Molecular Dynamics Simulations of Molecular Diffusion Equilibrium and Breakdown Mechanism of Oil-Impregnated Pressboard with Water Impurity



Abstract
:
1. Introduction
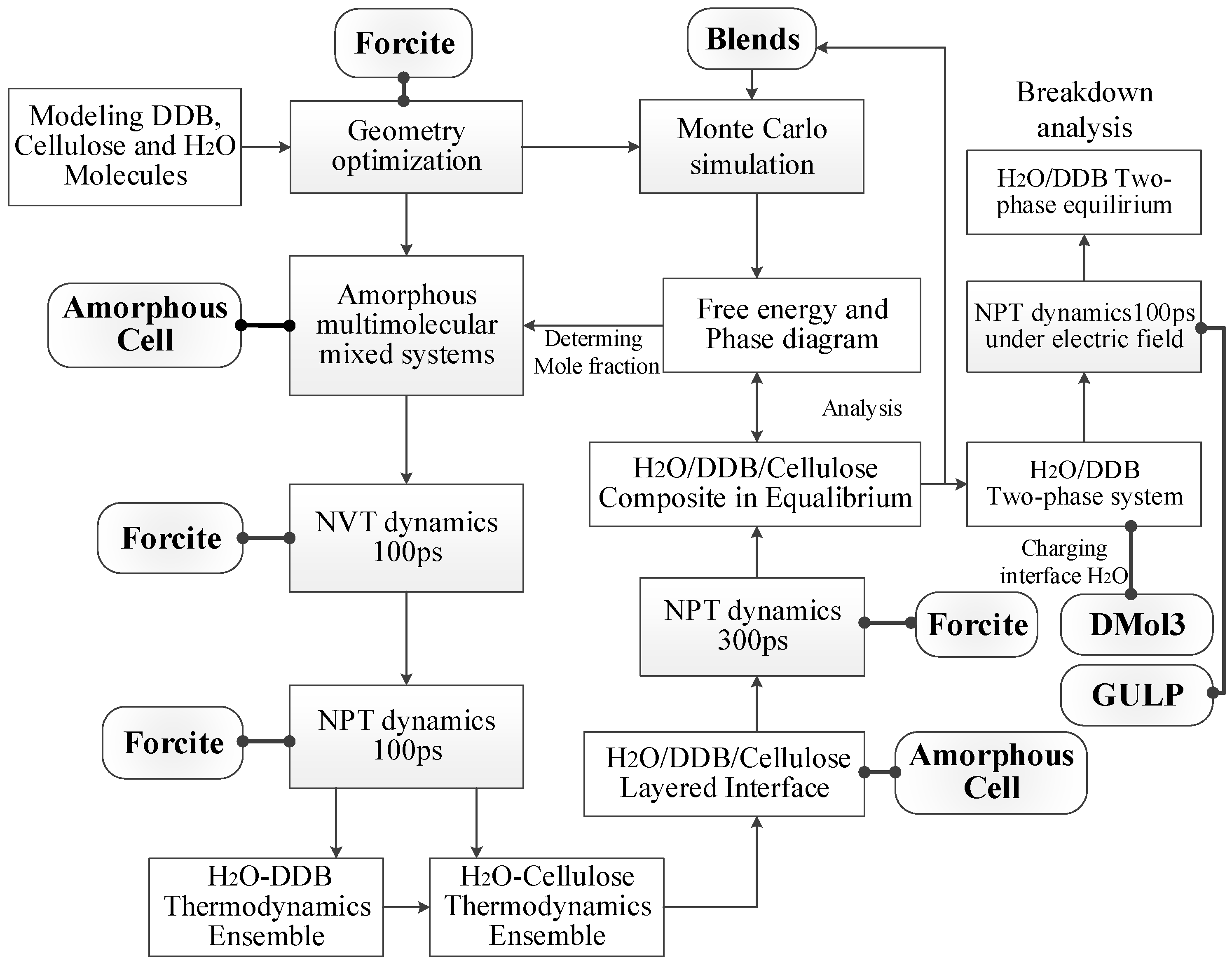
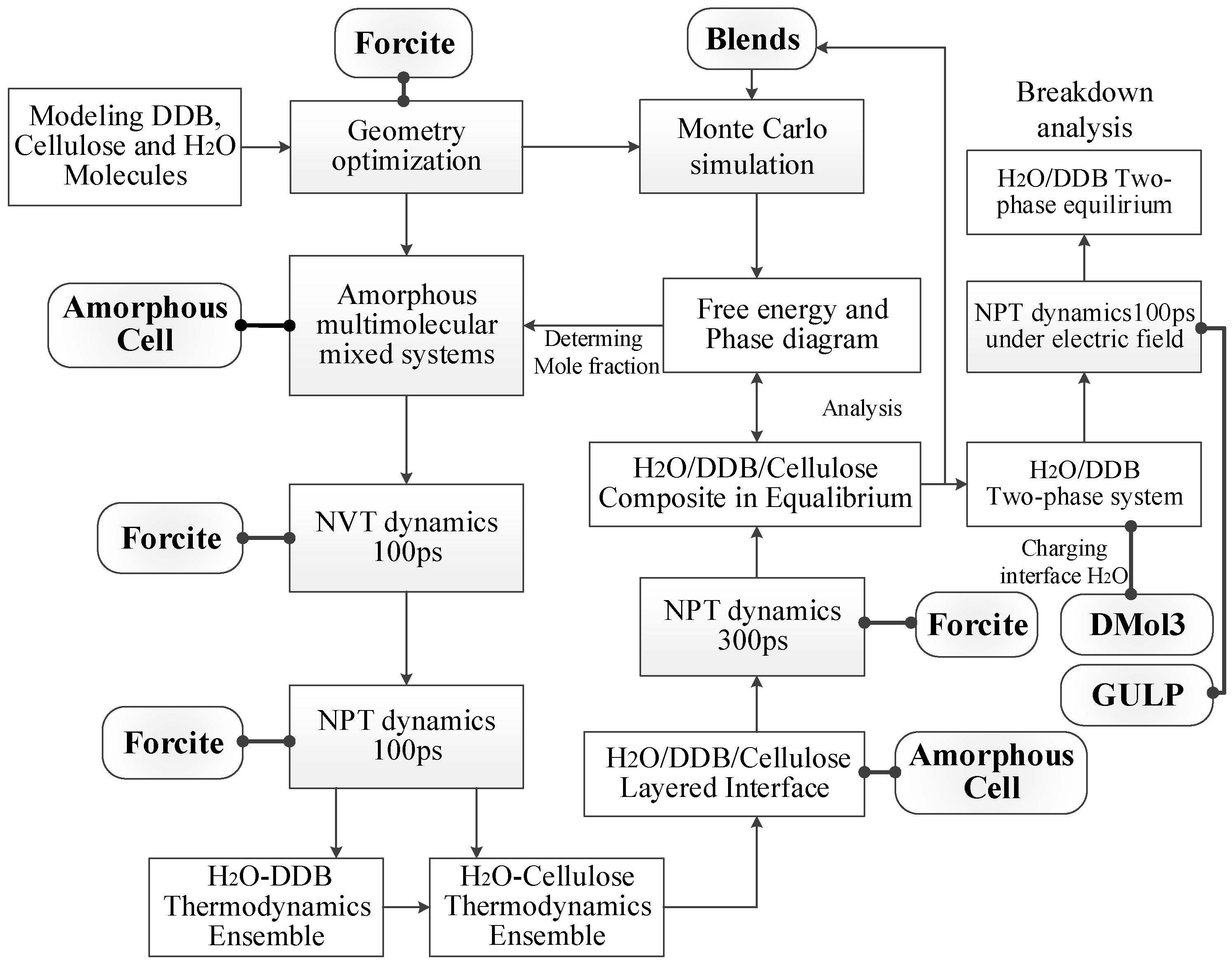
2. Theoretical Methodology
3. Results and Discussion
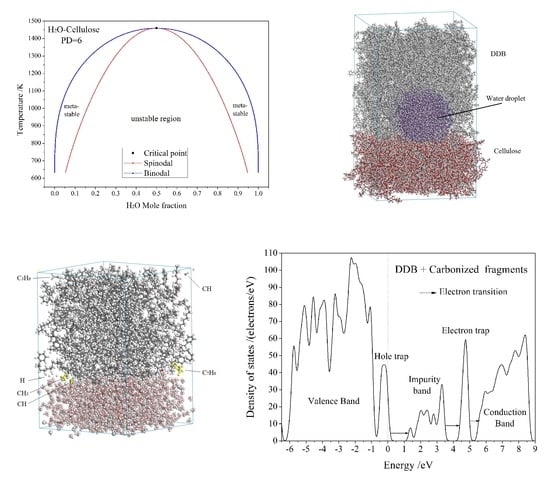
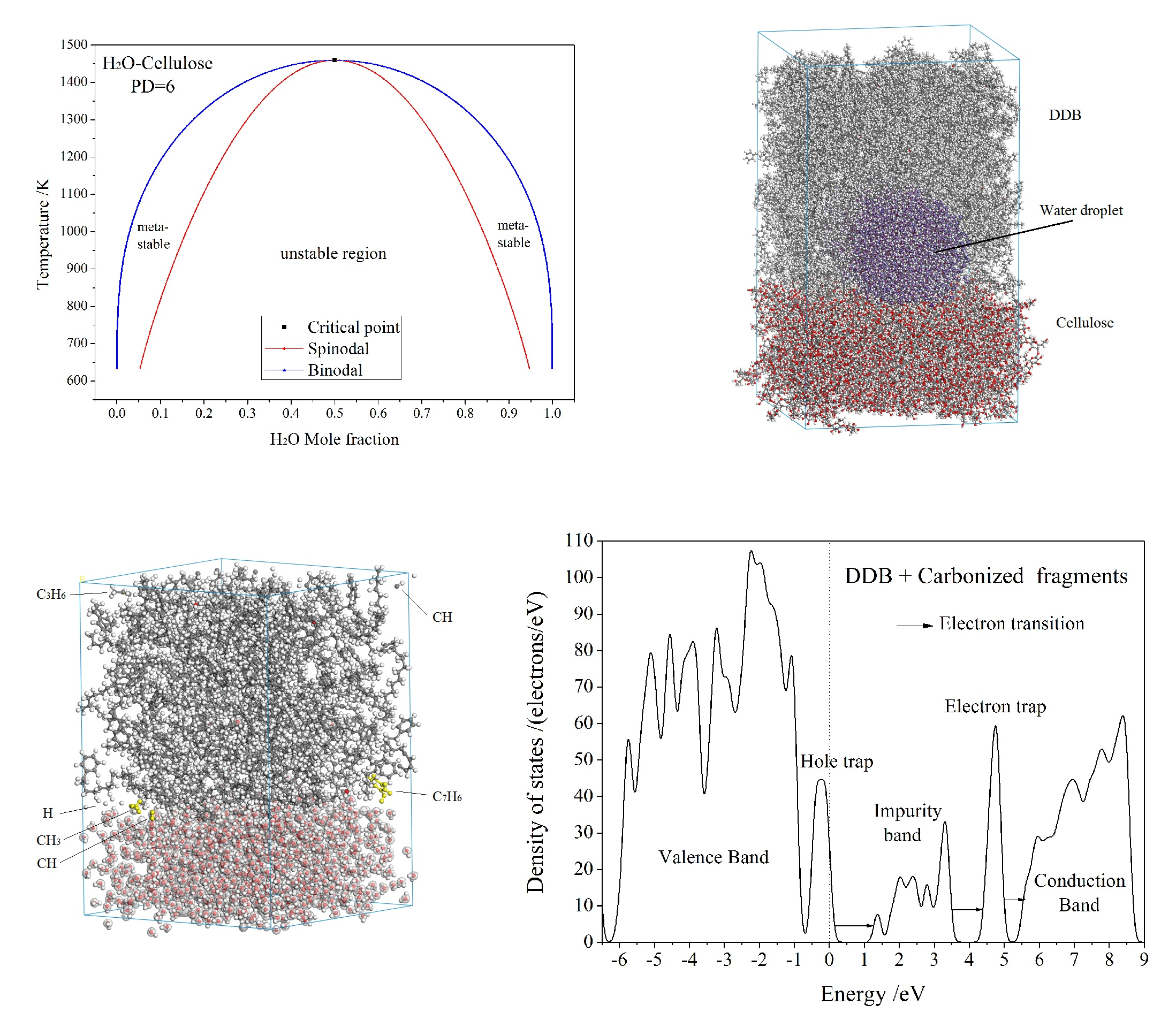
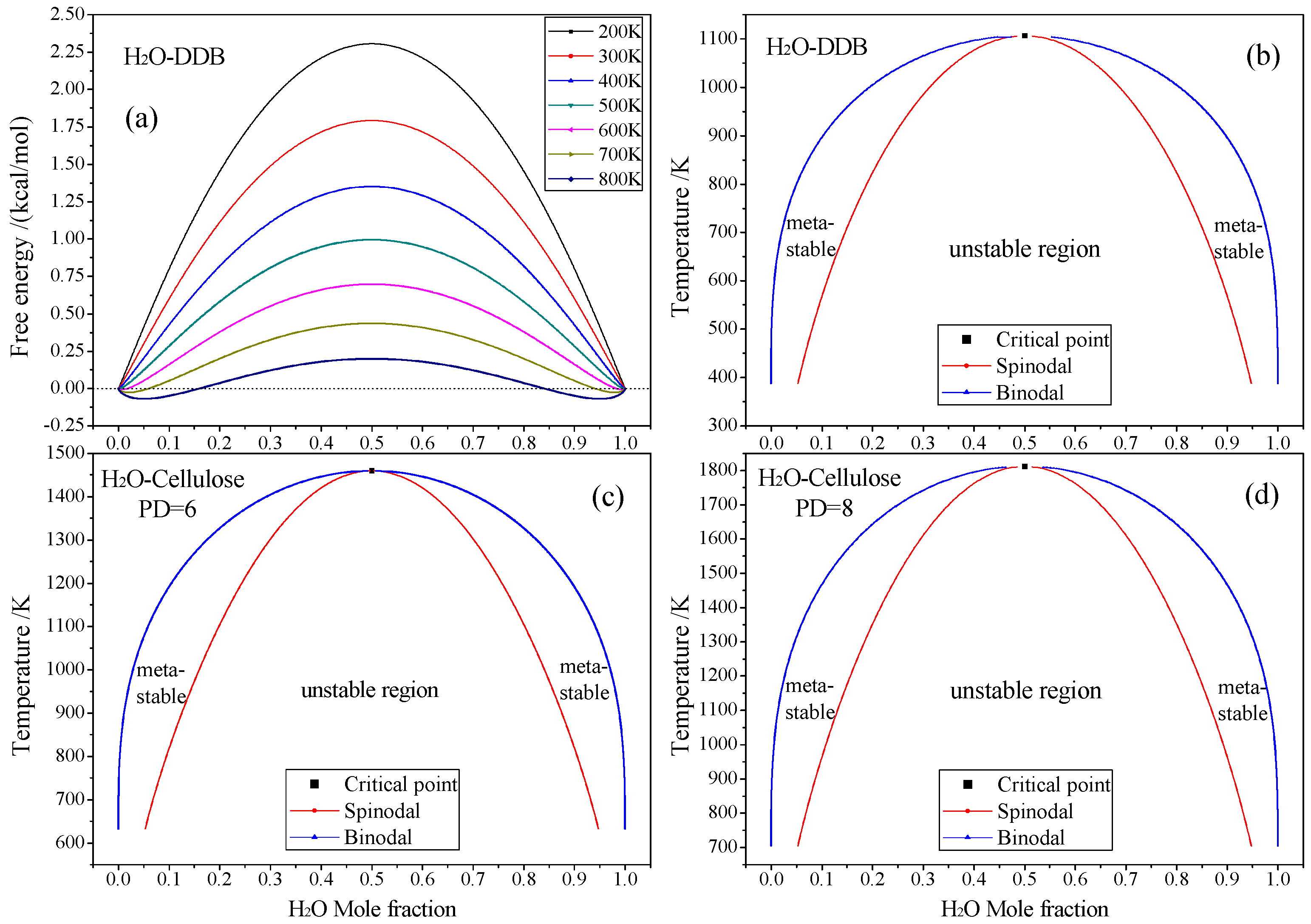
3.1. Free Energy and Phase Diagram
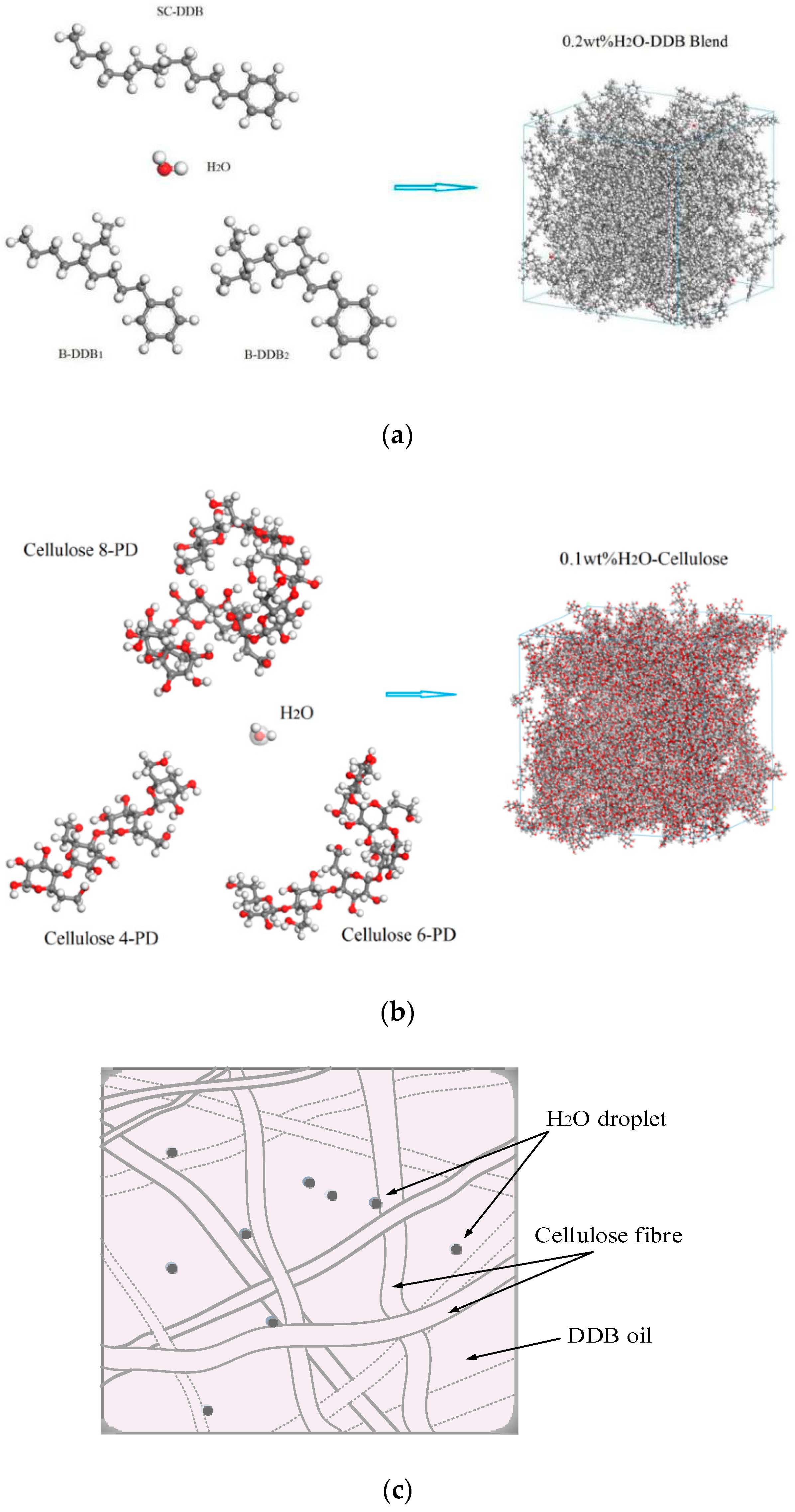
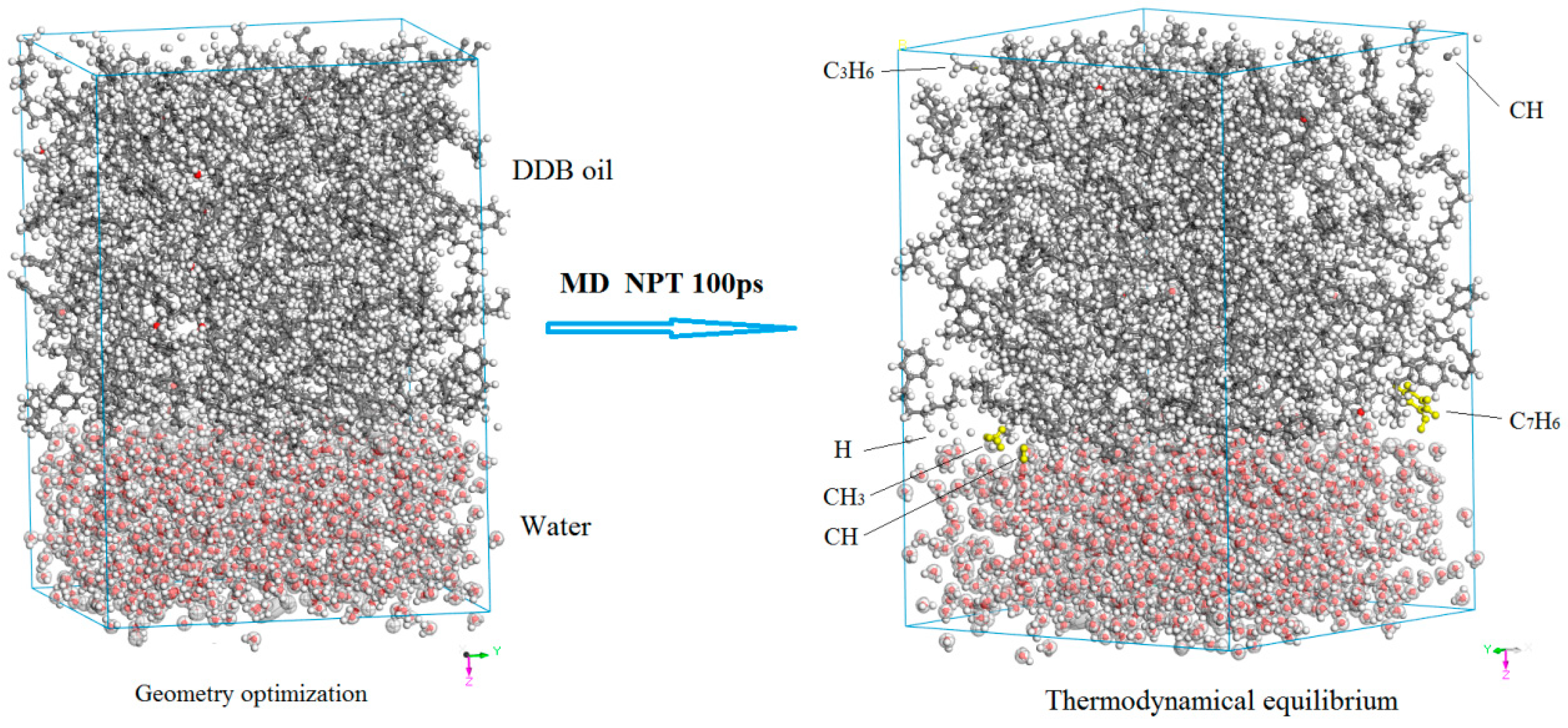
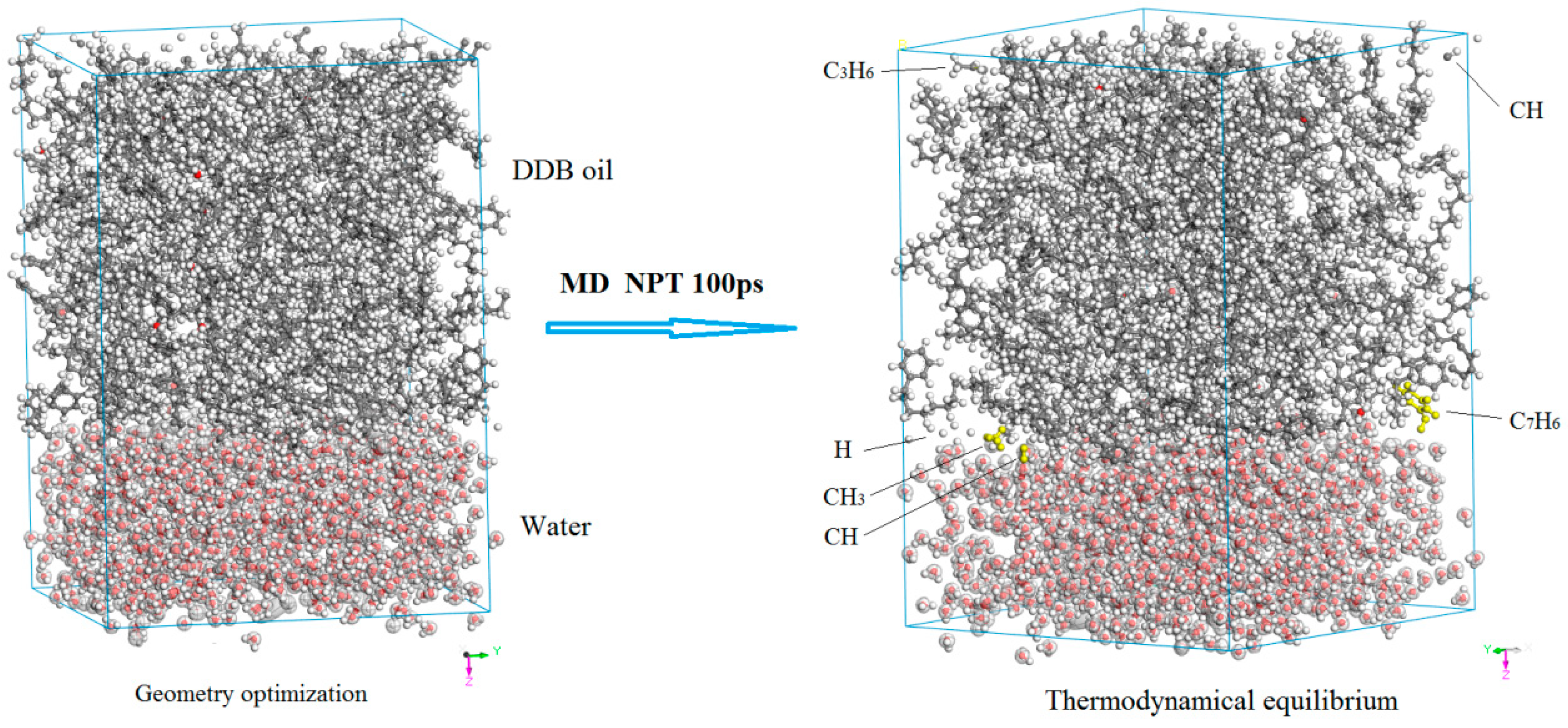
3.2. Diffusion and Phase Equilibrium
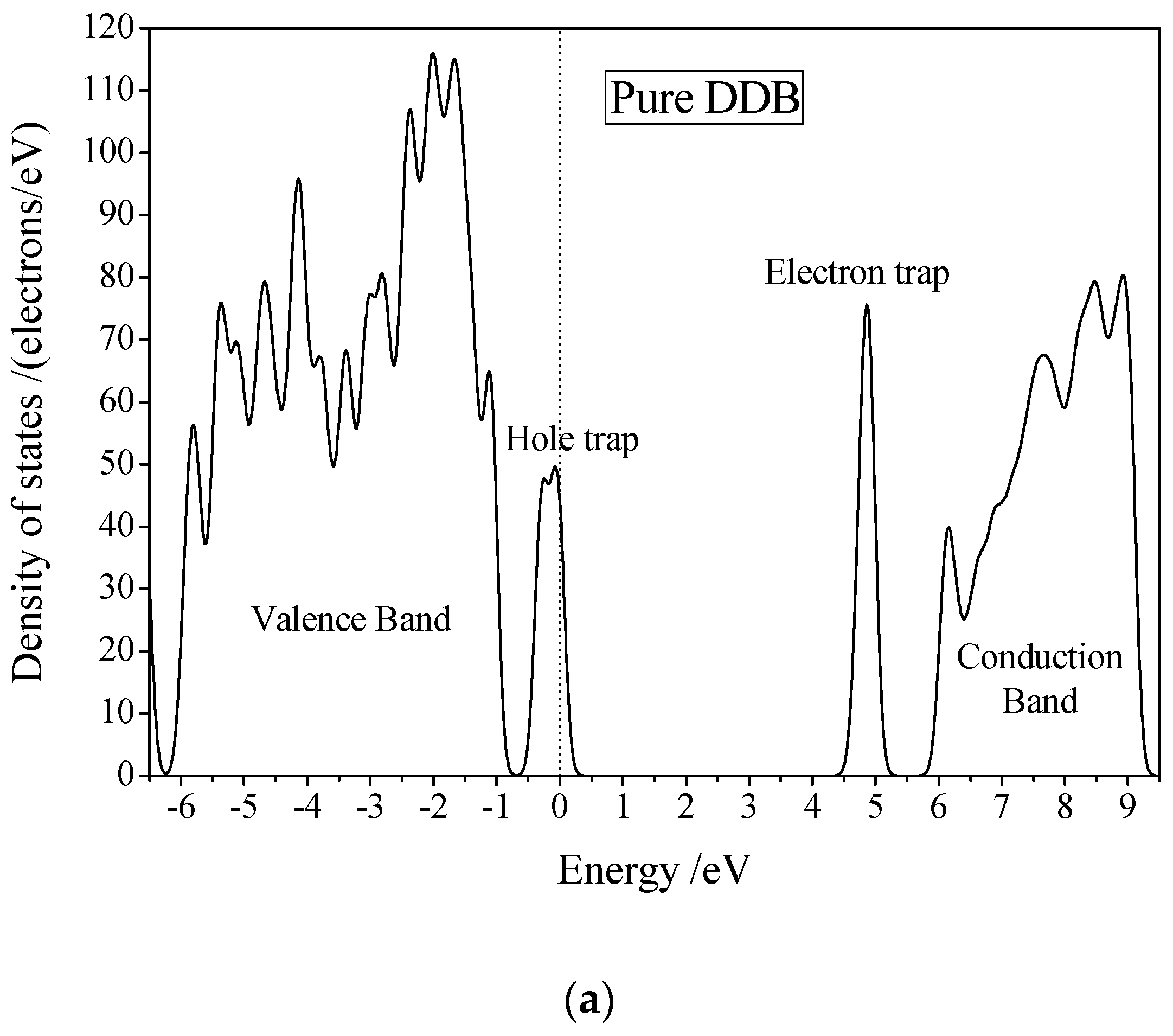
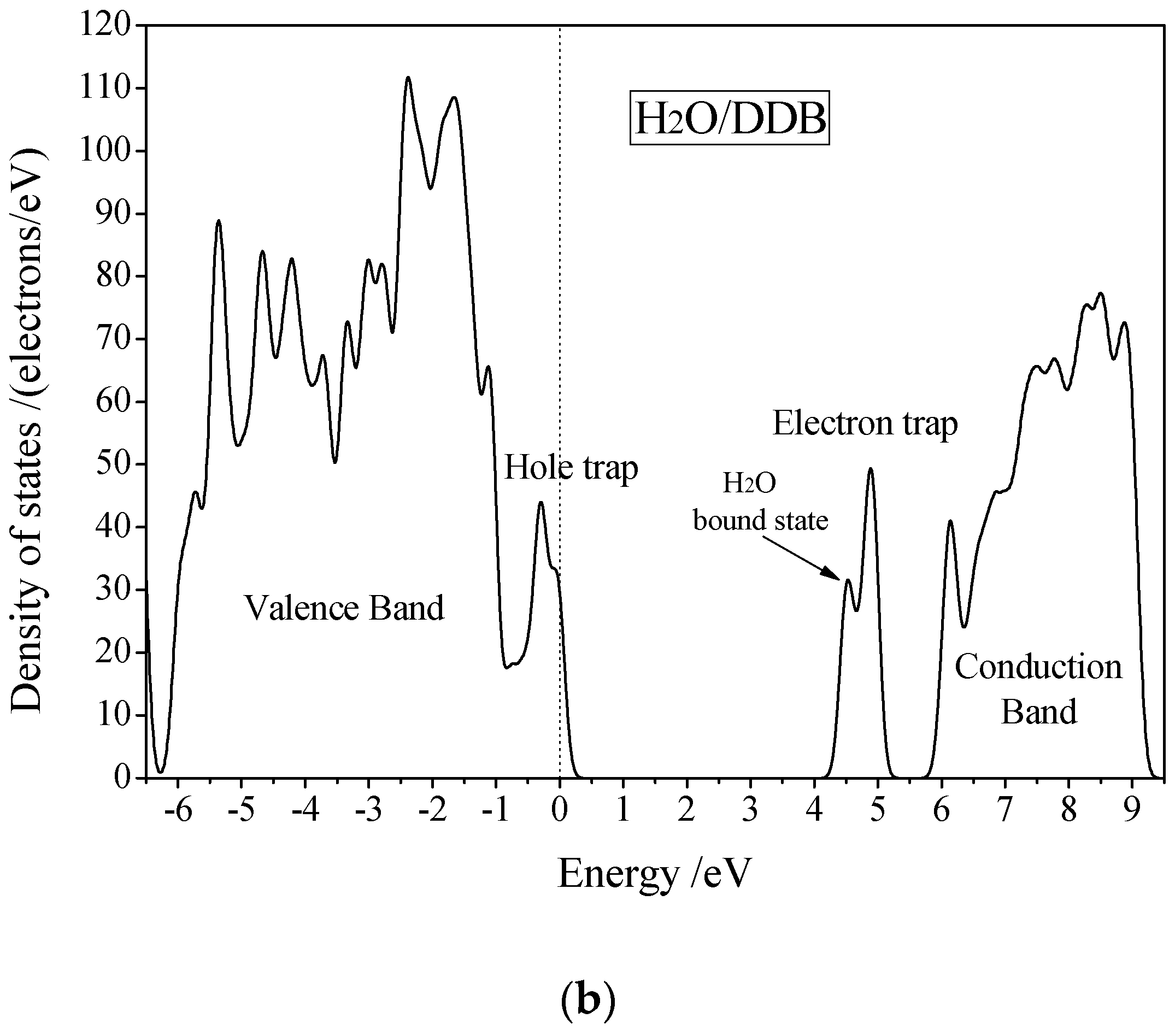
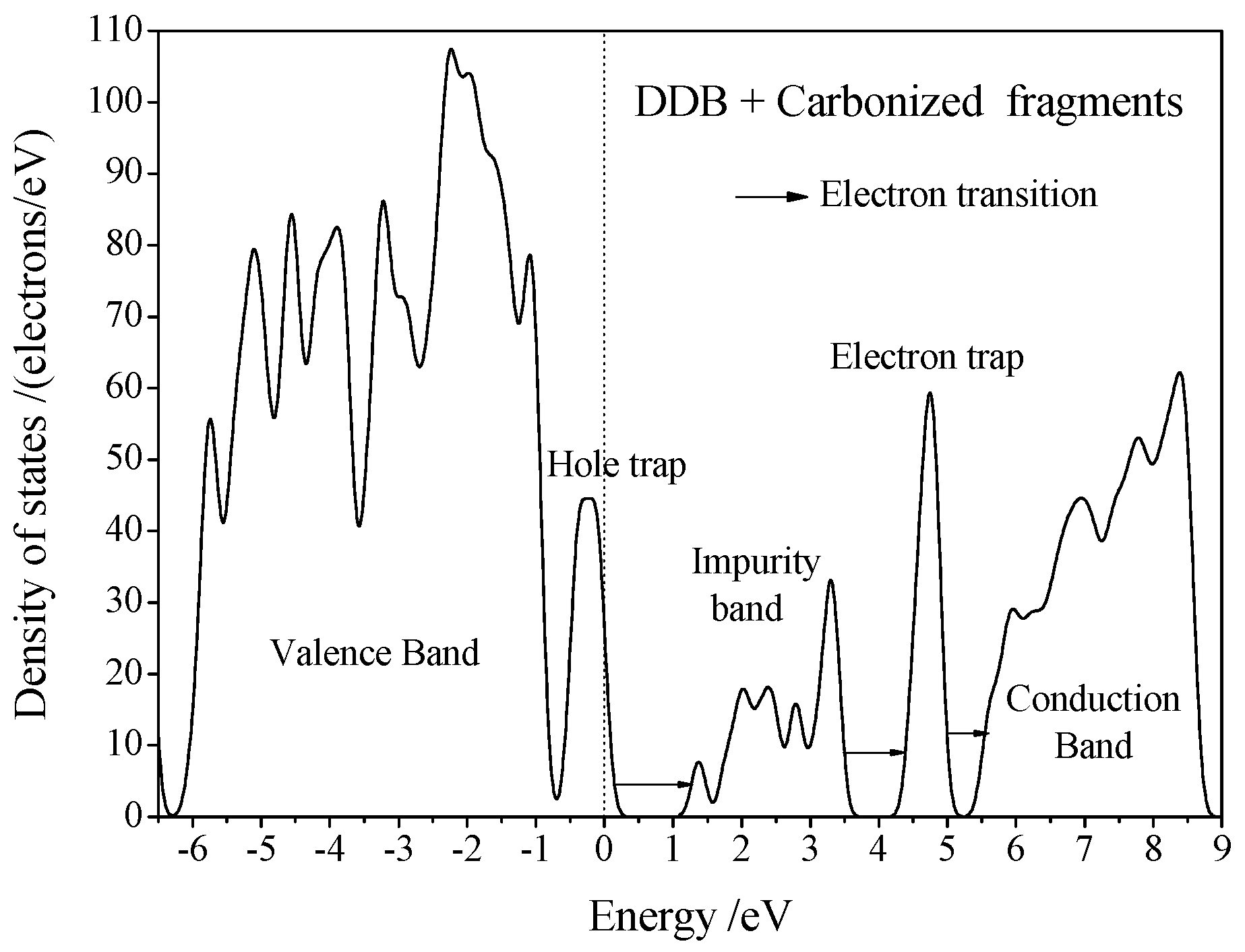
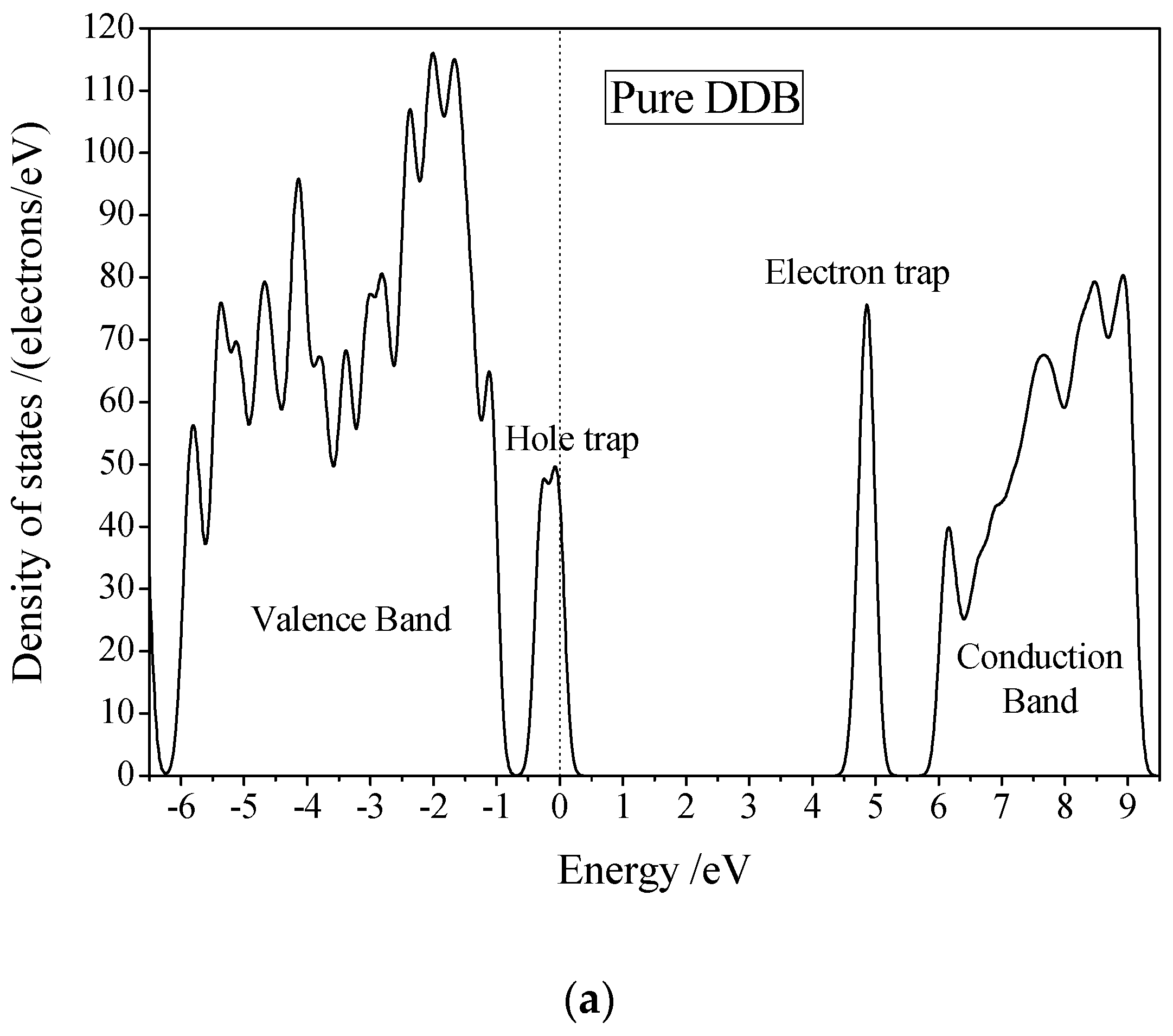
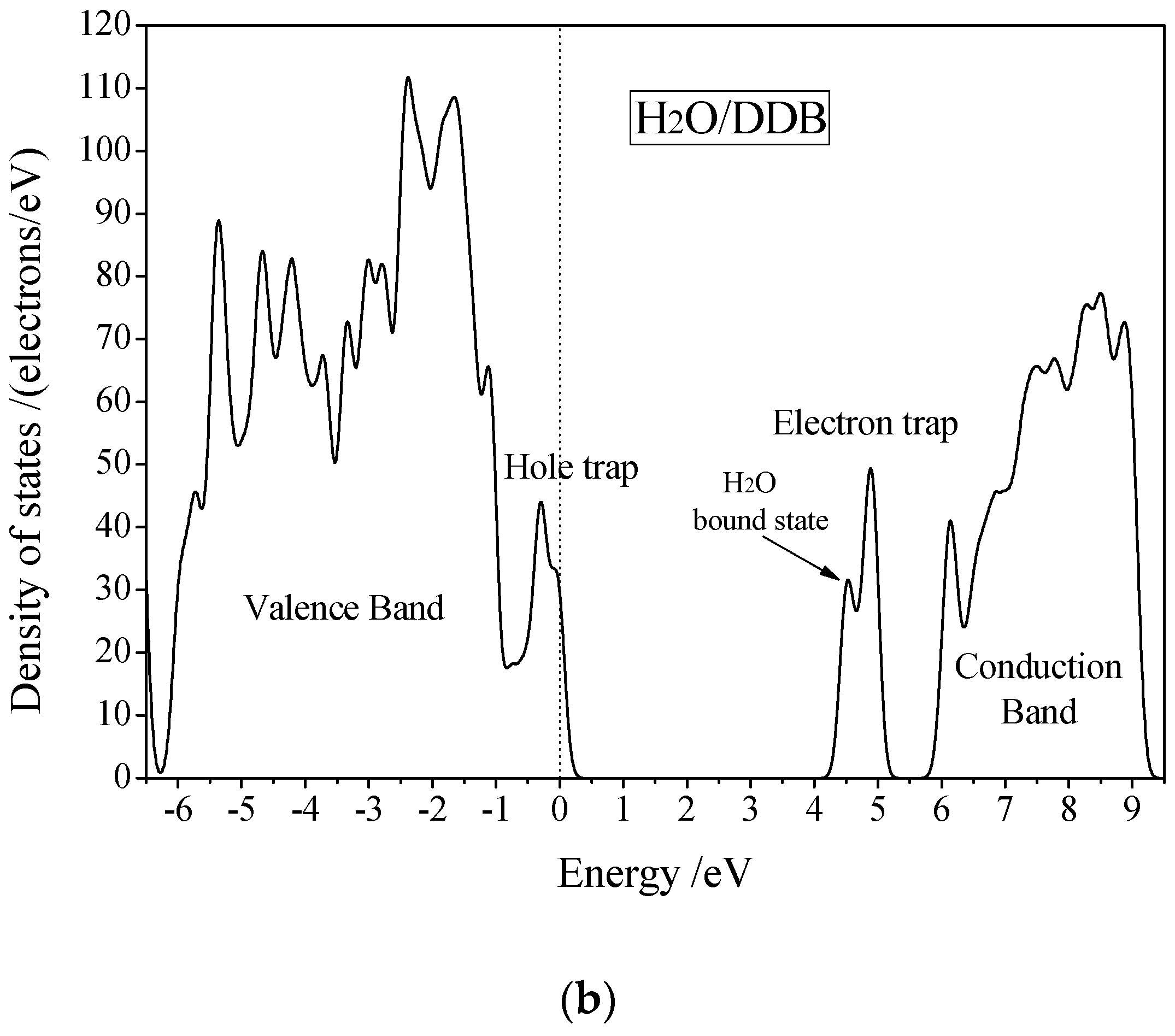
3.3. Electric Breakdown Process
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wang, Y.C.; Wang, F.P.; Li, J.; Liang, S.N.; Zhou, J.H. Electronic properties of typical molecules and the discharge mechanism of vegetable and mineral insulating oils. Energies 2018, 11, 523. [Google Scholar] [CrossRef]
- Yang, L.J.; Gao, S.H.; Deng, B.F.; Zhang, J.; Sun, W.D.; Hu, E.D. Inhibition method for the degradation of oil-paper insulation and corrosive sulphur in a transformer using adsorption treatment. IET Gener. Transm. Distrib. 2016, 10, 1893–1900. [Google Scholar] [CrossRef]
- Bolliger, D.; Pilania, G.; Boggs, S. The effect of oxidative and paper degradation impurities on partial discharge characteristics of hexadecane. IEEE Trans. Dielectr. Electr. Insul. 2013, 20, 1669–1682. [Google Scholar] [CrossRef]
- Hosier, I.L.; Koilraj, J.E.A.; Vaughan, A.S. Effect of aging on the physical, chemical and dielectric properties of dodecylbenzene. IEEE Trans. Dielectr. Electr. Insul. 2016, 23, 3389–3396. [Google Scholar] [CrossRef]
- Velusamy, S.; Punniyamurthy, T. Copper(II)-catalyzed C-H oxidation of alkylbenzenes and cyclohexane with hydrogen peroxide. Tetrahedron Lett. 2003, 44, 8955–8957. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Q.G.; Li, J.Z.; Wang, T.L.; Dong, W.L.; Ni, H.L. Study on micro bridge impurities in oil-paper insulation at DC voltage: Their generation, growth and interaction with partial discharge. IEEE Trans. Dielectr. Electr. Insul. 2016, 23, 2213–2222. [Google Scholar] [CrossRef]
- Zhang, D.N.; Zhao, H.X.; Yun, H.; Liu, X.W.; Han, Y.H.; Mu, H.B.; Zhang, G.J. Study on FDS characteristics of oil-immersed paper insulation bushing under non-uniform moisture content. IET Sci. Meas. Technol. 2018, 12, 691–697. [Google Scholar] [CrossRef]
- Rao, U.M.; Kumar, Y.N.; Jarial, R.K. Understanding the ageing behaviour of transformer oil-paper insulation with ester and mixed dielectric fluids. IET Sci. Meas. Technol. 2018, 12, 851–857. [Google Scholar] [CrossRef]
- Sun, W.F.; Wang, X. Molecular dynamics simulation study of polyimide/copper-nanoparticle composites. Acta Phys. Sin. 2013, 62, 186202. [Google Scholar]
- Rigby, D.; Roe, R.J. Molecular dynamics simulation of polymer liquid and glass. II. short range order and orientation correlation. J. Chem. Phys. 1988, 89, 5280–5290. [Google Scholar] [CrossRef]
- Forrest, B.M.; Suter, U.W. Hybrid monte carlo simulations of dense polymer systems. J. Chem. Phys. 1994, 101, 2616–2629. [Google Scholar] [CrossRef]
- Flory, P.J. Statistical Mechanics of Chain Molecules; Interscience: New York, NY, USA, 1969. [Google Scholar]
- Flory, P.J. Foundations of rotational isomeric state theory and general methods for generating configurational averages. Macromolecules 1974, 7, 381. [Google Scholar] [CrossRef]
- Mazeau, K.; Heux, K. Molecular dynamics simulations of bulk native crystalline and amorphous structures of cellulose. J. Phys. Chem. B 2003, 107, 2394–2403. [Google Scholar] [CrossRef]
- Blanco, M. Molecular silverware. I. general solutions to excluded volume constrained problems. J. Comput. Chem. 1991, 12, 237–247. [Google Scholar] [CrossRef]
- Fan, C.F.; Olafson, B.D.; Blanco, M.; Hsu, S.L. Application of molecular simulation to derive phase diagrams of binary mixtures. Macromolecules 1992, 25, 3667–3676. [Google Scholar] [CrossRef]
- Sun, H.; Mumby, S.J.; Maple, J.R.; Hagler, A.T. An ab initio CFF93 all-atom force field for polycarbonates. J. Am. Chem. Soc. 1994, 116, 2978–2987. [Google Scholar] [CrossRef]
- Samoletov, A.A.; Dettmann, C.P.; Chaplain, M.A.J. Thermostats for “slow” configurational modes. J. Stat. Phys. 2007, 128, 1321. [Google Scholar] [CrossRef]
- Souza, I.; Martins, J.L. Metric tensor as the dynamical variable for variable-cell-shape molecular dynamics. Phys. Rev. B 1997, 55, 8733. [Google Scholar] [CrossRef]
- Perdew, J.P.; Ruzsinszky, A.; Csonka, G.I.; Vydrov, O.A.; Scuseria, G.E.; Constantin, L.A.; Zhou, X.; Burke, K. Restoring the density-gradient expansion for exchange in solids and surfaces. Phys. Rev. Lett. 2008, 100, 136406. [Google Scholar] [CrossRef] [PubMed]
- Van Duin, A.C.T.; Dasgupta, S.; Lorant, F.; Goddard, W.A. ReaxFF: A reactive force field for hydrocarbons. J. Phys. Chem. A 2001, 105, 9396–9409. [Google Scholar] [CrossRef]



{kind=link}
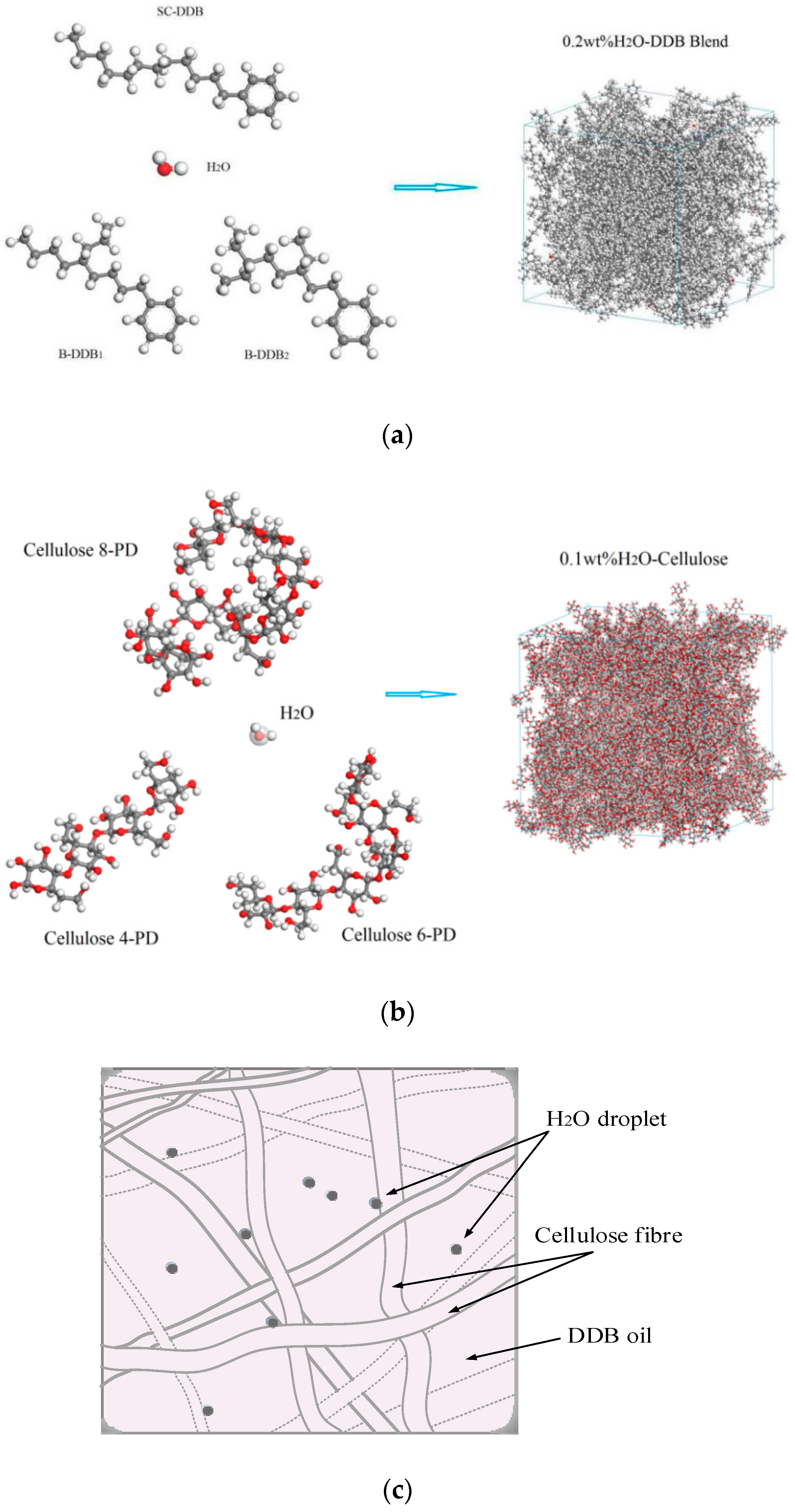{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Topics | Setting up | Methodology | Condition and Parameters |
|---|---|---|---|
| Energy | Forcefield | PCFF | |
| Electrostatic and van der Waals | Atom based summation | Cutoff distance: 18.50 Å Buffer width: 0.5 Å | |
| Geometry optimization | Optimization algorithm | Smart | Convergence:1 × 10−5 kcal/mol Maximum iterations: 2 × 104 |
| Ensemble | NVT | Constant volume/constant temperature | |
| Molecular dynamics | Ensemble | NPT | Constant pressure/constant temperature |
| Thermostatic control | Nosé-Hoover-Langevin (NHL) algorithm [18] | Temperature: 300 K, 400 K | |
| Barostatic control | Souza-Martins algorithm [19] | Pressure: 1 × 105 Pa | |
| Time integration | Total simulation time | 100.0 ps | |
| Time step and number | 0.5 fs and 2 × 105 | ||
| Simulation step control | Integration tolerance | Energy deviation: 5000.0 kcal/mol |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guan, Y.; Chi, M.-H.; Sun, W.-F.; Chen, Q.-G.; Wei, X.-L. Molecular Dynamics Simulations of Molecular Diffusion Equilibrium and Breakdown Mechanism of Oil-Impregnated Pressboard with Water Impurity. Polymers 2018, 10, 1274. https://doi.org/10.3390/polym10111274
Guan Y, Chi M-H, Sun W-F, Chen Q-G, Wei X-L. Molecular Dynamics Simulations of Molecular Diffusion Equilibrium and Breakdown Mechanism of Oil-Impregnated Pressboard with Water Impurity. Polymers. 2018; 10(11):1274. https://doi.org/10.3390/polym10111274
Chicago/Turabian StyleGuan, Yi, Ming-He Chi, Wei-Feng Sun, Qing-Guo Chen, and Xin-Lao Wei. 2018. "Molecular Dynamics Simulations of Molecular Diffusion Equilibrium and Breakdown Mechanism of Oil-Impregnated Pressboard with Water Impurity" Polymers 10, no. 11: 1274. https://doi.org/10.3390/polym10111274
APA StyleGuan, Y., Chi, M.-H., Sun, W.-F., Chen, Q.-G., & Wei, X.-L. (2018). Molecular Dynamics Simulations of Molecular Diffusion Equilibrium and Breakdown Mechanism of Oil-Impregnated Pressboard with Water Impurity. Polymers, 10(11), 1274. https://doi.org/10.3390/polym10111274