Green Processes for Green Products: The Use of Supercritical CO2 as Green Solvent for Compatibilized Polymer Blends



Abstract
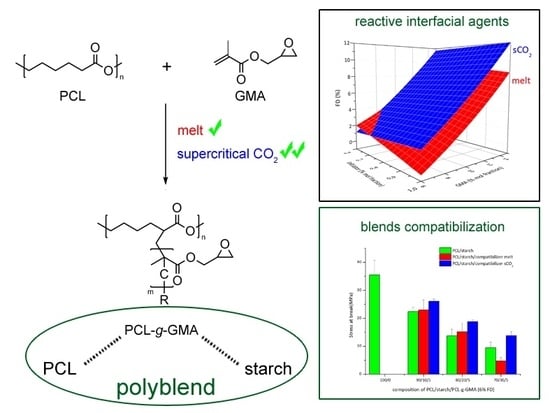
1. Introduction
2. Materials and Methods
2.1. Materials
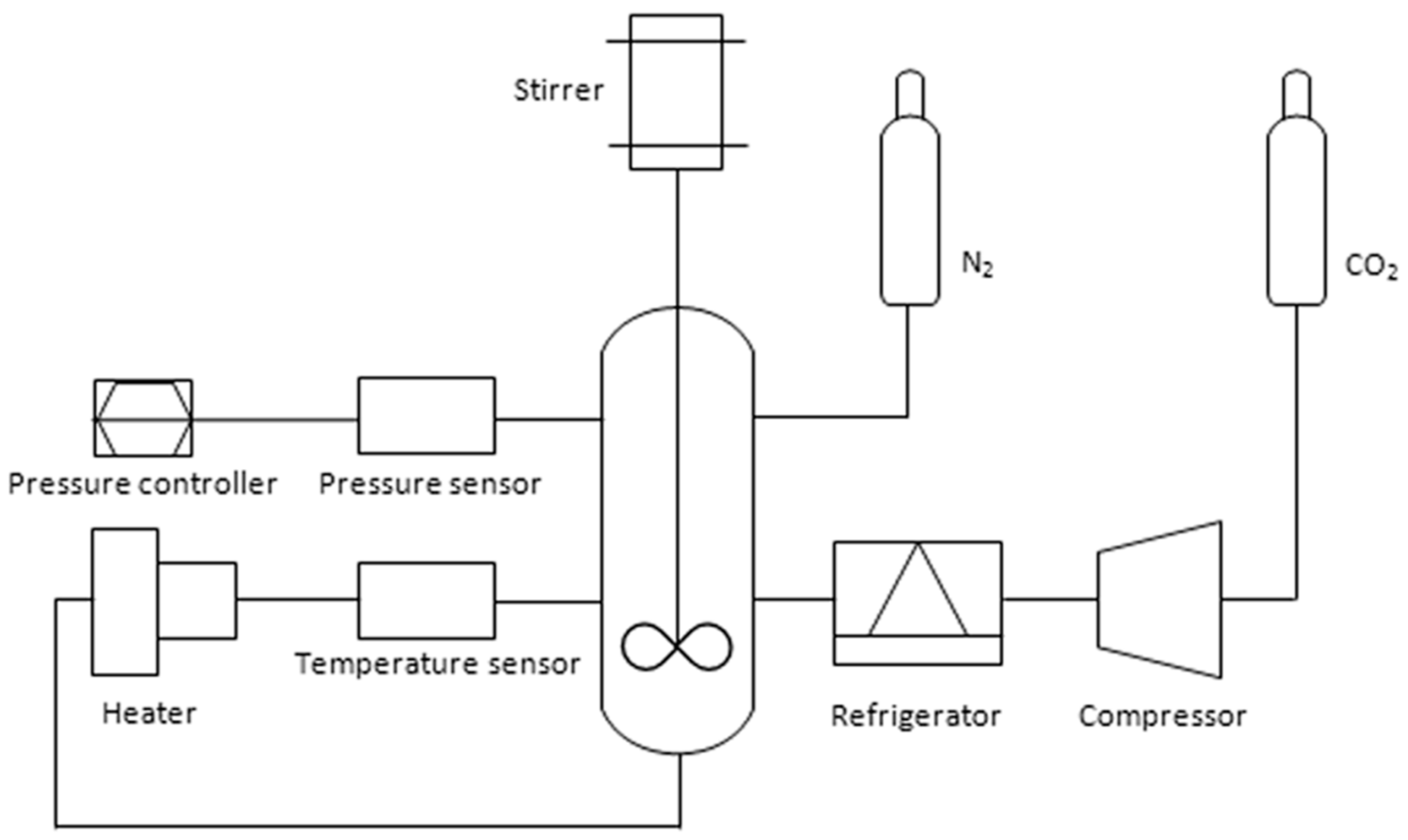
2.2. Synthesis and Purification of PCL-g-GMA
2.2.1. Purification of PCL-g-GMA
2.2.2. Ternary Blend of PCL/Starch/PCL-g-GMA
2.3. Analytical Methods
2.3.1. 1H-NMR
2.3.2. Gel Permeation Chromatography
2.3.3. Tensile Tests
2.3.4. Scanning Electronic Microscopy (SEM)
2.3.5. Selective Solvent Extraction
2.3.6. Differential Scanning Calorimetry (DSC)
3. Results
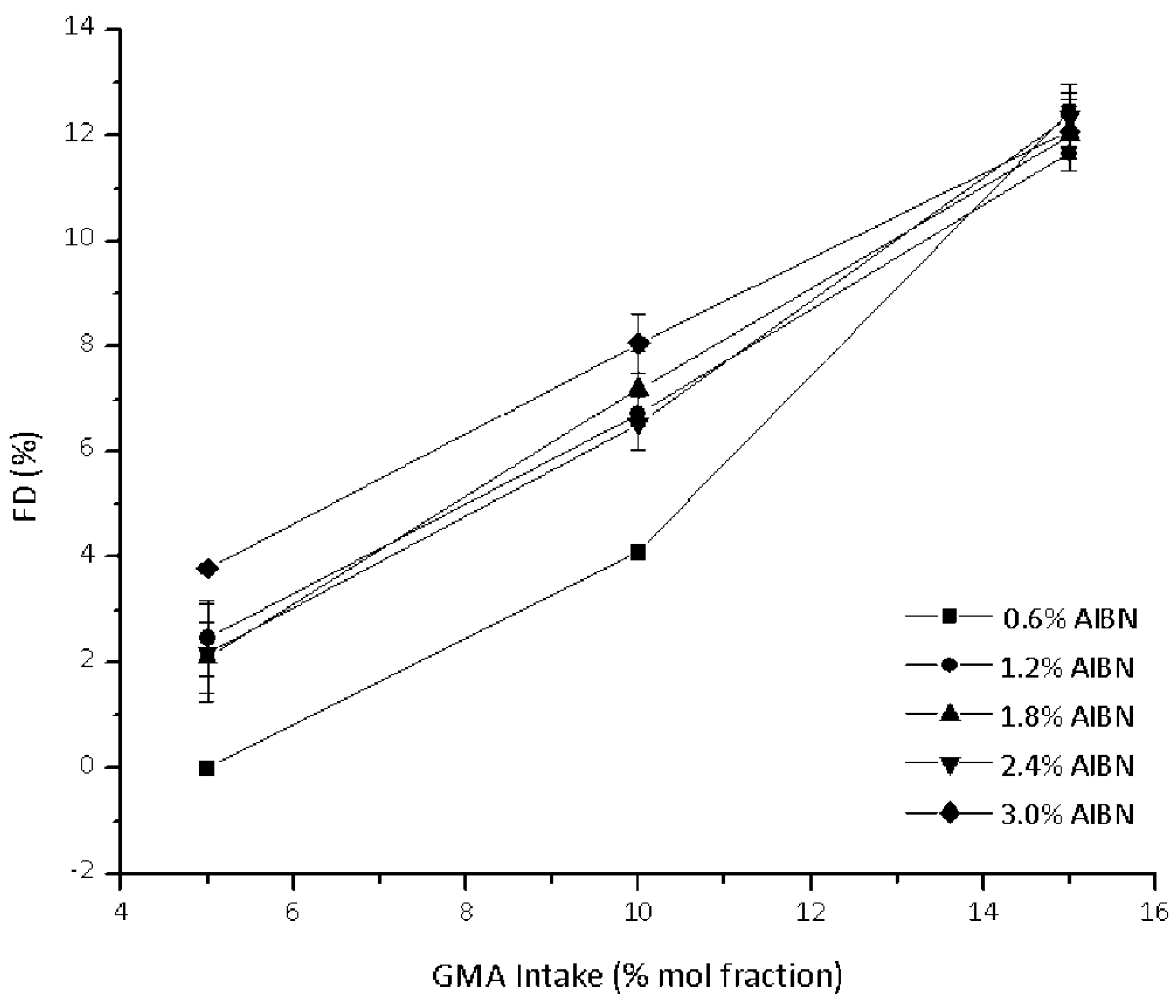
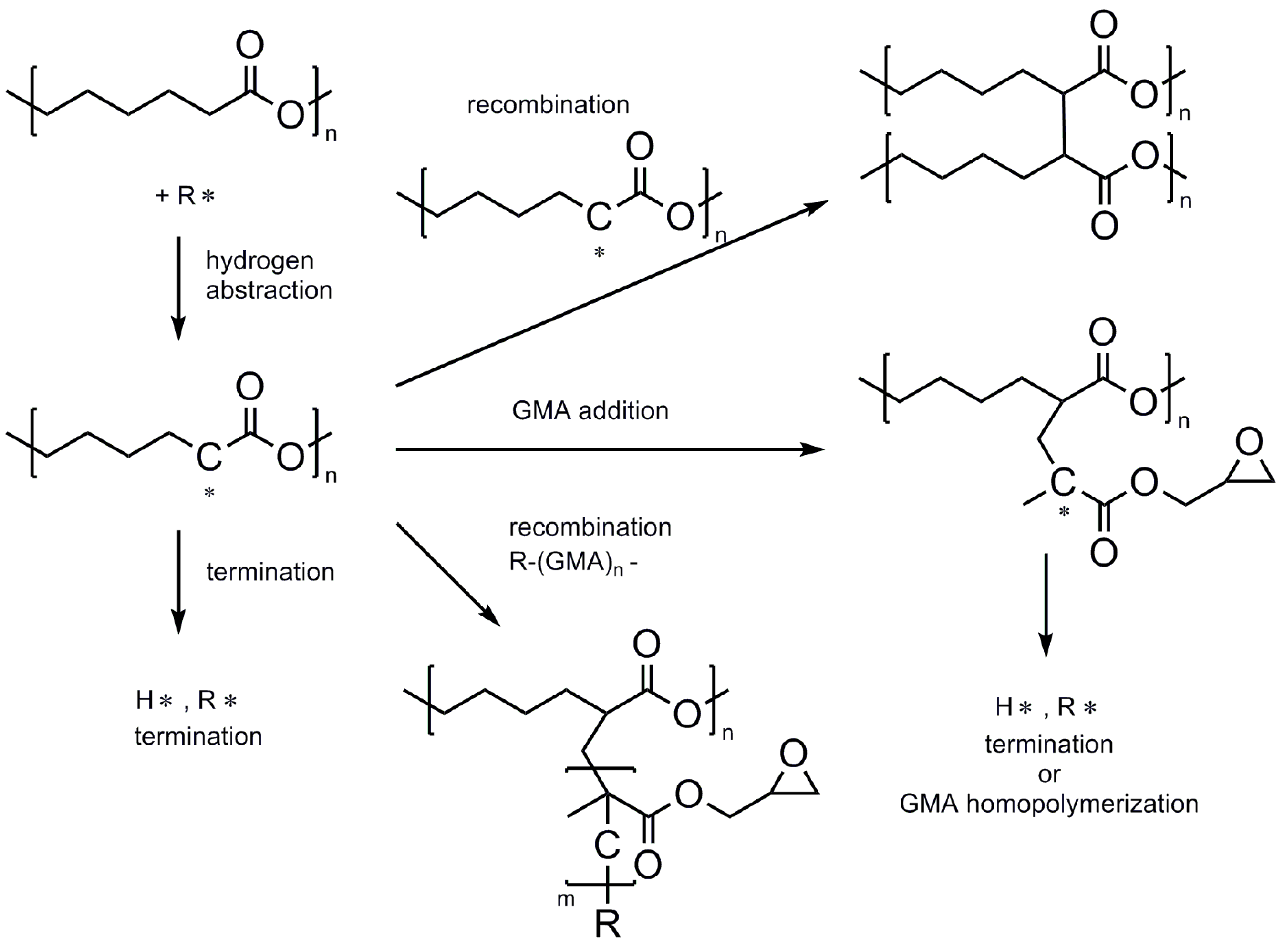
3.1. Functionalization Reaction of PCL-g-GMA
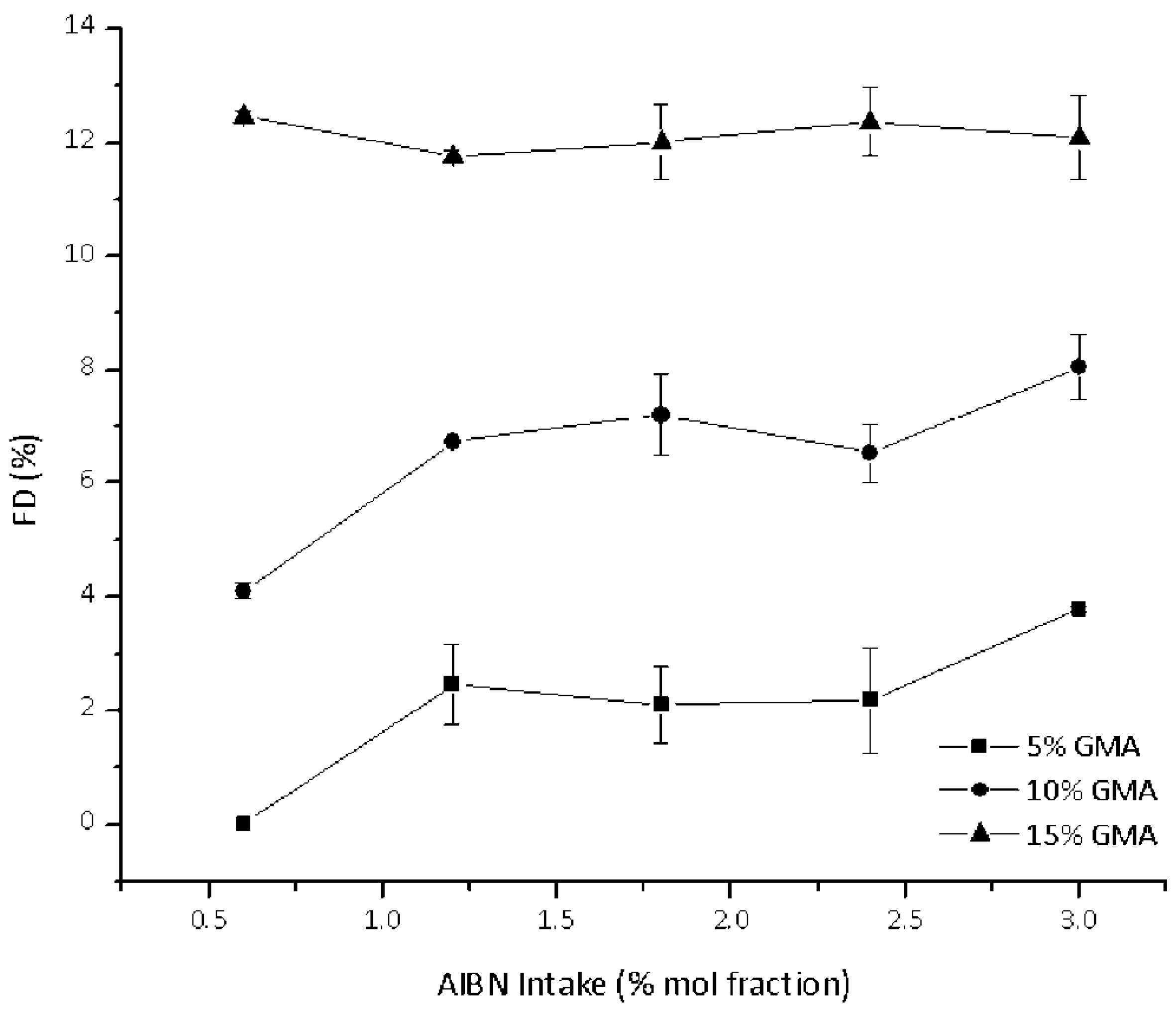
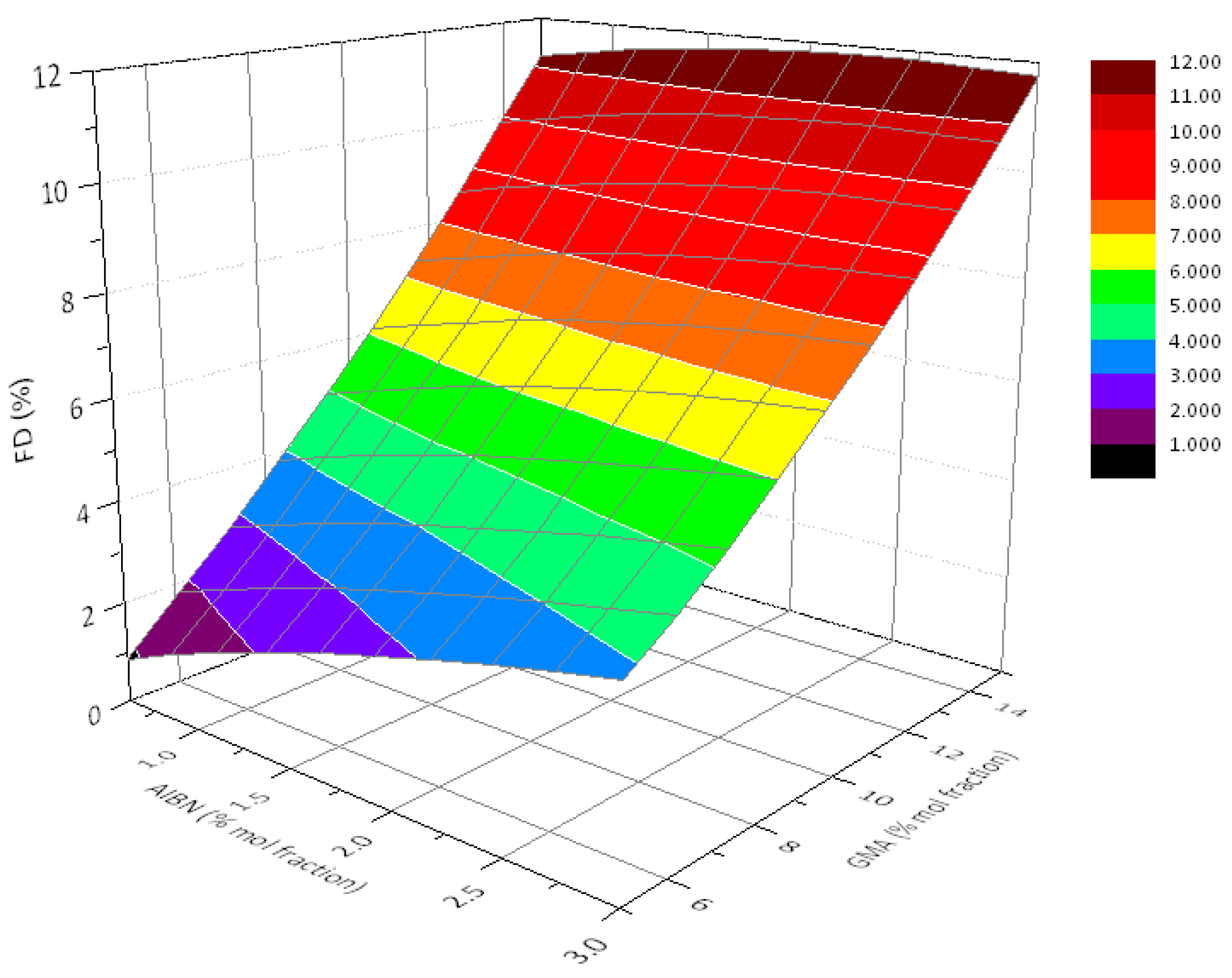
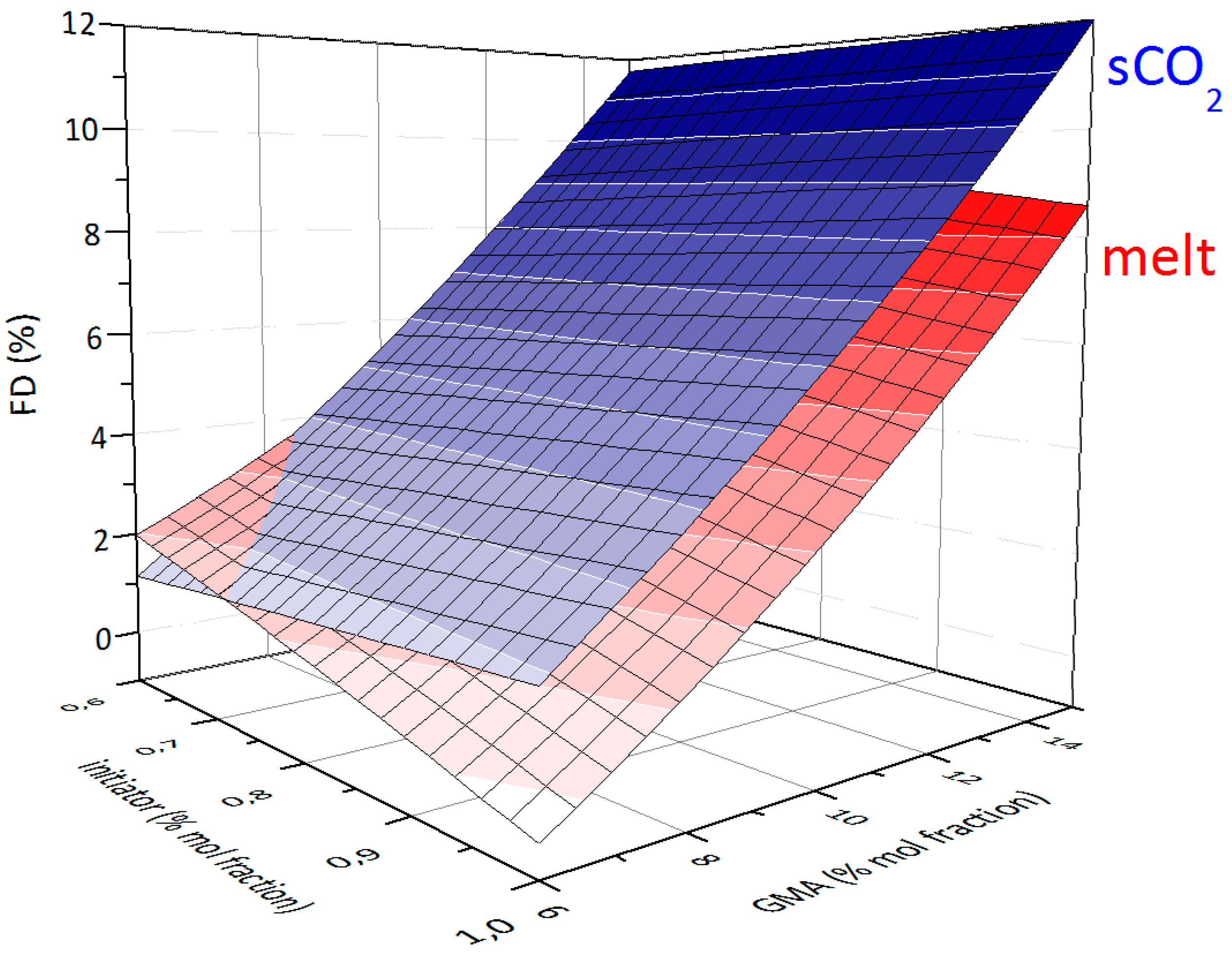
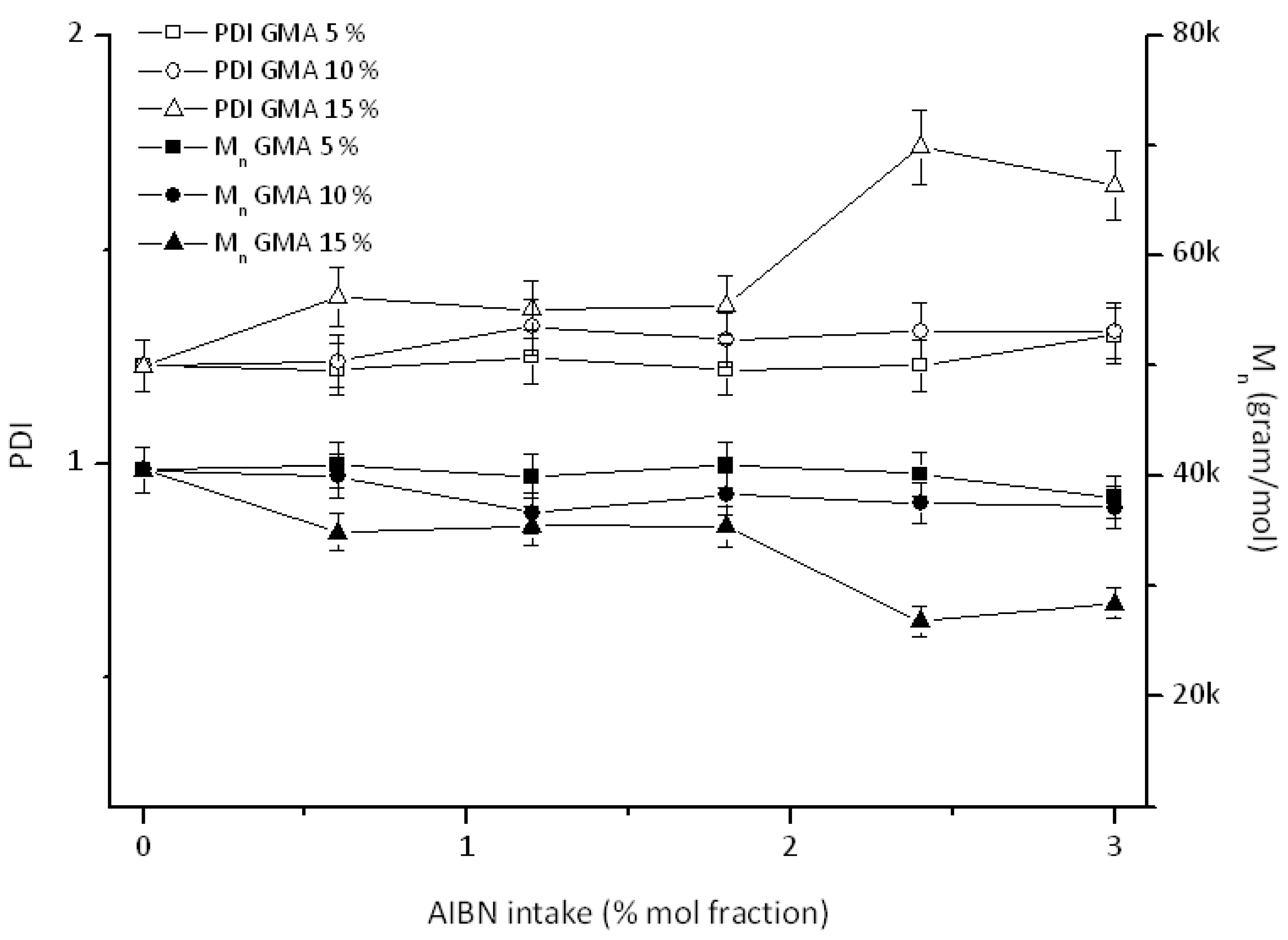
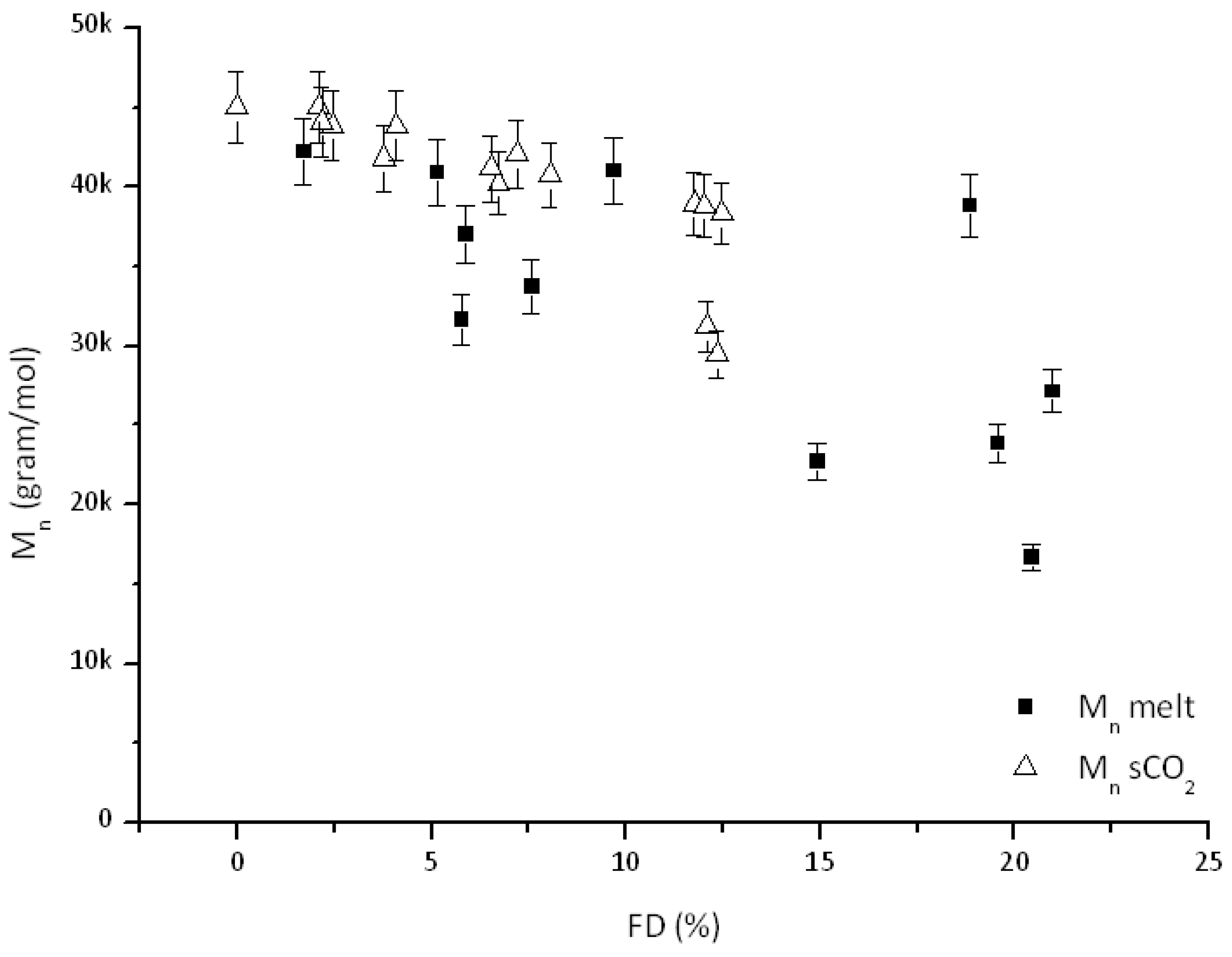
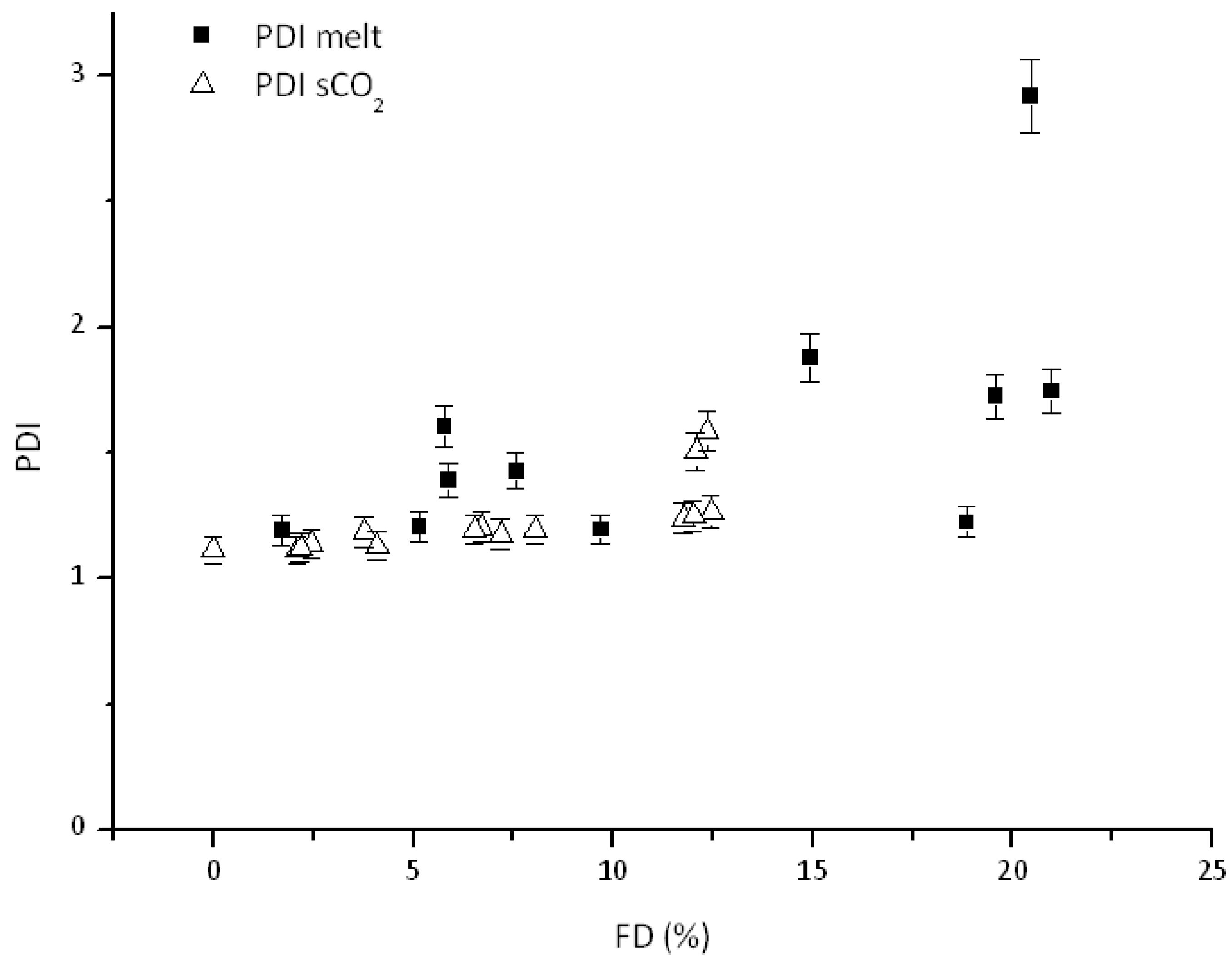
3.2. Effect of Feed Composition on Molecular Weight
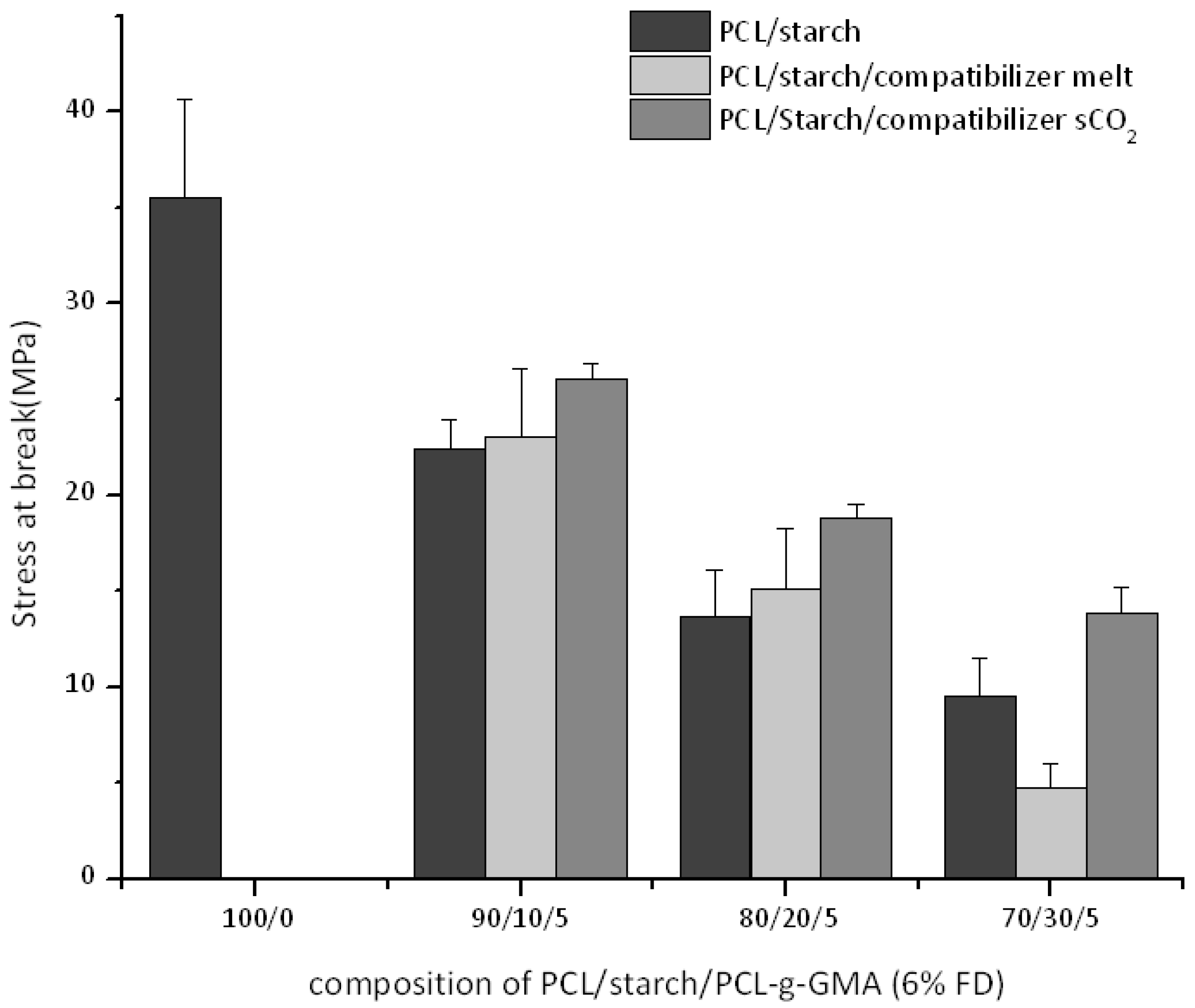
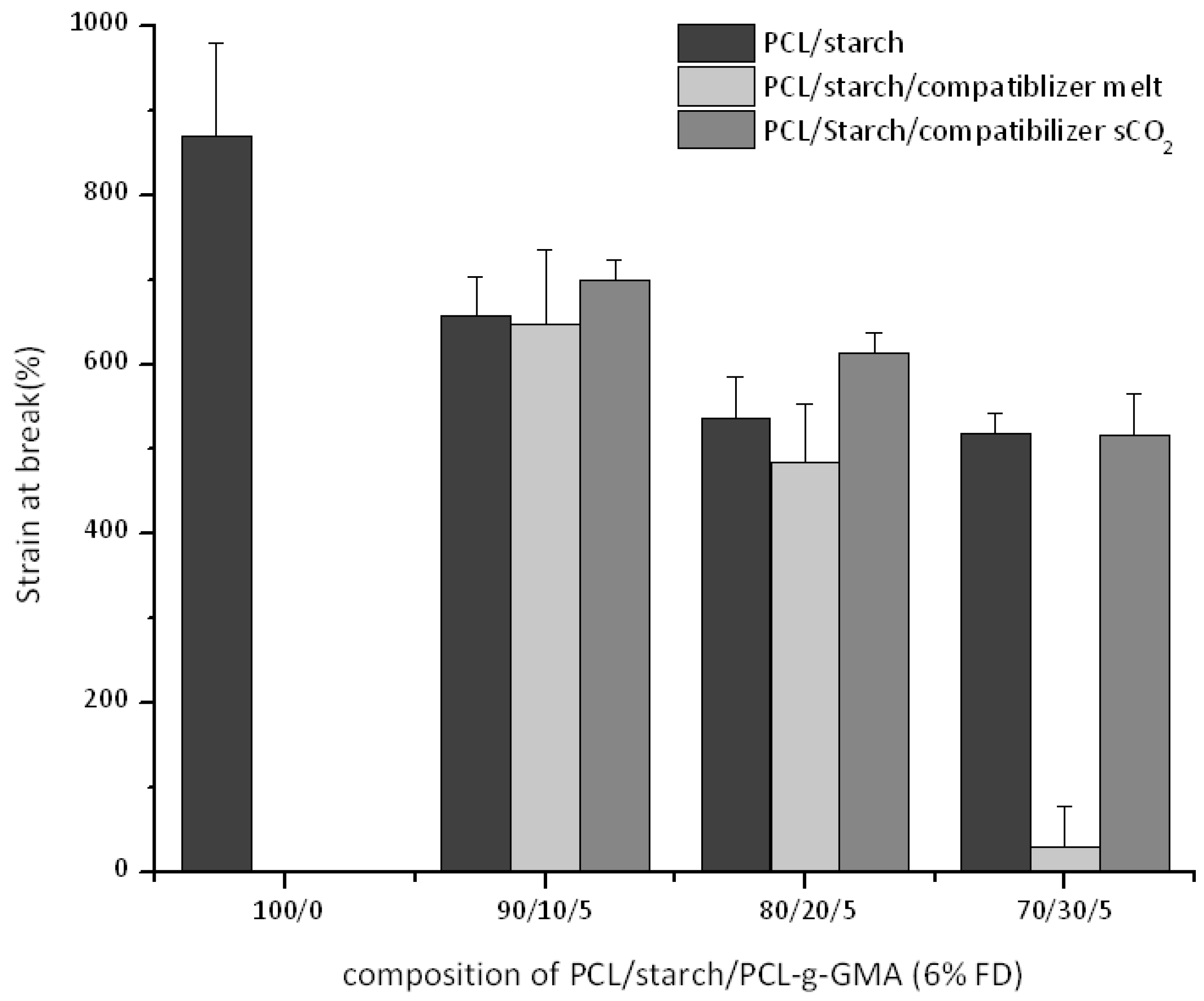
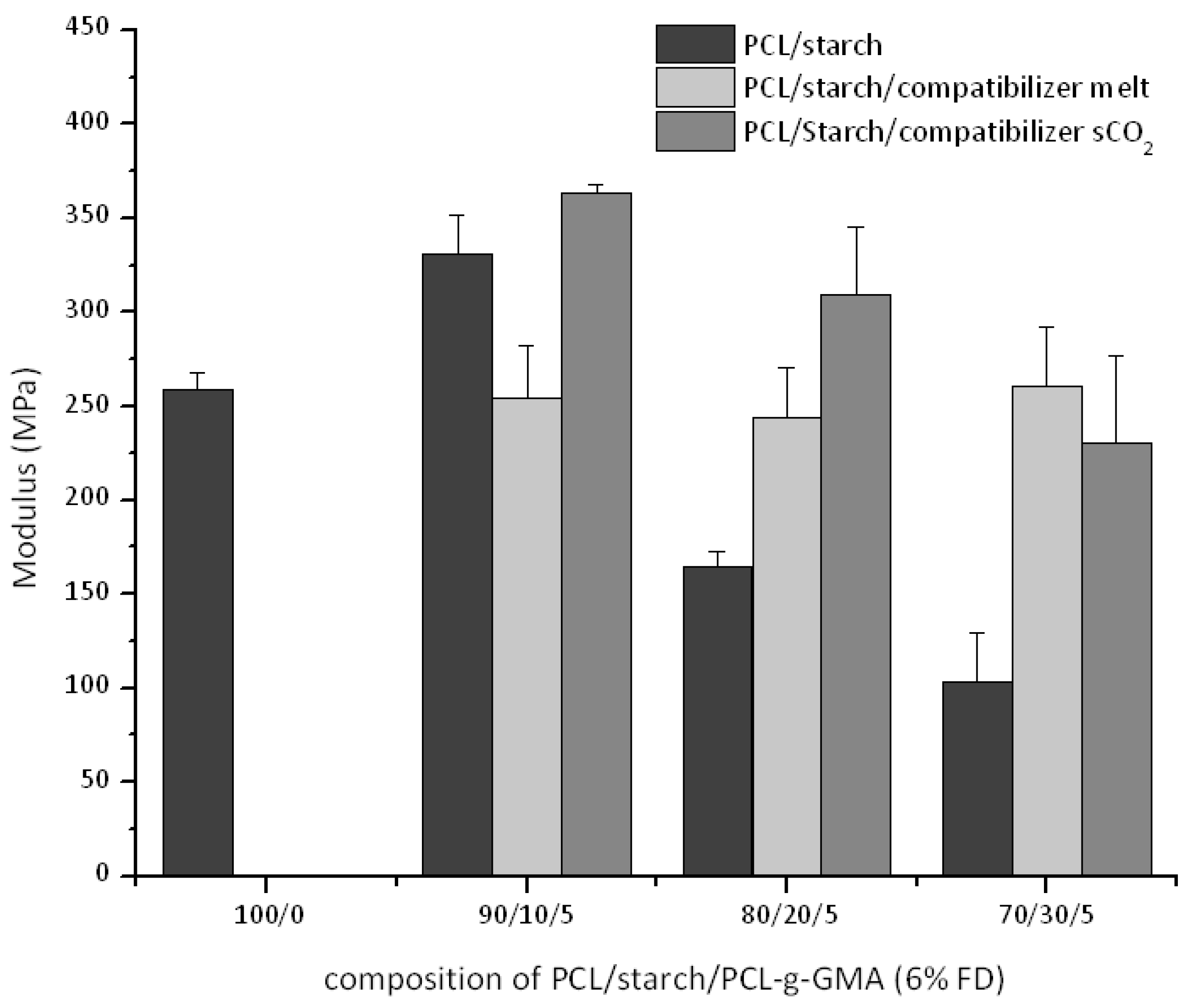
3.3. Thermal and Mechanical Properties for PCL-Starch Blends
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- PlasticEurope. Plastic—The Fact 2017: An Analysis of European Plastics Production, Demand and Waste Data; Plastic Europe: Brussels, Belgium, 2018. [Google Scholar]
- Flieger, M.; Kantorová, M.; Prell, A.; Řezanka, T.; Votruba, J. Biodegradable plastics from renewable sources. Folia Microbiol. 2003, 48, 27–44. [Google Scholar] [CrossRef]
- Averous, L.; Moro, L.; Dole, P.; Fringant, C. Properties of thermoplastic blends: Starch-polycaprolactone. Polymer 2000, 41, 4157–4167. [Google Scholar] [CrossRef]
- Sugih, A.K.; Picchioni, F.; Janssen, L.P.B.M.; Heeres, H.J. Synthesis of poly-(ε)-caprolactone grafted starch co-polymers by ring-opening polymerisation using silylated starch precursors. Carbohydr. Polym. 2009, 77, 267–275. [Google Scholar] [CrossRef]
- Matzinos, P.; Tserki, V.; Kontoyiannis, A.; Panayiotou, C. Processing and characterization of starch/polycaprolactone products. Polym. Degrad. Stab. 2002, 77, 17–24. [Google Scholar] [CrossRef]
- Avella, M.; Errico, M.E.; Laurienzo, P.; Martuscelli, E.; Raimo, M.; Rimedio, R. Preparation and characterisation of compatibilised polycaprolactone/starch composites. Polymer 2000, 41, 3875–3881. [Google Scholar] [CrossRef]
- Chen, L.; Ni, Y.; Bian, X.; Qiu, X.; Zhuang, X.; Chen, X.; Jing, X. A novel approach to grafting polymerization of ε-caprolactone onto starch granules. Carbohydr. Polym. 2005, 60, 103–109. [Google Scholar] [CrossRef]
- Dubois, P.; Krishnan, M.; Narayan, R. Aliphatic polyester-grafted starch-like polysaccharides by ring-opening polymerization. Polymer 1999, 40, 3091–3100. [Google Scholar] [CrossRef]
- Kim, C.H.; Jung, K.M.; Kim, J.S.; Park, J.K. Modification of aliphatic polyesters and their reactive blends with starch. J. Polym. Environ. 2004, 12, 179–187. [Google Scholar] [CrossRef]
- Pascente, C.; Márquez, L.; Balsamo, V.; Müller, A.J. Use of modified poly(ε-caprolactone) in the compatibilization of poly(ε-caprolactone)/maize starch blends. J. Appl. Polym. Sci. 2008, 109, 4089–4098. [Google Scholar] [CrossRef]
- Kim, C.H.; Cho, K.Y.; Park, J.K. Grafting of glycidyl methacrylate onto polycaprolactone: Preparation and characterization. Polymer 2001, 42, 5135–5142. [Google Scholar] [CrossRef]
- Kim, C.H.; Cho, K.Y.; Park, J.K. Reactive blends of gelatinized starch and polycaprolactone-g-glycidyl methacrylate. J. Appl. Polym. Sci. 2001, 81, 1507–1516. [Google Scholar] [CrossRef]
- Sugih, A.K.; Drijfhout, J.P.; Picchioni, F.; Janssen, L.P.B.M.; Heeres, H.J. Synthesis and properties of reactive interfacial agents for polycaprolactone-starch blends. J. Appl. Polym. Sci. 2009, 114, 2315–2326. [Google Scholar] [CrossRef]
- John, J.; Tang, J.; Yang, Z.; Bhattacharya, M. Synthesis and characterization of anhydride-functional polycaprolactone. J. Polym. Sci. Part A Polym. Chem. 1997, 35, 1139–1148. [Google Scholar] [CrossRef]
- Mani, R.; Bhattacharya, M.; Tang, J. Functionalization of polyesters with maleic anhydride by reactive extrusion. J. Polym. Sci. Part A Polym. Chem. 1999, 37, 1693–1702. [Google Scholar] [CrossRef]
- Wu, C.S. Physical properties and biodegradability of maleated-polycaprolactone/starch composite. Polym. Degrad. Stab. 2003, 80, 127–134. [Google Scholar] [CrossRef]
- Nalawade, S.P.; Picchioni, F.; Janssen, L.P.B.M. Supercritical carbon dioxide as a green solvent for processing polymer melts: Processing aspects and applications. Prog. Polym. Sci. 2006, 31, 19–43. [Google Scholar] [CrossRef]
- Kunita, M.H.; Rinaldi, A.W.; Girotto, E.M.; Radovanovic, E.; Muniz, E.C.; Rubira, A.F. Grafting of glycidyl methacrylate onto polypropylene using supercritical carbon dioxide. Eur. Polym. J. 2005, 41, 2176–2182. [Google Scholar] [CrossRef]
- Dorscht, B.M.; Tzoganakis, C. Reactive extrusion of polypropylene with supercritical carbon dioxide: Free radical grafting of maleic anhydride. J. Appl. Polym. Sci. 2003, 87, 1116–1122. [Google Scholar] [CrossRef]
- Dong, Z.; Liu, Z.; Han, B.; Pei, X.; Liu, L.; Yang, G. Modification of isotactic polypropylene films by grafting methyl acrylate using supercritical co2 as a swelling agent. J. Supercrit. Fluids 2004, 31, 67–74. [Google Scholar] [CrossRef]
- Liu, Z.; Song, L.; Dai, X.; Yang, G.; Han, B.; Xu, J. Grafting of methyl methylacrylate onto isotactic polypropylene film using supercritical CO2 as a swelling agent. Polymer 2002, 43, 1183–1188. [Google Scholar] [CrossRef]
- Tong, G.S.; Liu, T.; Hu, G.H.; Zhao, L.; Yuan, W.K. Supercritical carbon dioxide-assisted solid-state free radical grafting of methyl methacrylate onto polypropylene. J. Supercrit. Fluids 2007, 43, 64–73. [Google Scholar] [CrossRef]
- Nalawade, S.P.; Picchioni, F.; Marsman, J.H.; Janssen, L.P.B.M. The FT-IR studies of the interactions of CO2 and polymers having different chain groups. J. Supercrit. Fluids 2006, 36, 236–244. [Google Scholar] [CrossRef]
- Macosko, C.W.; Guégan, P.; Khandpur, A.K.; Nakayama, A.; Marechal, P.; Inoue, T. Compatibilizers for melt blending: Premade block copolymers. Int. J. Biol. Macromol. 1996, 29, 5590–5598. [Google Scholar] [CrossRef]
- Iqbal, M. Synthesis and Properties of Bio-Based and Renewable Polymeric Products; University of Groningen: Groningen, The Netherlands, 2014. [Google Scholar]
- Tong, G.S.; Liu, T.; Zhao, L.; Hu, L.; Yuan, W. Supercritical carbon dioxide-assisted preparation of polypropylene grafted acrylic acid with high grafted content and small gel percent. J. Supercrit. Fluids 2009, 48, 261–268. [Google Scholar]
- Montgomery, D.C. Design and Analysis of Experiment, 5th ed.; John Wiley & Sons Inc.: New York, NY, USA, 2001. [Google Scholar]
- Raquez, J.M.; Deléglise, M.; Lacrampe, M.F.; Krawczak, P. Thermosetting (bio)materials derived from renewable resources: A critical review. Prog. Polym. Sci. 2010, 35, 487–509. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Intake | PCL-g-GMA | ||
|---|---|---|---|---|
| GMA a | AIBN a | FD (%) | E (%) | |
| PCL-g-GMA 1 | 5 | 0.6 | 0.0 | 0.0 |
| PCL-g-GMA 2 | 5 | 1.2 | 2.5 | 46.2 |
| PCL-g-GMA 3 | 5 | 1.8 | 2.1 | 39.1 |
| PCL-g-GMA 4 | 10 | 0.6 | 4.1 | 36.6 |
| PCL-g-GMA 5 | 10 | 1.2 | 6.7 | 59.7 |
| PCL-g-GMA 6 | 10 | 1.8 | 7.2 | 63.5 |
| PCL-g-GMA 7 | 15 | 0.6 | 12.5 | 70.1 |
| PCL-g-GMA 8 | 15 | 1.2 | 11.8 | 65.7 |
| PCL-g-GMA 9 | 15 | 1.8 | 12.0 | 66.6 |
| PCL-g-GMA 10 | 5 | 2.4 | 2.2 | 40.4 |
| PCL-g-GMA 11 | 10 | 2.4 | 6.5 | 57.2 |
| PCL-g-GMA 12 | 15 | 2.4 | 12.7 | 68.1 |
| PCL-g-GMA 13 | 5 | 3 | 3.8 | 69.6 |
| PCL-g-GMA 14 | 10 | 3 | 8.1 | 70.1 |
| PCL-g-GMA 15 | 15 | 3 | 12.1 | 66.0 |
| PCL-g-GMA 16 | 8 | 1.2 | 6.0 | 68.1 |
| SS | DF | MS | F-Value | p-Value | ||
|---|---|---|---|---|---|---|
| Model | 262.794 | 5 | 52.559 | 86.961 | 6.079 × 10−7 | R2 = 0.978 |
| Error | 6.044 | 10 | 0.604 | R2(adj) = 0.969 | ||
| Total | 268.838 | 15 | R2(PRESS) = 0.933 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iqbal, M.; Mensen, C.; Qian, X.; Picchioni, F. Green Processes for Green Products: The Use of Supercritical CO2 as Green Solvent for Compatibilized Polymer Blends. Polymers 2018, 10, 1285. https://doi.org/10.3390/polym10111285
Iqbal M, Mensen C, Qian X, Picchioni F. Green Processes for Green Products: The Use of Supercritical CO2 as Green Solvent for Compatibilized Polymer Blends. Polymers. 2018; 10(11):1285. https://doi.org/10.3390/polym10111285
Chicago/Turabian StyleIqbal, Muhammad, Christiaan Mensen, Xiaohua Qian, and Francesco Picchioni. 2018. "Green Processes for Green Products: The Use of Supercritical CO2 as Green Solvent for Compatibilized Polymer Blends" Polymers 10, no. 11: 1285. https://doi.org/10.3390/polym10111285
APA StyleIqbal, M., Mensen, C., Qian, X., & Picchioni, F. (2018). Green Processes for Green Products: The Use of Supercritical CO2 as Green Solvent for Compatibilized Polymer Blends. Polymers, 10(11), 1285. https://doi.org/10.3390/polym10111285