying Together Multiscale Calculations for Charge Transport in P3HT: Structural Descriptors, Morphology, and Tie-Chains. Polymers. 2018; 10(12):1358. https://doi.org/10.3390/polym10121358
Chicago/Turabian StyleMiller, Evan D., Matthew L. Jones, and Eric Jankowski. 2018. "Tying Together Multiscale Calculations for Charge Transport in P3HT: Structural Descriptors, Morphology, and Tie-Chains" Polymers 10, no. 12: 1358. https://doi.org/10.3390/polym10121358
APA StyleMiller, E. D., Jones, M. L., & Jankowski, E. (2018). Tying Together Multiscale Calculations for Charge Transport in P3HT: Structural Descriptors, Morphology, and Tie-Chains. Polymers, 10(12), 1358. https://doi.org/10.3390/polym10121358