3.1. Online Microreactor/Mass Spectrometry Monitoring
Online mass spectrometry analysis of continuous flow processes provides real-time data and thus allows for rapid kinetic screening and in consequence efficient optimization of polymerization reactions. As schematically shown in
Figure 1, reagent mixtures were prepared in gastight syringes and injected in the microflow reactor. Screening of the reaction can be done in minimal time and by usage of only trace amounts of material due to the small reactor volume (19.5 µL). The microreactor residence time depends on the flow rate as preset by the employed syringe pumps. Reaction products that exit the microreactor are transferred from the flow microreactor to the electrospray ionization (ESI) nozzle, which is directly achieved under constant flow conditions that allow for nonstop monitoring of the polymerization reaction, without the requirement of a sampling method. In this way, an unmatched information density with respect to product compositions and reaction kinetics is directly accessed in very short time spans. This information can be used for the detailed investigation of reaction mechanisms, but also for the rapid optimization of synthesis procedures. Data was acquired in situ and feedback provided to optimize the reaction outcome. The setup and its use were previously described in detail [
23,
24].
A useful feature of the microreactor/mass spectrometry setup is the ability to perform time sweep experiments, as already applied in our previous work [
23]. In a time-sweep experiment, microreactor residence times are varied quickly by a sudden change in the preset flow rates of the embedded syringe pumps, followed by monitoring the outlet of the reactor. Typically, a range of different microreactor residence times (fixed flow rates) are screened in an online monitoring experiment. Different parameters (temperature, concentration and flow rate) can be chosen to comprehensively screen reaction efficiency. This way, the on-line setup allows for continuous ‘nonstop’ analysis of the reaction mixture at any given set of reaction conditions and reactor residence times. However, it only becomes really interesting if these flow rates are varied during the course of a microreactor polymerization without interrupting the continuous mass spectrometry measurement. Thus, a polymerization can be screened within a predetermined microreactor residence time interval. Time sweeps enable the operator to move forward or go back in time (as increased flow rates correspond to shorter reaction times), a feature that cannot be achieved in any batch reaction monitoring setup (see
Figure 2). Practically any instance in time of a reaction can be directly imaged by sweeping over a range of reactor residence times, giving access to continuous data sampling with respect to product structure and composition under synthesis conditions. As example, when operating the setup at a flow rate of 0.975 µL/min (20 min residence time), flow rate can then be adjusted to 19.5 µL/min (1 min residence time) to perform a time sweep experiment from 20 to 1 min residence time. If now applied to a polymerization reaction, residence times can be sweeped forward and backwards (molecular weights will increase and decrease) until optimal conditions are identified. Note that a full sweep experiment always only takes as long as the lower flow rate that was chosen. Hence in the present example, a full kinetic screening is carried out within 20 min.
3.2. Real Time Monitoring Dithiobenzoate-Mediated RAFT Polymerizations
Dithiobenzoate-mediated RAFT polymerization processes were targeted via online microreactor/mass spectrometry monitoring. As a first demonstration on how the online monitoring setup can be employed, a reversible addition-fragmentation chain transfer (RAFT) polymerization (
Scheme 2) of
n-butyl acrylate (
nBA) with the RAFT agent 2-cyano-2-propyl dithiobenzoate (CPDB) and 2-2′-azoisobutyronitrile (AIBN) as thermal initiator was screened.
To investigate the kinetics and species formed during the early stages of the RAFT polymerization, typically, samples are taken during the course of polymerization whereby endgroup patterns are checked offline via soft ionization mass spectrometry methods. Here, the polymerization was continuously monitored by the online setup for different conditions (temperature, residence time and reagent concentrations) and ESI-MS spectra were analyzed accordingly.
Table 1 highlights 8 screening results for the CPDB RAFT polymerization reactions with 1 equivalent of the monomer
n-butyl acrylate and 0.1 or 0.25 equivalents of the thermal initiator AIBN. Screening equimolar RAFT to monomer concentrations allows us to monitor species formed during the early stages of the RAFT polymerization, before the main equilibrium of the process is reached. Polymerizations were screened at a microreactor temperature of 80 and 100 °C and microreactor residence times of 5 and 10 min. Product yields (%) for the species identified in
Table 1 were determined by ESI-MS peak intensities (relative abundances) according to the following equation:
All individual species are calculated relative to the sum of the ESI-MS intensities of all species observed, an evaluation method that has been used and validated for series of polymer product analysis before [
25,
26]. It has to be noted though that ESI-MS is a qualitative analysis technique due to mass and ionization biases and is therefore not representative for absolute concentrations of species in the reaction mixture. This can be overcome by a complex labor-intensive calibration of the mass spectrometer, as shown previously. However, in this manuscript, ESI-MS results are described based on the relative abundance of the species rather than absolute concentrations.
After screening equimolar conditions of CPDB RAFT agent and
nBA monomer, single unit monomer insertion (SUMI) products and the CPDB RAFT agent are identified in the reaction mixture for all screening conditions (
Table 1). Furthermore, irreversible termination products (so-called cross-termination, CT) of the intermediate (macro)RAFT agent with propagating radicals present in the reaction mixture were observed. In
Table 1, single unit monomer insertion products are indicated as SUMI
x (x = 1, 2, …) according to the amount of monomers inserted respectively and cross-termination products as CT
x (x = 0, 1, …) with CT
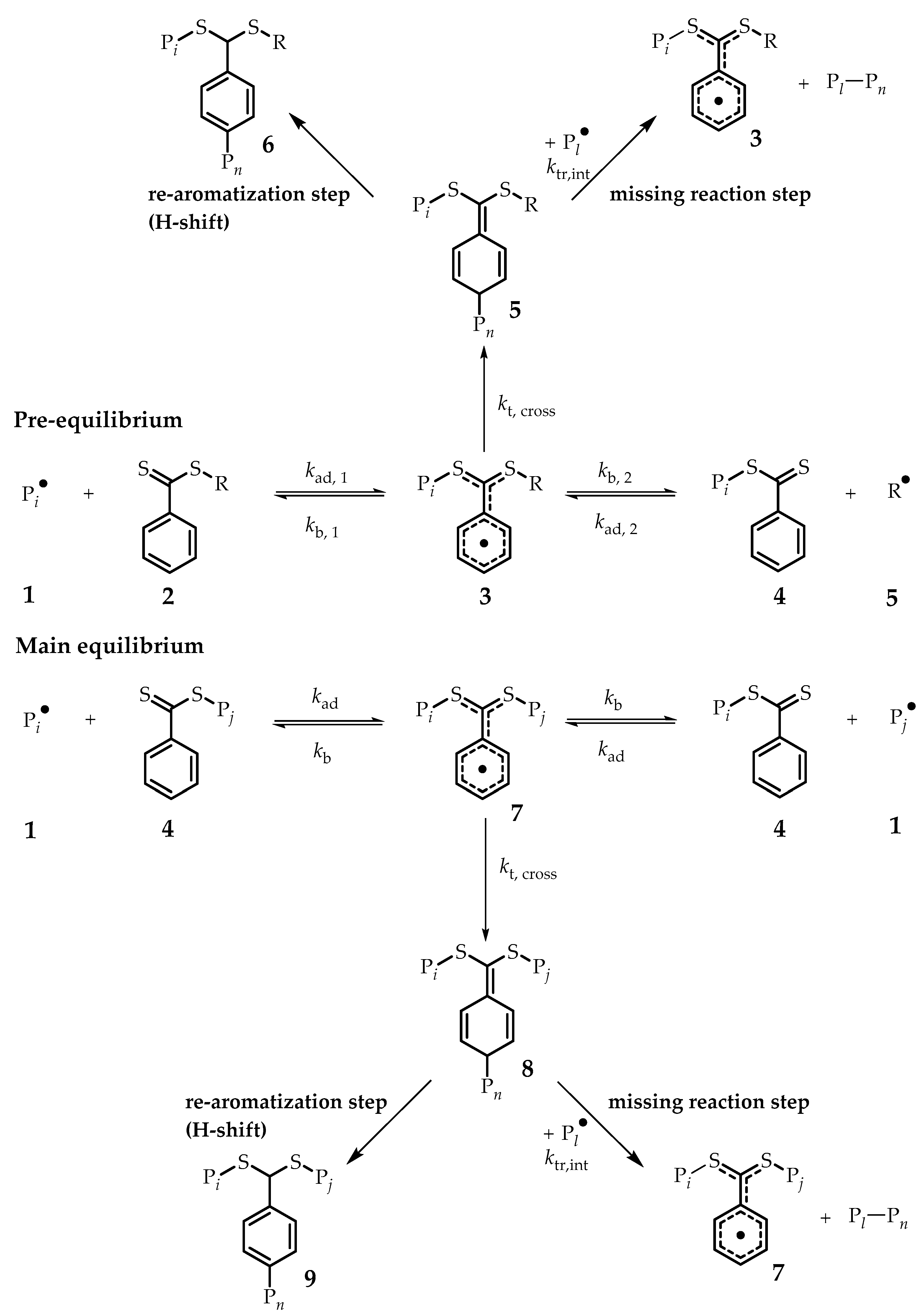
0 being the irreversibly terminated intermediate CPDB RAFT by AIBN radical fragments during the pre-equilibrium stage (species
5,
Scheme 1). Unexpected rate retardation is a well-known phenomenon in dithiobenzoate-mediated RAFT polymerizations as discussed before. Observing irreversible termination in the early stages of the process is a direct proof of this cause. Rate constants of the RAFT equilibrium are not accessible via online mass spectrometry monitoring; however, kinetic studies were published that support the slow fragmentation of the intermediate RAFT radical (species
3 or
7,
Scheme 1). Chernikova et al. used an electron spin resonance (ESR) spin-trapping technique for quantitative detection of radicals (to measure addition and fragmentation rate coefficients for the RAFT equilibrium) to obtain an equilibrium constant in the order of 10
8 L mol
−1 [
27]. In another contribution, Ranieri et al. showed via a ESR spin-trapping methodology that the stable RAFT intermediate radicals are formed in the early stages of the RAFT polymerization when dithiobenzoates are employed as controlling agents as stipulated by the so-called slow fragmentation theory [
28].
Scheme 3 shows the chemical structures of the expected species with their corresponding monoisotopic masses observed during MS screening as discussed in
Table 1. CT
1 represents termination of the intermediate (macro)RAFT species where one
n-butyl acrylate monomer is inserted. However, mass spectrometry cannot distinguish where the monomer unit is built in as both possible structures are isobaric (see
Scheme 3). CPDB RAFT agent, SUMI
1, CT
0 and CT
1 are identified for all conditions screened in
Table 1. Very high amounts of AIBN were used in the study to allow for observation of significant peak intensities in the ESI-MS. Overall, as expected for higher residence times and reactor temperature a higher intensity of SUMI
1 species is observed and less CPDB RAFT agent is present is the mixture. A direct relation is observed between the amount of CT
0 species formed and the number of AIBN radicals generated. CT products are directly related to the number of radicals generated by AIBN so more CT is observed in the reaction mixture if the AIBN concentration is increased from 0.1 to 0.25 equivalents. Since the decomposition rate of AIBN (half-life time) is directly related to the reaction temperature more CT products are formed at 100 °C compared to 80 °C, simply due to the increased radical concentration and therefore cross-termination, and termination events in general, are more likely to occur.
Figure 3 shows the ESI-MS spectra for condition
3 (0.1 equiv. AIBN) and
7 (0.25 equiv. AIBN) in
Table 1 at 100 °C and 5 min residence time. For these conditions, a high amount of CT
0 product is formed which again is related to the radical concentration in the reaction mixture. In the first 5 min of the polymerization (during first half-life time of AIBN, t
1/2 = ~8 min), more radicals are generated and radicals are more likely to terminate. Over the course of the polymerization, less AIBN radicals are generated and the relative abundance of CT
0 decreases over time. This effect is nicely illustrated in the relative abundance of SUMI
1 and CT
0 in
Table 1. Furthermore, SUMI
2 products are not observed at the conditions screened in
Table 1; higher SUMI products are only expected once the CPDB RAFT is fully consumed (so-called inhibition period). In addition, CT
1 species are only observed with increasing radical concentration (thus high initiator concentration and temperature) or upon increasing the residence time (condition
8). It has to be noted that by referring to a decrease in CT
x products in the results reported it actually refers to an increase of other (mostly likely SUMI) species in the polymerization mixture, which relies on the assumption of irreversible cross-termination; however, the reaction of propagating radicals with the irreversible cross-termination product has been proposed in literature [
16].
Table 2 shows results for screening the same dithiobenzoate-mediated polymerization reaction as shown in
Scheme 2 with five times increased monomer concentration to identify higher SUMI and CT species. Here, the same behavior is observed and similar conclusions can be drawn for condition
9,
10 and
11 compared to the conditions in
Table 1 (as discussed earlier). The RAFT polymerization shows inhibition until all CPDB RAFT agent is consumed. However, at 100 °C for 20 min (condition
12) a clear transition is observed from the pre -to the main equilibrium in the RAFT polymerization process compared to condition
11. At this stage all RAFT agent is converted into (macro)RAFT and higher SUMI and CT products (SUMI
2, SUMI
3, CT
1 and CT
2) are observed in high amounts. Thus, cross-termination does not only occur in the pre-equilibrium stage. However, reactions are only screened for lower chain lengths, which may explain why in previous studies these species could not be identified. At chain lengths above 10 these might not occur in significant abundancies anymore, as severe chain length effects of the RAFT equilibrium constant have indeed been proposed both from ab-initio calculations, but also based on kinetic modelling [
14,
15,
29].
3.3. Time Sweeping the Early Stages of Dithiobenzoate-Mediated Polymerizations
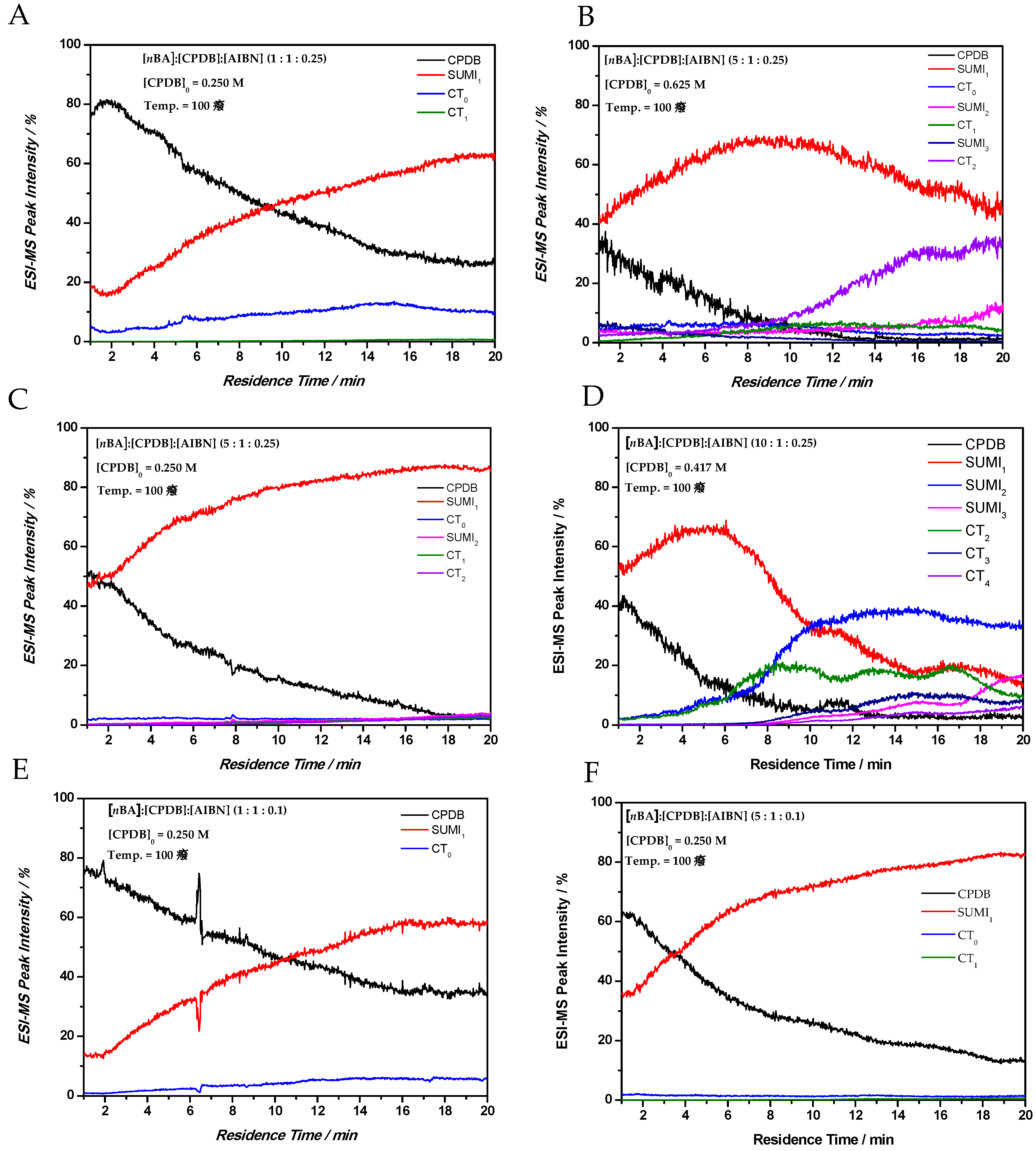
After identifying polymerization species formed during the early stages of dithiobenzoate-mediated polymerization, time sweep experiments were performed (see
Section 3.1) to investigate the change in CT product formation in time (and chain length). Here, the polymerization was continuously monitored by ESI-MS while sweeping over a given residence time range. Solutions of CPDB RAFT agent, nBA monomer and AIBN in butyl acetate (BuOAc) were prepared in gastight syringes and employed in the microreactor/MS setup. Initial conditions applied to the microreactor were set to 1 min microreactor residence time (flow rate = 19.5 µL/min) and at a reactor temperature of 100 °C. The polymerization reaction is then recorded by increasing the residence time in the microreactor from 1 to 20 min (flow rate from 19.5 to 0.975 µL/min). Spectra were continuously acquired for the next 20 min and a continuous set of kinetic data is obtained based on the specific product patterns recorded for each individual residence time (MS spectra acquired every 2.2 s). Note that the dead volume between the microreactor exit and ESI-MS nozzle is not included (in practice data acquisition happens for 20 min + dead time of ~6 min). Such intense screening is not feasible with any other offline sampling method since the polymer sample needs to be quenched at the exact aimed residence time, purified and manually measured by ESI-MS, assuming all products are stable. Care has to be taken when performing these measurements, as the amount of data acquired in a short time interval (20 min) for each polymerization is enormous. As an example to illustrate this further, in this manuscript an ESI-MS spectrum from the polymer mixture that exits the microreactor was measured every 2.2 s. In total, 545 ESI-MS spectra are acquired in a time sweep from 1 to 20 min. To obtain information on a single species present in the spectra, peak intensities for all species present have to be extracted.
Via this technique, the formation of cross-termination products over time for a range of reaction conditions (varying reagent concentrations and reactor temperature) is easily followed. In
Figure 4 time sweep experiments are shown for different
nBA:CPDB:AIBN ratios. Furthermore, the absolute concentration of the CPDB RAFT agent was varied and a clear effect on the reaction kinetics was observed. The difference in reagent concentrations can be explained by the microreactor setup whether the reaction mixture is prepared in one gastight syringe or, in case of screening different equivalents of monomer, two gastight syringes were applied. Overall, a similar trend is observed compared to the conditions screened in
Table 1 and
Table 2. CT products increase with increasing radical concentration in the reaction mixture, thus with increasing initiator concentration, higher SUMI and CT products are only formed when the CPDB RAFT agent is almost fully consumed (
Figure 4D). In
Figure 4A,C,E,F, the initial RAFT agent concentration is 0.25 M, therefore, by varying the [
nBA] and [AIBN] concentration, a clear effect on the reaction rates of SUMI
x and CT
x could be observed. All reactions were performed at 100 °C in butyl acetate. In
Figure 4C, a 5-times higher monomer concentration was screened compared to
Figure 4A (initiator and RAFT concentration was kept constant). As expected, the formation of the SUMI
1 product happens faster in 4C; however, almost no CT products were observed, while 4A shows the formation of CT
0 in significant amounts from the start. The same effect is observed in
Figure 4E,F for a lower overall initiator concentration. In
Figure 4B,D, a higher initial RAFT agent concentration is used, which clearly influences the kinetic behavior in the pre- and main equilibrium. The higher RAFT agent concentration in
Figure 4B causes the formation of CT
2 compared to no CT products in
Figure 4C. This can be explained by the higher initial concentration of the AIBN initiator (0.0625 M in
Figure 4C compared to 0.1563 M in
Figure 4B); cross-termination is therefore much more favored. In
Figure 4B,D, the RAFT agent is fully consumed after approximately 12 min residence time and higher SUMI products were observed.
3.4. Proposed Chemical Structure of the Cross-Termination Product
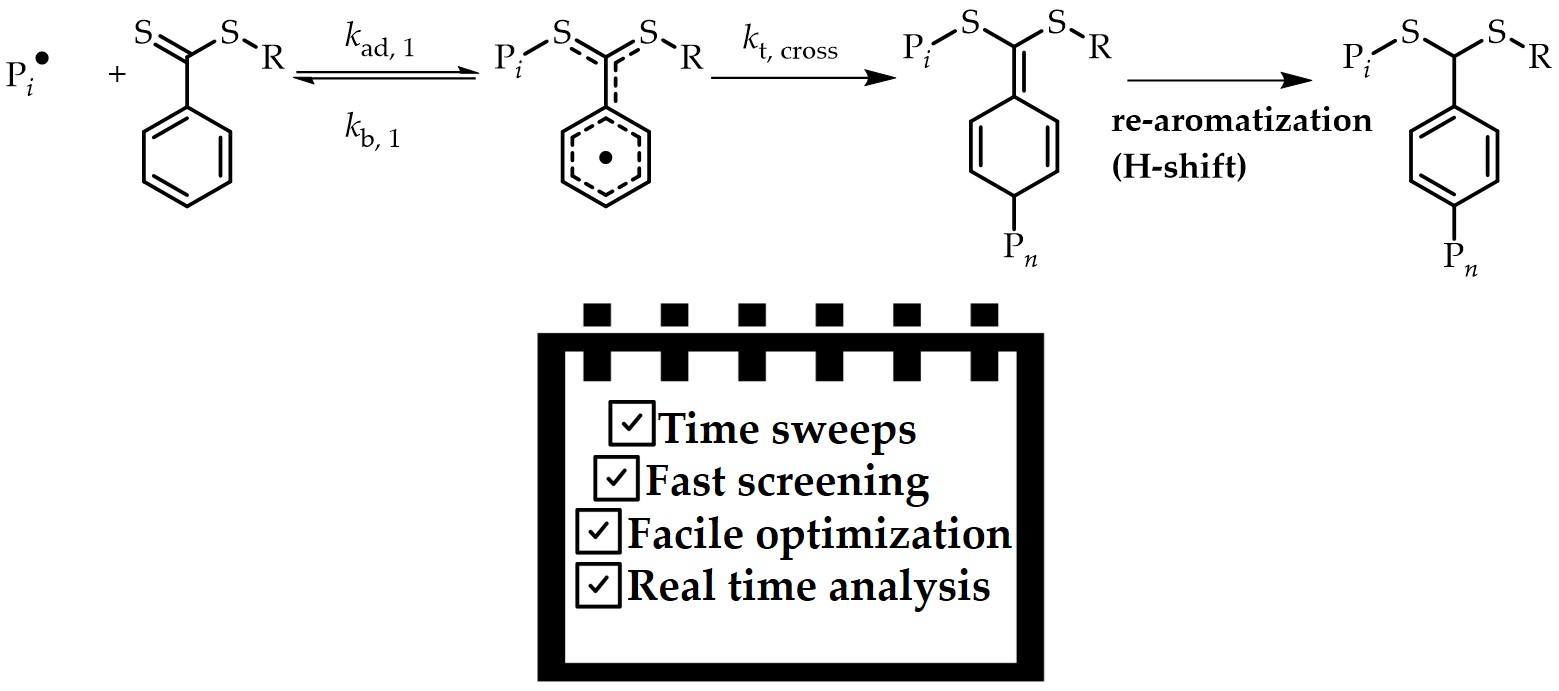
RAFT polymerizations mediated by dithiobenzoates experience rate retardation, mainly due to resonance stabilization of the intermediate radical (
3 and
7,
Scheme 1). Delocalization of the radical functionality affords resonance structures with reduced steric hindrance for the formation of cross-termination products by radical-radical coupling, as shown in
Scheme 4. Previous discussions in literature focused on the cross-termination between propagating radicals and the delocalized radical (
Scheme 4 to end up with a mixture of several regioisomers [
13]. Observation of these products demonstrates that CT products are stable, and not prone to transfer reactions, where a CT product is transformed back into a regular RAFT polymer (see the “missing reaction step”) [
16,
30]. If such transfer is not occurring at low chain lengths, it is not evident why it should occur at higher degrees of polymerization. Observation of CT products in this study for early stage RAFT polymerization would thus underpin that the kinetic situation at the early stage of the main equilibrium is significantly different from later stages and that pronounced chain length effects do exist. Since termination is diffusion controlled, it is straightforward to assume that cross-termination will become less significant with increasing radical lengths. However, the equilibrium constant might also be prone to change. This is an effect that the current monitoring technique is, however, not able to catch.
In a last step, we aimed at confirming the exact structure of the CT product, as ESI-MS does not allow to discern between several possible isobaric product structures (structural isomers). To reach this aim, a simple and straightforward batch experiment was designed. CPDB RAFT agent was heated in the presence of high amounts of AIBN (0.5 equivalents) dissolved in butyl acetate. The high concentration of AIBN radicals generated upon heating the mixture for 2 h at 90 °C will undergo radical termination events by radical combination, while still also taking part in the degenerative transfer equilibrium with the RAFT agent. In such a scenario, intermediate radical species (
3 and
7,
Scheme 1) are constantly formed upon addition of radical fragments, and concomitantly very likely to be terminated with further cyanoisopropyl fragments. Although this experiment is not performed under real polymerization conditions, it gives good insight into the processes that cause a loss of radicals, and in consequence rate retardation, during dithiobenzoate-mediated polymerization. After the reaction, the product mixture was purified by flash column chromatography and the cross-termination product (
Figure 5B) and CPDB RAFT agent (
Figure 5A) were isolated.
Figure 5 shows NMR and ESI-MS analysis of the crude and purified reaction mixture. Details about the synthesis is provided in
Section 2. Surprisingly, the main CT product (over 95%) observed is the irreversible radical-radical coupling on the para-position (Species
5 and
8,
Scheme 1), followed by a re-aromatization (H-shift) to regain its aromaticity (Species
6 and
9,
Scheme 1). Thus, it can be safely concluded that radical-radical coupling occurs exclusively on the para position as expected but re-aromatization via an H-shift occurs, which is not fully unexpected since it is much more energetically stable. HETCOR NMR (
Figure 5C) analysis confirmed the re-aromatization by coupling between the
13C-
1H of the dithioester moiety. No other coupling patterns were observed in heteronuclear correlation (HETCOR) NMR spectroscopy for this particular proton shift. The isolated re-aromatized para terminated cross-coupling product has a purity of +95% according to
1H NMR analysis. Termination in ortho position can be excluded, since no corresponding coupling was observed in NMR analysis (
1H + HETCOR NMR).
Figure 5D,E show MS of the crude and isolated re-aromatized cross-termination product, respectively.














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}