α,ω-Epoxide, Oxetane, and Dithiocarbonate Telechelic Copolyolefins: Access by Ring-Opening Metathesis/Cross-Metathesis Polymerization (ROMP/CM) of Cycloolefins in the Presence of Functional Symmetric Chain-Transfer Agents


 and
and 
Abstract
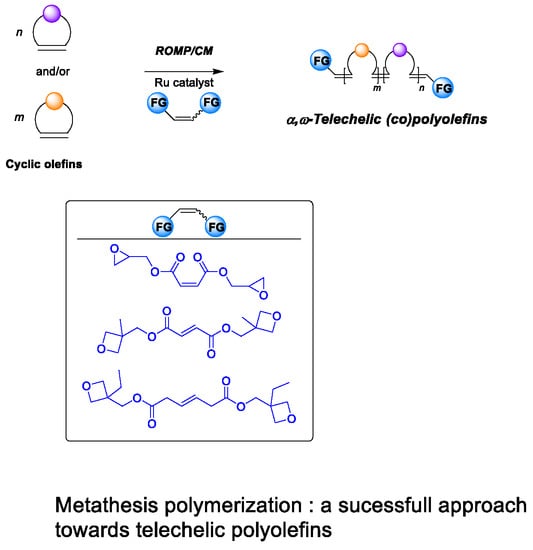
1. Introduction
2. Experimental Section
2.1. Materials
2.2. Instrumentation and Measurements
2.3. Bis((3-methyloxetan-3-yl)methyl) Fumarate (CTA 2)
2.4. Bis((3-ethyloxetan-3-yl)methyl) (E)-Hex-3-enedioate (CTA 3)
2.5. General Procedure for the Polymerization of COE
2.6. General Procedure for the Copolymerization of Cyclic Olefins
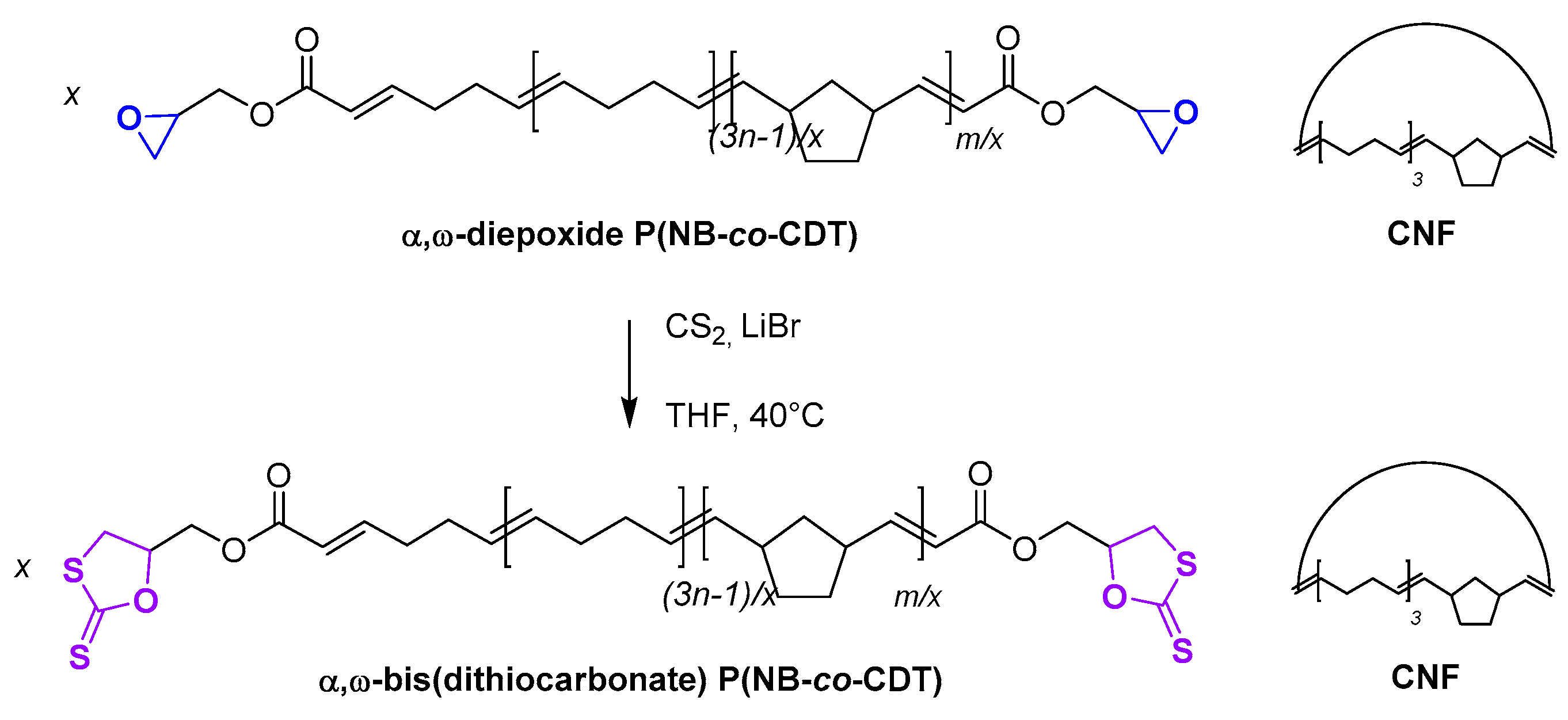
2.7. Chemical Modification of α,ω-Diepoxide Telechelic P(NB-co-CDT) into α,ω-Bis(dithiocarbonate) Telechelic P(NB-co-CDT)
2.8. Quantification of Cyclic Nonfunctional (CNF) Copolymers within the Recovered Chemically Modified P(NB-co-CDT) Crude Copolymers
3. Results and Discussion
3.1. Synthesis of CTAs 1–3
3.2. Metathesis of COE in the Presence of CTAs 1–3
3.3. Ring-Opening Metathesis Copolymerization of Cyclic Olefins in the Presence of CTAs 1 or 3
3.4. α,ω-Diepoxide Telechelic P(COE-co-NB) and P(COE-co-NBCOOMe) Copolymers
3.5. α,ω-Diepoxide and -Dioxetane Telechelic P(NB-co-CDT) Copolymers
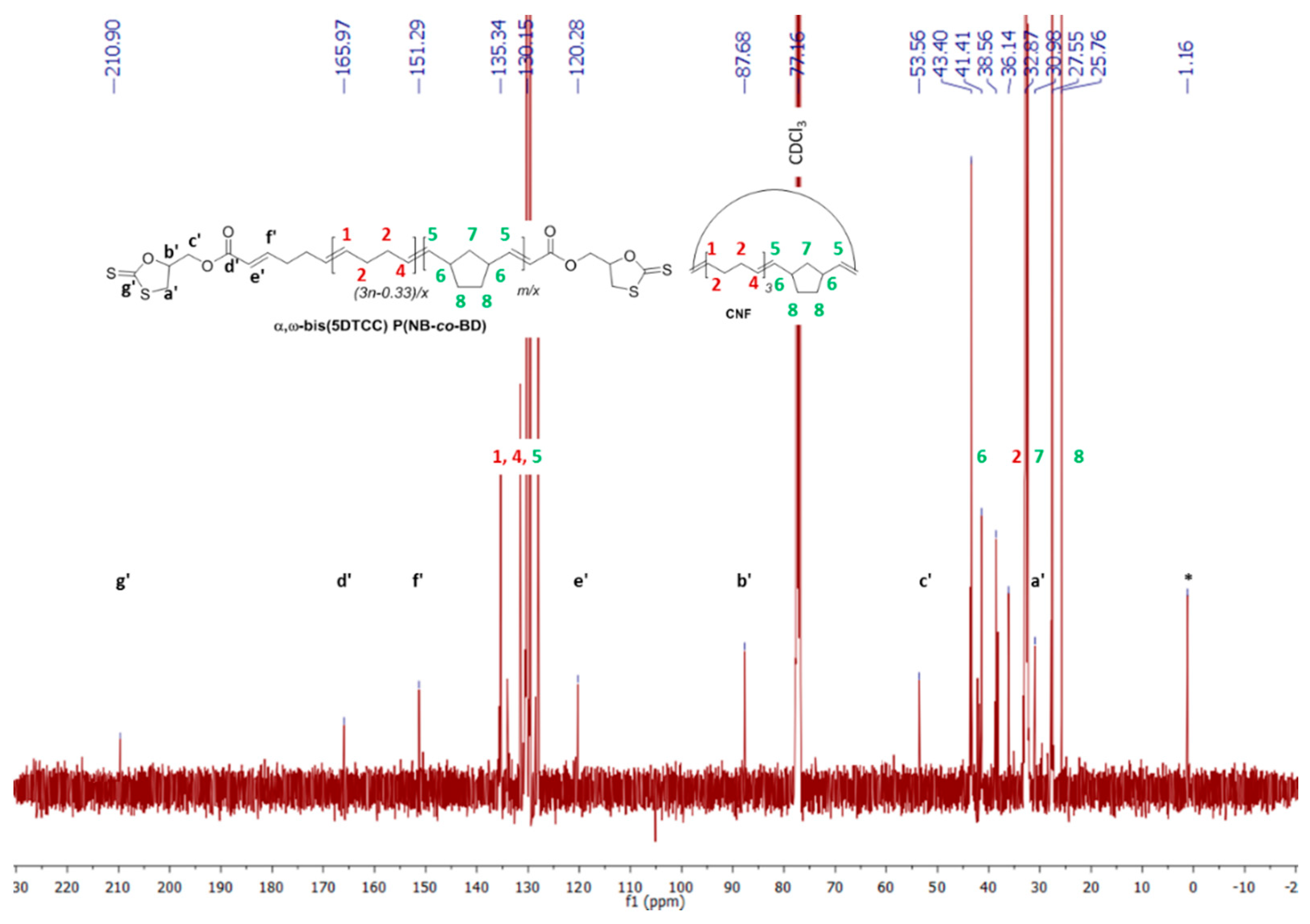
3.6. Post-Polymerization Chemical Modification of α,ω-Di(epoxide) Telechelic P(NB-co-CDT) into α,ω-Dithiocarbonate Telechelic P(NB-co-CDT)
3.7. Thermal Properties of the Polymers
3.8. Viscosity of the Copolymers
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Goethals, E.J. Telechelic Polymers: Synthesis and Applications; CRC Press: Boca Raton, FL, USA, 1989. [Google Scholar]
- Odian, G. Principles of Polymerization, 4th ed.; Wiley-Interscience: New York, NY, USA, 2004. [Google Scholar]
- Hanik, N.; Kilbinger, A.F.M. Telechelic Polymers. In Handbook of Metathesis, 2nd ed.; Grubbs, R.H., Wenzel, A.G., O’Leary, D.J., Khosravi, E., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2015; Volume 3. [Google Scholar]
- Guillaume, S.M. Handbook of Telechelic Polyesters, Polycarbonates and Polyethers; Pan Stanford Publishing Pte. Ltd.: Singapore, 2017. [Google Scholar]
- Chung, T.C. Functionalization of Polyolefins; Academic Press: London, UK, 2002. [Google Scholar]
- Yanjarappa, M.J.; Sivaram, S. Recent developments in the synthesis of functional poly (olefin)s. Prog. Polym. Sci. 2002, 27, 1347–1398. [Google Scholar] [CrossRef]
- Boaen, N.K.; Hillmyer, M.A. Post-polymerization functionalization of polyolefins. Chem. Soc. Rev. 2005, 34, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Amin, S.B.; Marks, T.J. Versatile pathways for in situ polyolefin functionalization with heteroatoms: catalytic chain transfer. Angew. Chem. Int. Ed. 2008, 47, 2006–2025. [Google Scholar] [CrossRef] [PubMed]
- Hilf, S.; Kilbinger, A.F.M. Functional end groups for polymers prepared using ring-opening metathesis polymerization. Nat. Chem. 2009, 1, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Franssen, N.M.G.; Reek, J.N.H.; de Bruin, B. Synthesis of functional ‘polyolefins’: State of the art and remaining challenges. Chem. Soc. Rev. 2013, 42, 5809–5832. [Google Scholar] [CrossRef] [PubMed]
- Bielawski, C.W.; Hillmyer, M.A. Telechelic Polymers from Olefins Meztathesis Methodologies. In Handbook of Metathesis, 2nd ed.; Grubbs, R.H., Wenzel, A.G., O’Leary, D.J., Khosravi, E., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2015; Volume 3. [Google Scholar]
- Hutley, T.J.; Ouederni, M. Polyolefin Compounds and Materials—Fundamentals and Industrial Applications; AlMa’adeed, M.A.A., Krupa, I., Eds.; Springer: Heidelberg, Germany, 2016. [Google Scholar]
- Guillaume, S.M. Advances in the synthesis of silyl-modified polymers (SMPs). Polym. Chem. 2018, 9, 1911–1926. [Google Scholar] [CrossRef]
- Wang, Y.; Hillmyer, M. Hydroxy-telechelic poly (ethylene-co-isobutylene) as a soft segment for thermoplastic polyurethanes. Polym. Chem. 2015, 6, 6806–6811. [Google Scholar] [CrossRef]
- Martinez, H.; Hillmyer, M. Carboxy-telechelic polyolefins in cross-linked elastomers. Macromolecules 2014, 47, 479–485. [Google Scholar] [CrossRef]
- Amendt, M.A.; Pitet, L.M.; Moench, S.; Hillmyer, M. Reactive triblock polymers from tandem ring-opening polymerization for nanostructured vinyl thermosets. Polym. Chem. 2012, 3, 1827–1837. [Google Scholar] [CrossRef]
- LPitet, M.; Hillmyer, M. Carboxy-telechelic polyolefins by ROMP using maleic acid as a chain transfer agent. Macromolecules 2011, 44, 2378–2381. [Google Scholar]
- Pitet, L.M.; Chamberlain, M.M.; Hauser, A.W.; Hillmyer, M. Synthesis of linear, H-shaped, and arachnearm block copolymers by tandem ring-opening polymerizations. Macromolecules 2010, 43, 8018–8025. [Google Scholar] [CrossRef]
- Thomas, M.R.; Grubbs, R.H. Synthesis of telechelic polyisoprene via ring-opening metathesis polymerization in the presence of chain transfer agent. Macromolecules 2010, 43, 3705–3709. [Google Scholar] [CrossRef]
- Pitet, L.M.; Hillmyer, M. Combining Ring-Opening Metathesis Polymerization and Cyclic Ester Ring-Opening Polymerization To Form ABA Triblock Copolymers from 1,5-Cyclooctadiene and d,l-Lactide. Macromolecules 2009, 42, 3674–3680. [Google Scholar] [CrossRef]
- Ji, S.; Hoye, T.R.; Macosko, C.W. Diamino telechelic polybutadienes for solventless styrene–butadiene–styrene (SBS) triblock copolymer formation. Polymer 2008, 49, 5307–5313. [Google Scholar] [CrossRef] [PubMed]
- Ji, S.; Hoye, T.R.; Macosko, C.W. Controlled synthesis of high molecular weight telechelic polybutadienes by ring-opening metathesis polymerization. Macromolecules 2004, 37, 5485–5489. [Google Scholar] [CrossRef]
- Scherman, O.A.; Rutenberg, I.M.; Grubbs, R.H. Direct synthesis of soluble, end-functionalized polyenes and polyacetylene block copolymers. J. Am. Chem. Soc. 2003, 125, 8515–8522. [Google Scholar] [CrossRef] [PubMed]
- Bielawski, C.W.; Scherman, O.A.; Grubbs, R.H. Highly efficient syntheses of acetoxy-and hydroxy-terminated telechelic poly (butadiene) s using ruthenium catalysts containing N-heterocyclic ligands. Polymer 2001, 42, 4939–4945. [Google Scholar] [CrossRef]
- Bielawski, C.W.; Benitez, D.; Morita, T.; Grubbs, R.H. Synthesis of end-functionalized poly (norbornene) s via ring-opening metathesis polymerization. Macromolecules 2001, 34, 8610–8618. [Google Scholar] [CrossRef]
- Gibson, V.C.; Okada, T. Synthesis of end-functionalized polynorbornenes and polynorbornanes via metathesis: novel macromonomers for polycondensation reactions. Macromolecules 2000, 33, 655–656. [Google Scholar] [CrossRef]
- Morita, T.; Maughon, B.R.; Bielawski, C.W.; Grubbs, R.H. A Ring-Opening Metathesis Polymerization (ROMP) Approach to Carboxyl- and Amino-Terminated Telechelic Poly(butadiene)s. Macromolecules 2000, 33, 6621–6623. [Google Scholar] [CrossRef]
- Maughon, B.R.; Morita, T.; Bielawski, C.W.; Grubbs, R.H. Synthesis of cross-linkable telechelic poly (butenylene) s derived from ring-opening metathesis polymerization. Macromolecules 2000, 33, 1929–1935. [Google Scholar] [CrossRef]
- Hillmyer, M.A.; Nguyen, S.B.T.; Grubbs, R.H. Utility of a ruthenium metathesis catalyst for the preparation of end-functionalized polybutadiene. Macromolecules 1997, 30, 718–721. [Google Scholar] [CrossRef]
- Fraser, C.; Hillmyer, M.A.; Gutierrez, E.; Grubbs, R.H. Degradable Cyclooctadiene/Acetal Copolymers: Versatile Precursors to 1,4-Hydroxytelechelic Polybutadiene and Hydroxytelechelic Polyethylene. Macromolecules 1995, 28, 7256–7261. [Google Scholar] [CrossRef]
- Hillmyer, M.A.; Grubbs, R.H. Chain transfer in the ring-opening metathesis polymerization of cyclooctadiene using discrete metal alkylidenes. Macromolecules 1995, 28, 8662–8667. [Google Scholar] [CrossRef]
- Chung, T.C.; Chasmawala, M. Synthesis of telechelic 1, 4-polybutadiene by metathesis reactions and borane monomers. Macromolecules 1992, 25, 5137–5144. [Google Scholar] [CrossRef]
- Saetung, N.; Campistron, I.; Pascual, S.; Pilard, J.-F.; Fontaine, L. One-Pot Synthesis of Natural Rubber-Based Telechelic cis-1,4-Polyisoprenes and Their Use To Prepare Block Copolymers by RAFT Polymerization. Macromolecules 2011, 4, 784–794. [Google Scholar] [CrossRef]
- Annunziata, L.; Fouquay, S.; Michaud, G.; Simon, F.; Guillaume, S.M.; Carpentier, J.-F. Mono- and di-cyclocarbonate telechelic polyolefins synthesized from ROMP using glycerol carbonate derivatives as chain-transfer agents. Polym. Chem. 2013, 4, 1313–1316. [Google Scholar] [CrossRef]
- Diallo, A.K.; Annunziata, L.; Fouquay, S.; Michaud, G.; Simon, F.; Brusson, J.-M.; Guillaume, S.M.; Carpentier, J.-F. Ring-opening metathesis polymerization of cyclooctene derivatives with chain transfer agents derived from glycerol carbonate. Polym. Chem. 2014, 5, 2583–2591. [Google Scholar] [CrossRef]
- Annunziata, L.; Diallo, A.K.; Fouquay, S.; Michaud, G.; Simon, F.; Brusson, J.-M.; Carpentier, J.-F.; Guillaume, S.M. α, ω-Di (glycerol carbonate) telechelic polyesters and polyolefins as precursors to polyhydroxyurethanes: an isocyanate-free approach. Green Chem. 2014, 16, 1947–1956. [Google Scholar] [CrossRef]
- Diallo, A.K.; Michel, X.; Fouquay, S.; Michaud, G.; Simon, F.; Brusson, J.-M.; Guillaume, S.M.; Carpentier, J.-F. α-Trialkoxysilyl functionalized polycyclooctenes synthesized by chain-transfer ring-opening metathesis polymerization. Macromolecules 2015, 48, 7453–7465. [Google Scholar] [CrossRef]
- Michel, X.; Fouquay, S.; Michaud, G.; Simon, F.; Brusson, J.-M.; Carpentier, J.-F.; Guillaume, S.M. α, ω-Bis (trialkoxysilyl) difunctionalized polycyclooctenes from ruthenium-catalyzed chain-transfer ring-opening metathesis polymerization. Polym. Chem. 2016, 7, 4810–4823. [Google Scholar] [CrossRef]
- Vanbiervliet, E.; Fouquay, S.; Michaud, G.; Simon, F.; Carpentier, J.-F.; Guillaume, S.M. From Epoxide to Cyclodithiocarbonate Telechelic Polycyclooctene through Chain-Transfer Ring-Opening Metathesis Polymerization (ROMP): Precursors to Non-Isocyanate Polyurethanes (NIPUs). Macromolecules 2017, 50, 69–82. [Google Scholar] [CrossRef]
- Michel, X.; Fouquay, S.; Michaud, G.; Simon, F.; Brusson, J.-M.; Carpentier, J.-F.; Guillaume, S.M. Tuning the properties of α, ω-bis (trialkoxysilyl) telechelic copolyolefins from ruthenium-catalyzed chain-transfer ring-opening metathesis polymerization (ROMP). Polym. Chem. 2017, 8, 1177–1187. [Google Scholar] [CrossRef]
- Michel, X.; Fouquay, S.; Michaud, G.; Simon, F.; Brusson, J.-M.; Carpentier, J.-F.; Guillaume, S.M. Simple access to alkoxysilyl telechelic polyolefins from ruthenium-catalyzed cross-metathesis depolymerization of polydienes. Eur. Polym. J. 2017, 96, 403–413. [Google Scholar] [CrossRef]
- Chauveau, C.; Vanbiervliet, E.; Fouquay, S.; Michaud, G.; Simon, F.; Carpentier, J.-F.; Guillaume, S.M. Azlactone Telechelic Polyolefins as Precursors to Polyamides: A Combination of Metathesis Polymerization and Polyaddition Reactions. Macromolecules 2018. [Google Scholar] [CrossRef]
- Zhang, Y.; Sudo, A.; Endo, T. Syntheses of bisphenol-type oligomers having five-membered dithiocarbonate groups at the terminals and their application as accelerators to epoxy-amine curing system. J. Polym. Sci. A Polym. Chem. 2008, 46, 1907–1912. [Google Scholar] [CrossRef]
- Rauleder, G.; Roxo, B.; Waldmann, H.; Bottenbruch, L.; Traenckner, H.J.; Gau, W. Method for the preparation and separation of polyglycidyl compounds. U.S. Patent 4,562,274, 31 December 1985. [Google Scholar]
- Otton, J.; Colleuille, Y.; Varagnat, J. Metathesis of functionalised olefins: homogeneous cross-metathesis of cycloolefin and ethylenic esters. J. Mol. Cat. 1980, 313–324. [Google Scholar] [CrossRef]
- Darensbourg, D.J.; Horn, A., Jr.; Moncadra, A.I. A facile catalytic synthesis of trimethylene carbonate from trimethylene oxide and carbon dioxide. Green Chem. 2010, 12, 1376–1379. [Google Scholar] [CrossRef]
- Kobayashi, T. Lipase-catalyzed syntheses of sugar esters in non-aqueous media. Biotechnol. Lett. 2011, 33, 1911–1919. [Google Scholar] [CrossRef] [PubMed]
- Rouillard, H. Etude de Résolutions Catalysées par des Lipases sous Irradiation Micro-Onde. Ph.D. Dissertation, Université de La Rochelle, La Rochelle, France, January 2012. [Google Scholar]
- Martinez, H.; Ren, N.; Matta, M.E.; Hillmyer, M.A. Ring-opening metathesis polymerization of 8-membered cyclic olefins. Polym. Chem. 2014, 5, 3507–3532. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
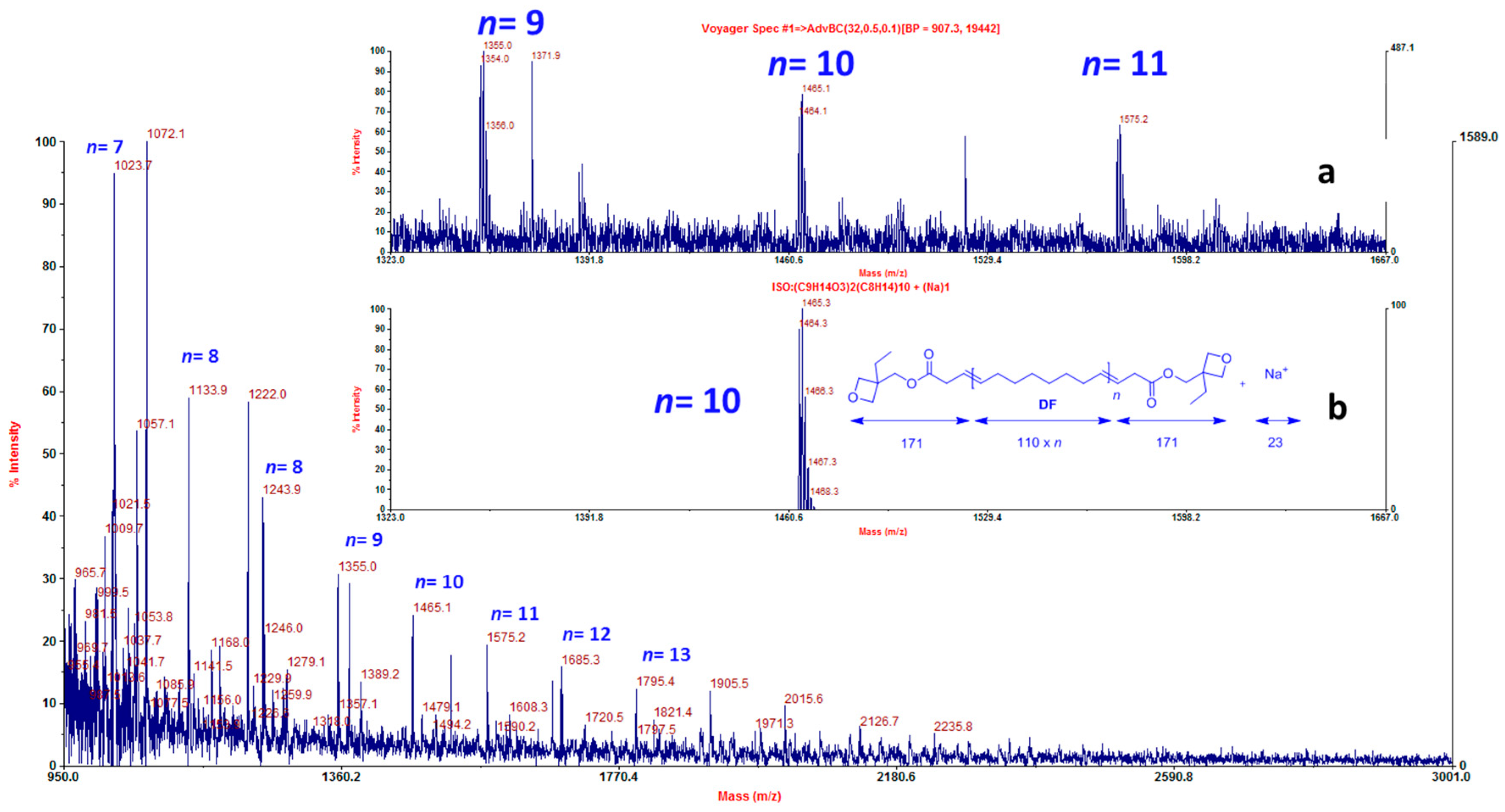{kind=link}
{kind=link}
{kind=link}
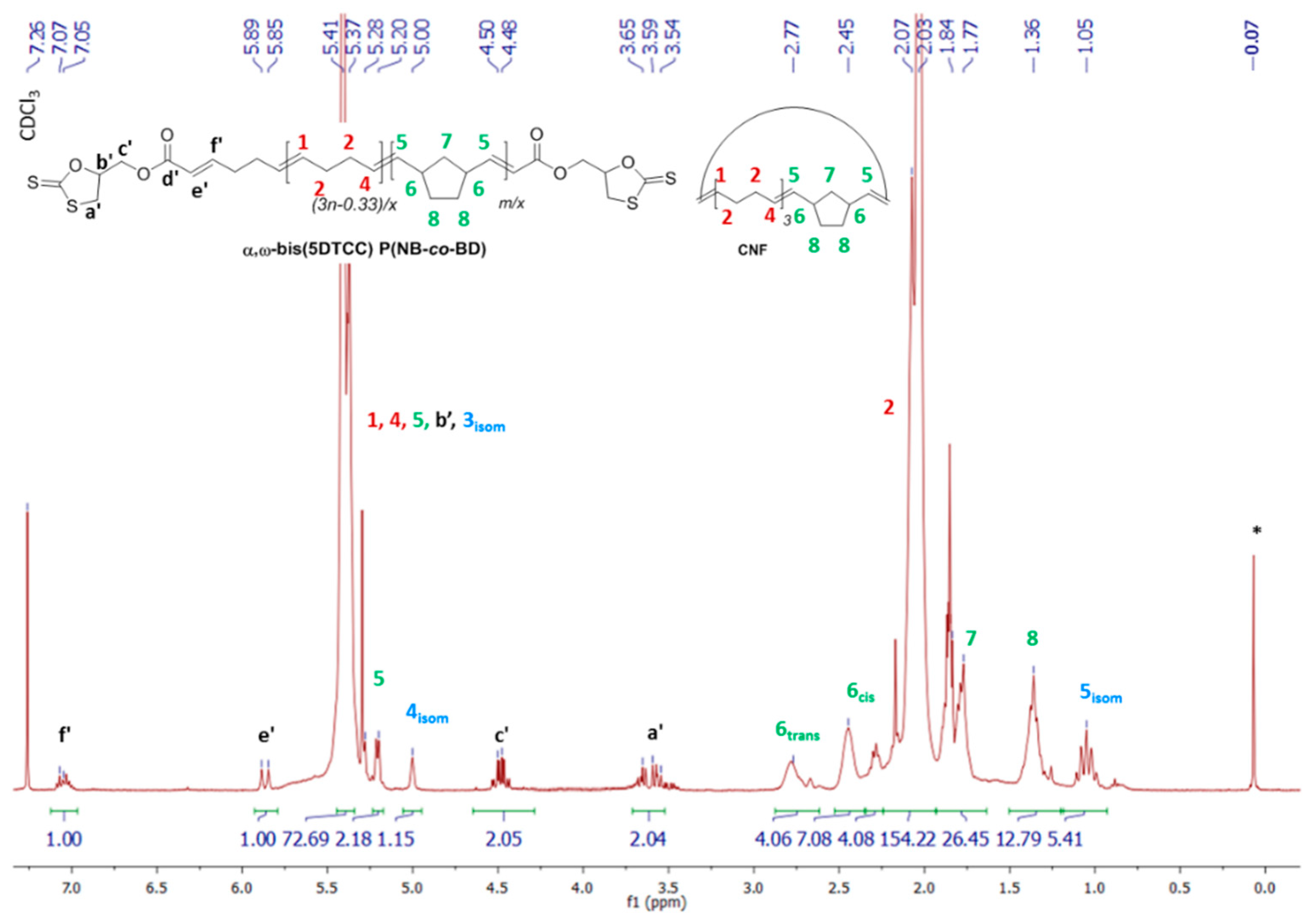{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | CTA | [COE]0/[CTA]0/[G2]0 | CTA Conv. b (%) | Mn,theoc (g·mol−1) | Mn,NMRd (g·mol−1) | Mn,SECe (g·mol−1) | ÐMe |
|---|---|---|---|---|---|---|---|
| 1 | 2 | 1000:20:1 | 20 | 27,800 | 30,300 | 56,800 | 1.6 |
| 2 | 2 | 2000:20:1 | 20 | 55,300 | 55,800 | 95,600 | 1.5 |
| 3 | 2 | 2000:100:1 | <5 | >44,300 | 69,700 | 127,600 | 1.6 |
| 4 | 3 | 1000:30:1 | 100 | 4000 | 3700 | 13,300 | 1.5 |
| 5 | 3 | 1000:40:1 | 100 | 3100 | 2900 | 7500 | 1.5 |
| Entry | NB or NBCOOMe | [NB(COOMe)]0/[COE]0/[CTA 1]0/[G2]0 a | CTA Conv. b (%) | Mn,theoc (g·mol−1) | Mn,NMRd (g·mol−1) | Mn,SECe (g·mol−1) | ÐMe |
|---|---|---|---|---|---|---|---|
| 1 | NB | 500:500:20:1 | 75 | 7000 | 9100 | 20,100 | 1.5 |
| 2 | NB | 500:500:30:1 | 70 | 5100 | 6600 | 17,500 | 1.4 |
| 3 | NB | 500:500:50:1 | 78 | 2800 | 3600 | 9700 | 1.3 |
| 4 | NB | 1000:1000:200:1 | 31 | 3500 | 5500 | 12,400 | 1.5 |
| 5 | NBCOOMe | 1000:1000:20:1 | 56 | 23,600 | 22,700 | 52,400 | 1.3 |
| 6 | NBCOOMe | 1000:1000:50:1 | 40 | 13,300 | 23,000 | 51,700 | 1.4 |
| Entry | CTA | Solvent | Cat. | Temp. (°C) | [NB]0/[CDT]0/ [CTA]0/[Cat.]0 | CTA Conv. b (%) | Mn,theoc (g·mol−1) | Mn,NMRd (g·mol−1) | Mn,SECe (g·mol−1) | ÐMe |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1 | CH2Cl2 | G2 | 40 | 1000:1000:100:1 | 0 | 256,000 f | - | - | - |
| 2 | 1 | CH2Cl2 | G2 | 40 | 2000:2000:200:1 | 0 | 512,000f | - | - | - |
| 3 | 1 | CH2Cl2 | HG2 | 40 | 500:500:20:1 | 8 | 80,300 | - | - | - |
| 4 | 1 | THF | HG2 | 60 | 500:500:25:1 | 80 | 6600 | 6900 | 17,500 | 1.5 |
| 5 | 1 | THF | HG2 | 60 | 500:500:100:1 | 45 | 3200 | 3400 | 10,100 | 1.3 |
| 6 | 1 | THF | HG2 | 60 | 500:500:50:1 | 50 | 5350 | 5700 | 17,300 | 1.5 |
| 7 | 1 | THF | HG2 | 60 | 1000:1000:20:1 | 75 | 17,300 | 17,900 | 26,800 | 1.5 |
| 8 | 3 | CH2Cl2 | G2 | 40 | 1000:1000:50:1 | 100 | 5500 | 5900 | 12,600 | 2.1 |
| 9 | 3 | CH2Cl2 | G2 | 40 | 1000:1000:100:1 | 100 | 2900 | 2900 | 8700 | 1.7 |
| α,ω-Diepoxide P(NB-co-CDT) | α,ω-Bis(dithiocarbonate) P(NB-co-CDT) | |||||
|---|---|---|---|---|---|---|
| Entry | Mn,NMRa (g·mol−1) | Mn,secb (g·mol−1) | ÐMb | Mn,NMRa (g·mol−1) | Mn,secb (g·mol−1) | ÐMb |
| 1 | 3400 | 10,100 | 1.3 | 3400 | 10,200 | 1.5 |
| 2 | 5700 | 17,300 | 1.5 | 6100 | 19,100 | 1.8 |
| 3 | 6600 | 19,900 | 1.5 | 8700 | 24,700 | 1.8 |
| Entry | Polymer | CTA/Chain-End Group | Mn,NMRd (g·mol−1) | Tga (°C) | Tma (°C) | ∆Hfus a (J/g) | Tca (°C) |
|---|---|---|---|---|---|---|---|
| 1 | PCOE | 1/epoxide | 3900 | n.o. | 58 | 21 | 45 |
| 2 | PCOE | 3/oxetane | 3700 | −45 | 54 | 23 | 41 |
| 3 | P(NB-co-CDT) | 1/epoxide | 6900 | −75 | 5 | 5 | −14 |
| 4 | P(NB-co-CDT) | 3/oxetane | 5900 | −72 | 4 | 5 | −16 |
| 5 | P(NB-co-CDT) | -/dithiocarbonate | 3400 | −75 | 5 | 5 | −14 |
| 6 | P(NB-co-CDT) | -/dithiocarbonate | 8100 | −74 | 5 | 5 | −12 |
| 7 | P(NB-co-CDT) | -/dithiocarbonate | 8700 | n.o. | 4 | 5 | −10 |
| Entry | Telechelic Copolymer | CTA/Chain-End Group | Mn,NMRa (g·mol−1) | Newtonian Viscosity at 25 °C a (Pa·s) |
|---|---|---|---|---|
| 1 | P(COE-co-NB) | 1/epoxide | 6600 | 750 ± 50 |
| 2 | P(COE-co-NBCOOMe) | 1/epoxide | 22,700 | 800 ± 50 |
| 3 | P(NB-co-CDT) | 1/epoxide | 6900 | 14 ± 3 |
| 4 | P(NB-co-CDT) | 3/oxetane | 5900 | 13 ± 5 |
| 5 | P(NB-co-CDT) | -/dithiocarbonate | 3400 | 14 ± 5 |
| 6 | P(NB-co-CDT) | -/dithiocarbonate | 6100 | 15 ± 5 |
| 7 | P(NB-co-CDT) | -/dithiocarbonate | 8700 | 17 ± 3 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vanbiervliet, E.; Fouquay, S.; Michaud, G.; Simon, F.; Carpentier, J.-F.; Guillaume, S.M. α,ω-Epoxide, Oxetane, and Dithiocarbonate Telechelic Copolyolefins: Access by Ring-Opening Metathesis/Cross-Metathesis Polymerization (ROMP/CM) of Cycloolefins in the Presence of Functional Symmetric Chain-Transfer Agents. Polymers 2018, 10, 1241. https://doi.org/10.3390/polym10111241
Vanbiervliet E, Fouquay S, Michaud G, Simon F, Carpentier J-F, Guillaume SM. α,ω-Epoxide, Oxetane, and Dithiocarbonate Telechelic Copolyolefins: Access by Ring-Opening Metathesis/Cross-Metathesis Polymerization (ROMP/CM) of Cycloolefins in the Presence of Functional Symmetric Chain-Transfer Agents. Polymers. 2018; 10(11):1241. https://doi.org/10.3390/polym10111241
Chicago/Turabian StyleVanbiervliet, Elise, Stéphane Fouquay, Guillaume Michaud, Frédéric Simon, Jean-François Carpentier, and Sophie M. Guillaume. 2018. "α,ω-Epoxide, Oxetane, and Dithiocarbonate Telechelic Copolyolefins: Access by Ring-Opening Metathesis/Cross-Metathesis Polymerization (ROMP/CM) of Cycloolefins in the Presence of Functional Symmetric Chain-Transfer Agents" Polymers 10, no. 11: 1241. https://doi.org/10.3390/polym10111241
APA StyleVanbiervliet, E., Fouquay, S., Michaud, G., Simon, F., Carpentier, J.-F., & Guillaume, S. M. (2018). α,ω-Epoxide, Oxetane, and Dithiocarbonate Telechelic Copolyolefins: Access by Ring-Opening Metathesis/Cross-Metathesis Polymerization (ROMP/CM) of Cycloolefins in the Presence of Functional Symmetric Chain-Transfer Agents. Polymers, 10(11), 1241. https://doi.org/10.3390/polym10111241