Polypropylene-Based Porous Membranes: Influence of Polymer Composition, Extrusion Draw Ratio and Uniaxial Strain

Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Compounding
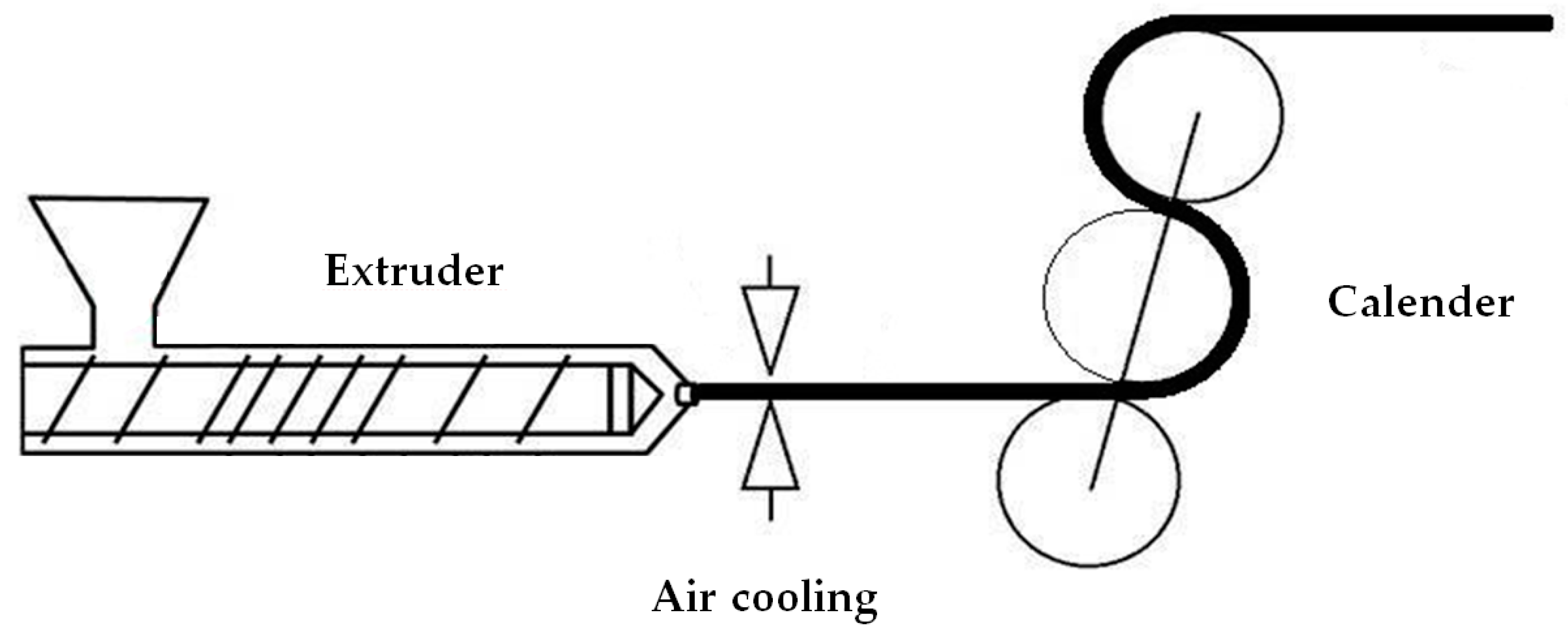
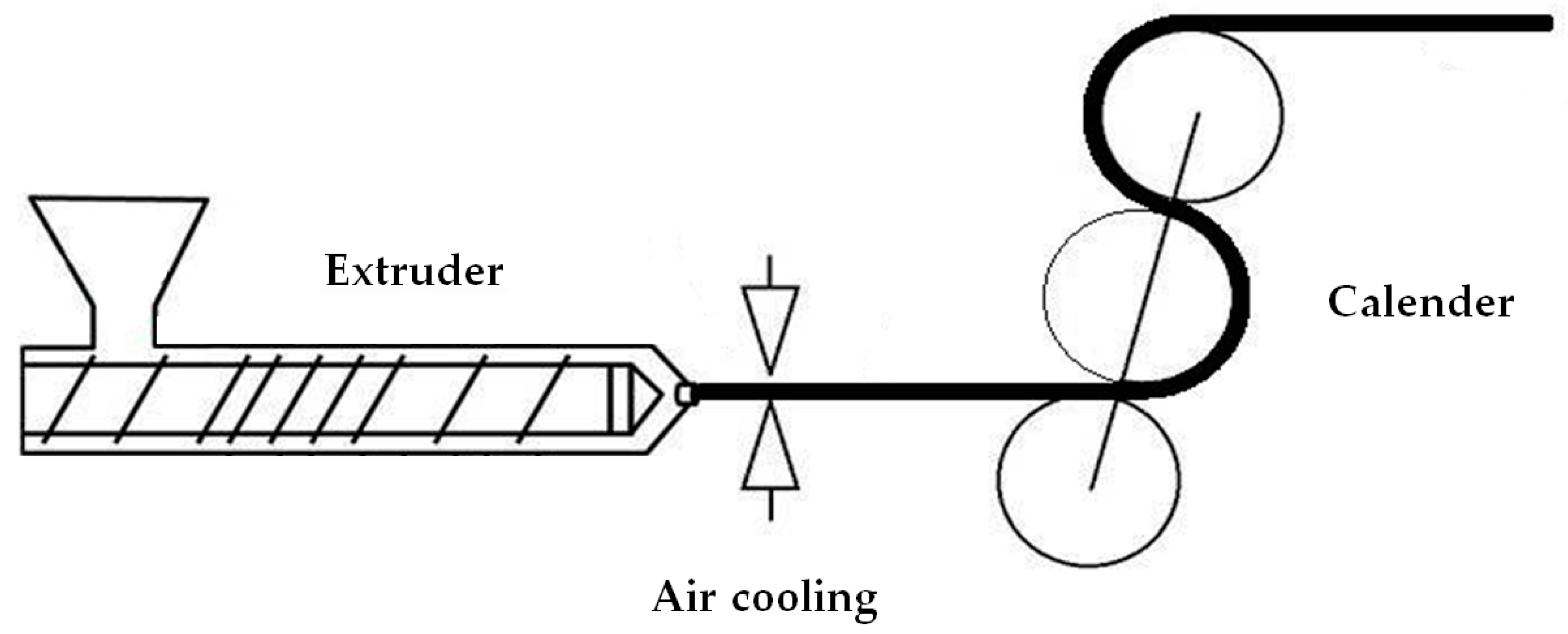
2.2. Extrusion and Annealing of Precursor Films
2.3. Uniaxial Strain of Annealed Precursor Films
2.4. Membrane Characterization
3. Results
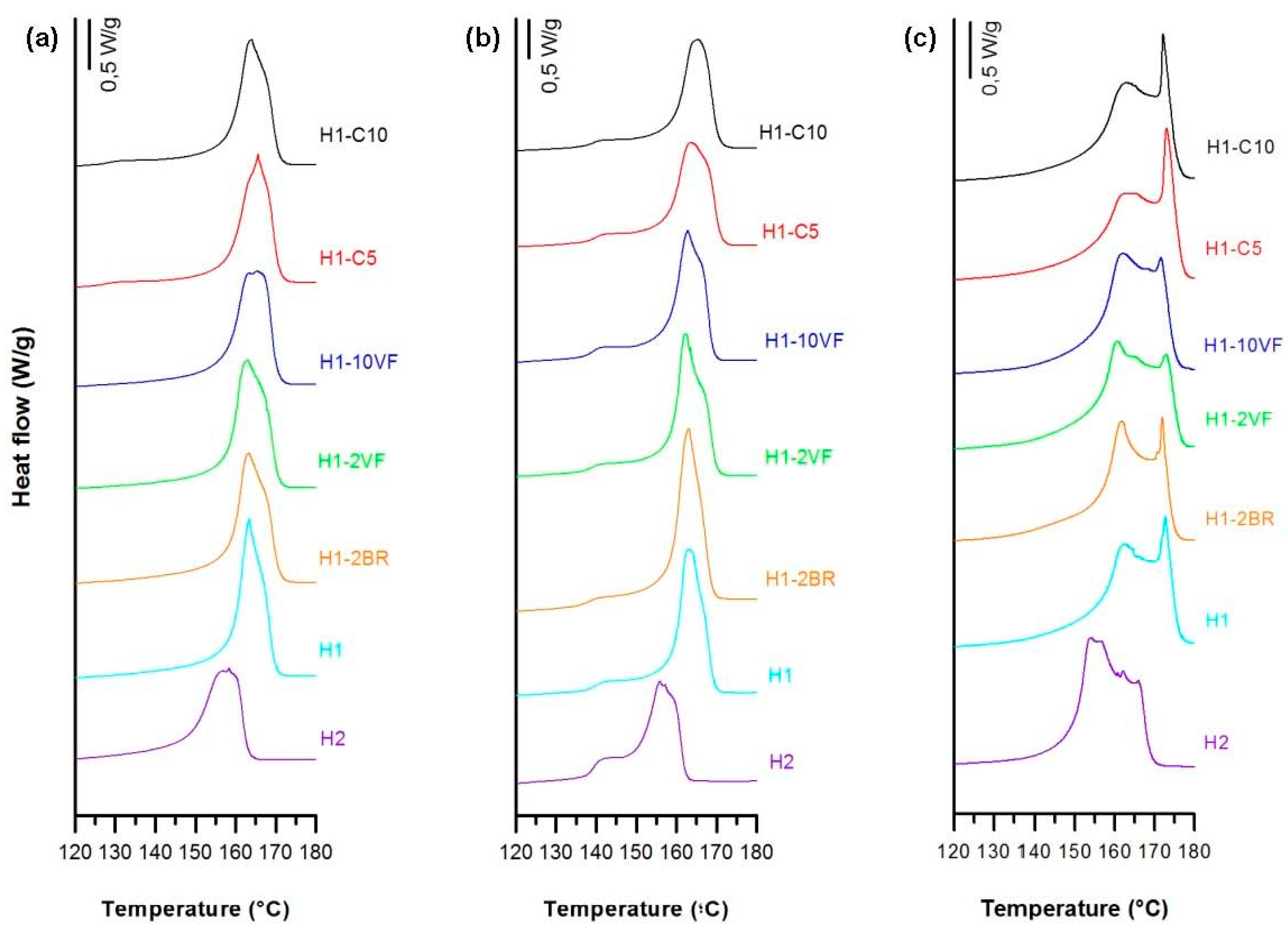
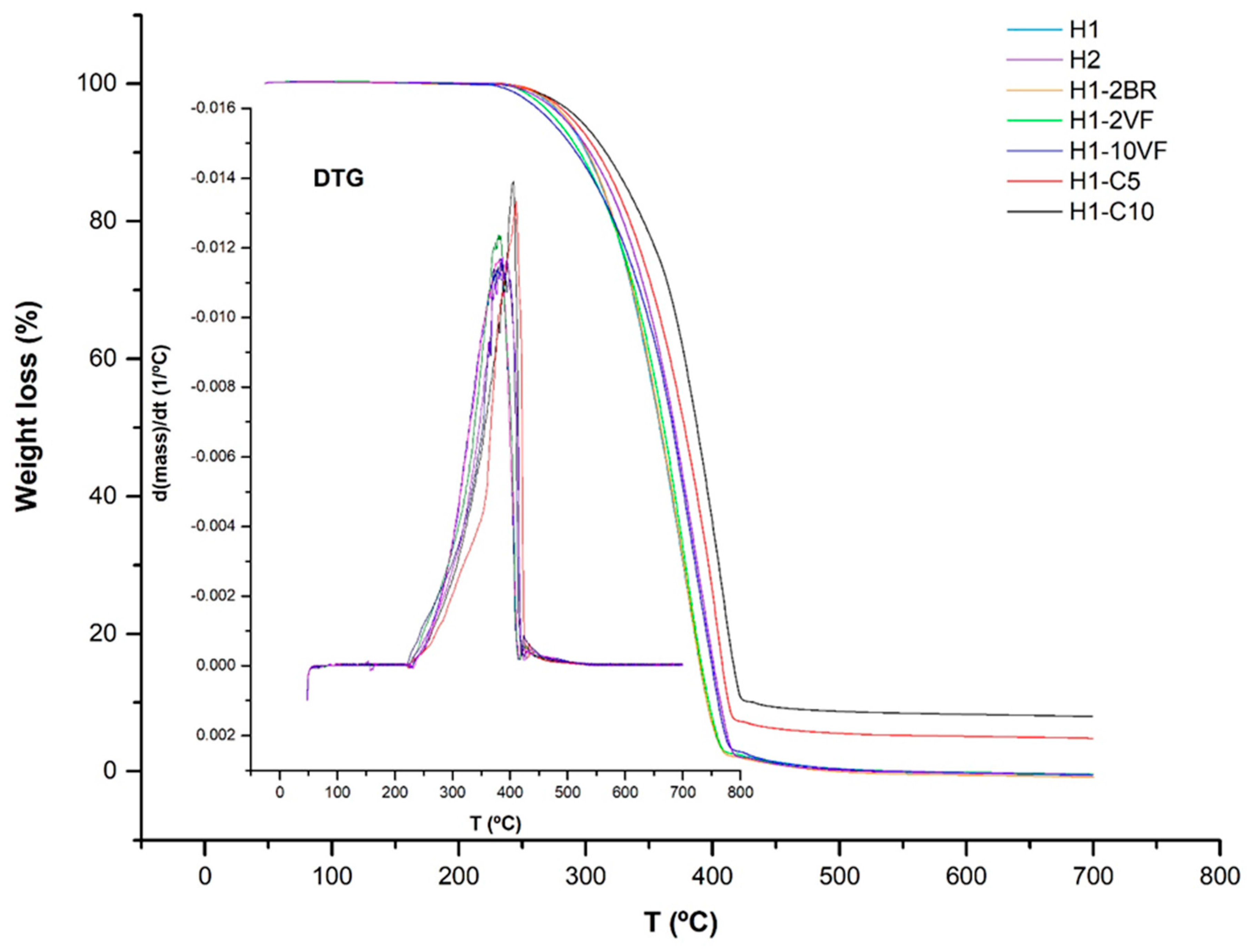
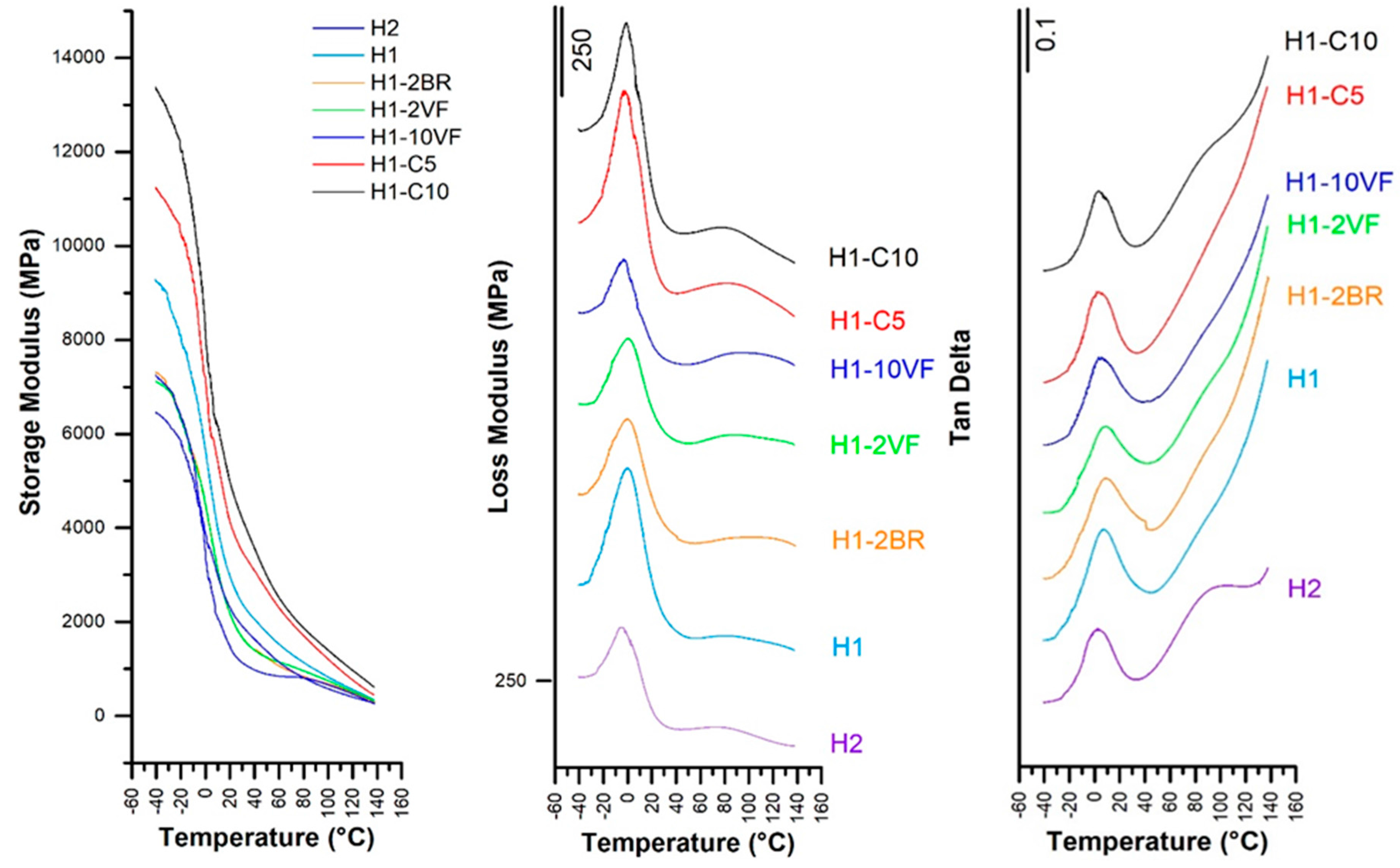
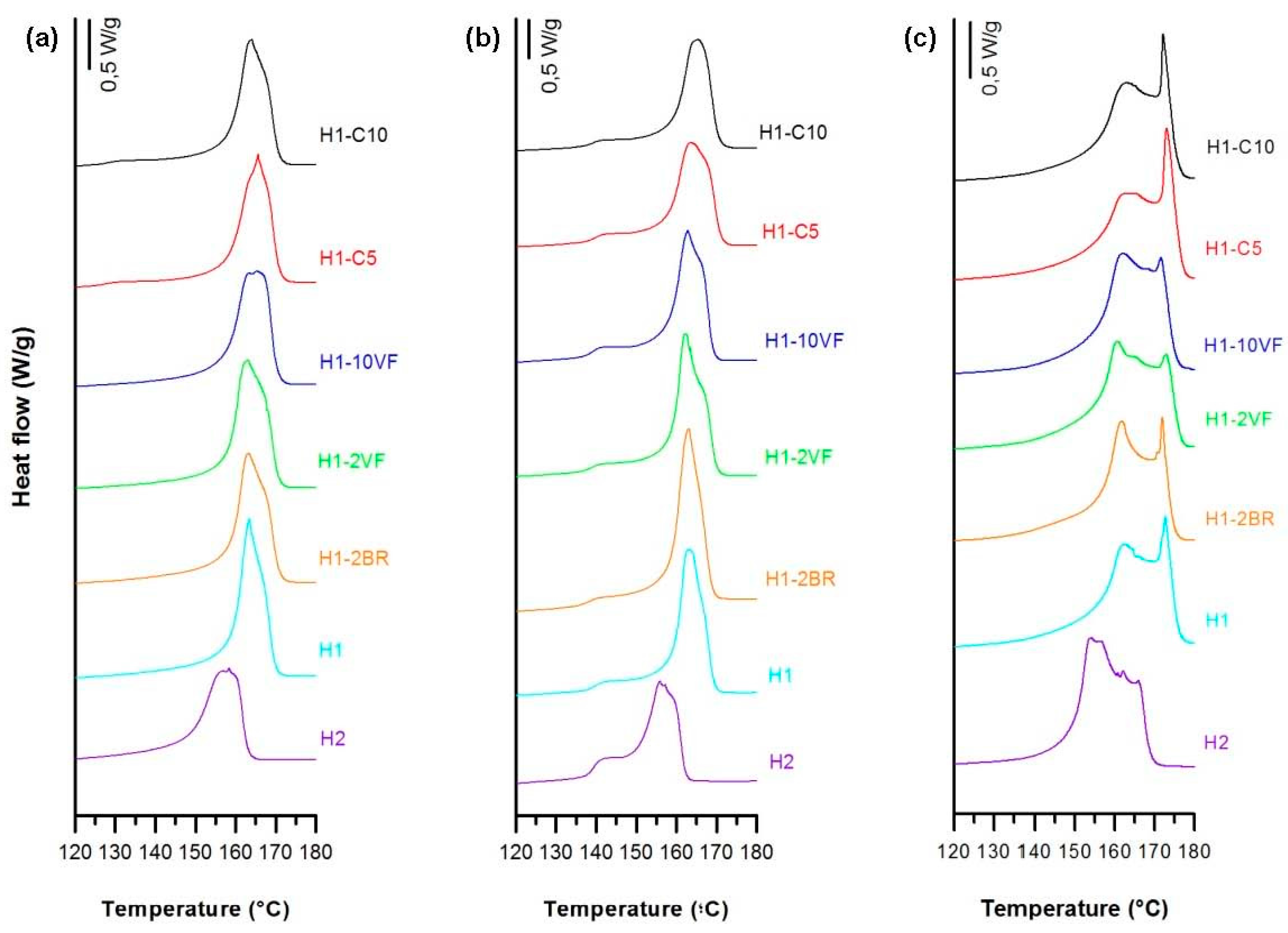
3.1. Influence of Polymer Composition
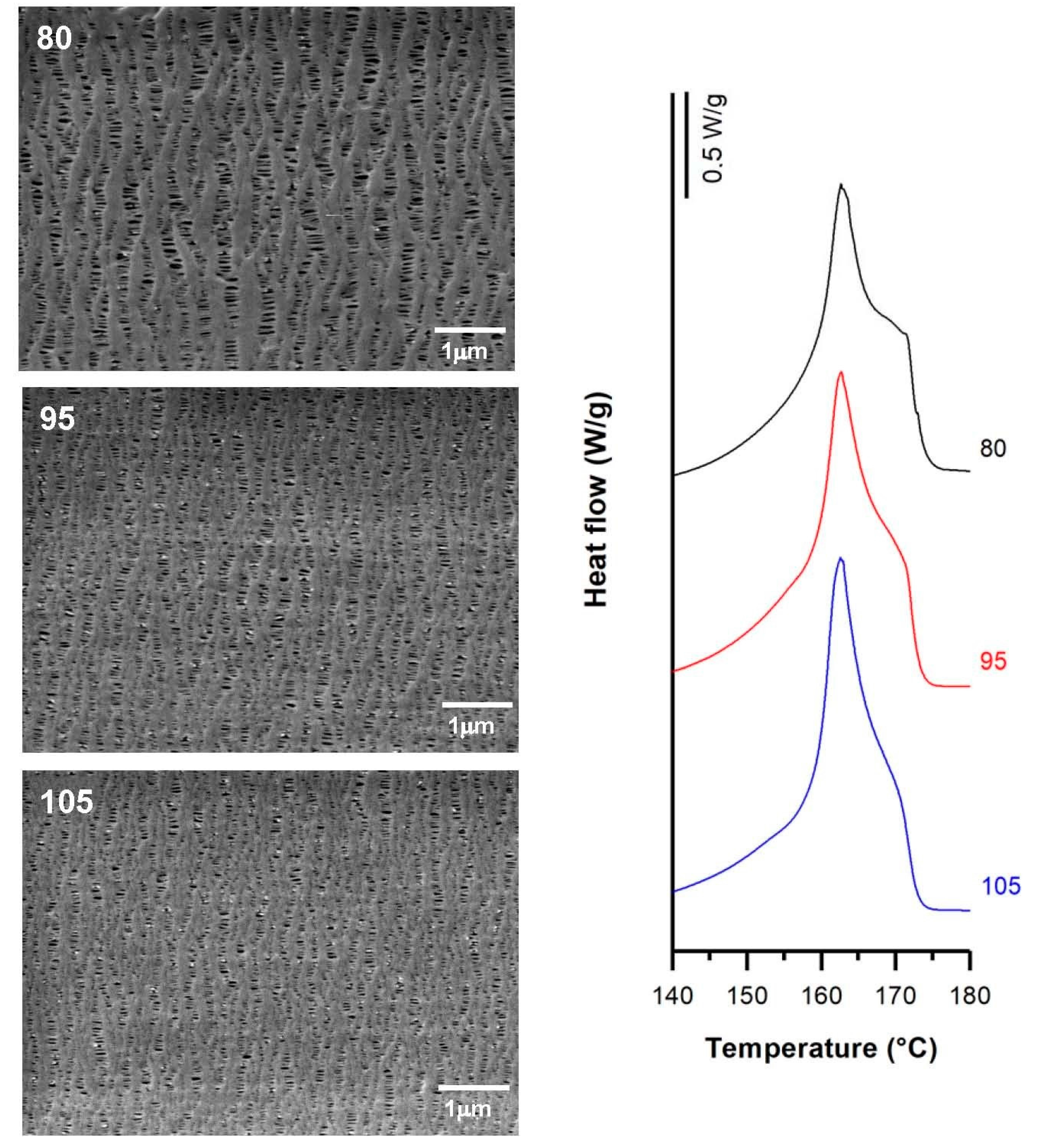
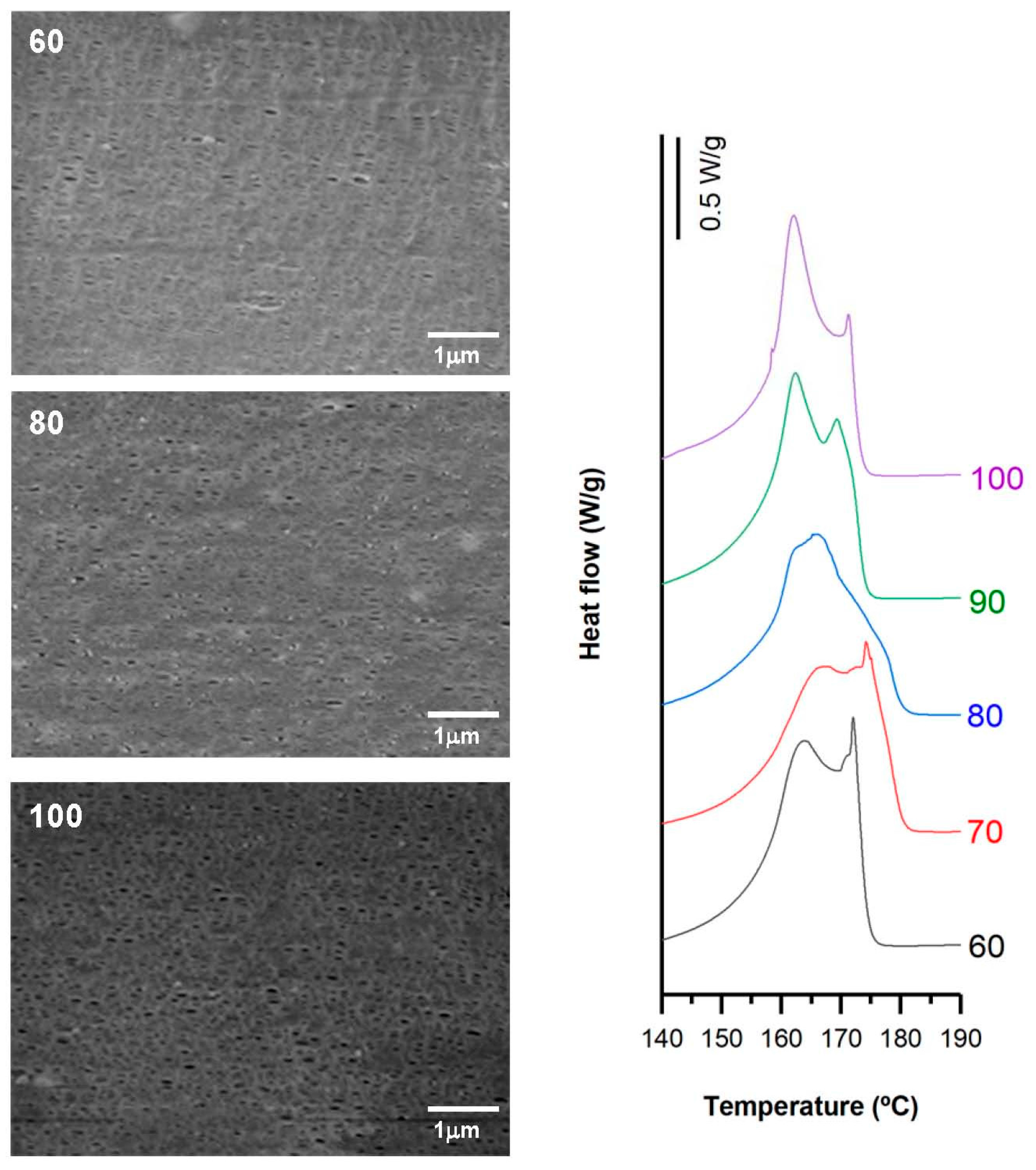
3.2. Influence of Draw Ratio
3.3. Influence of Uniaxial Strain Experimental Parameters
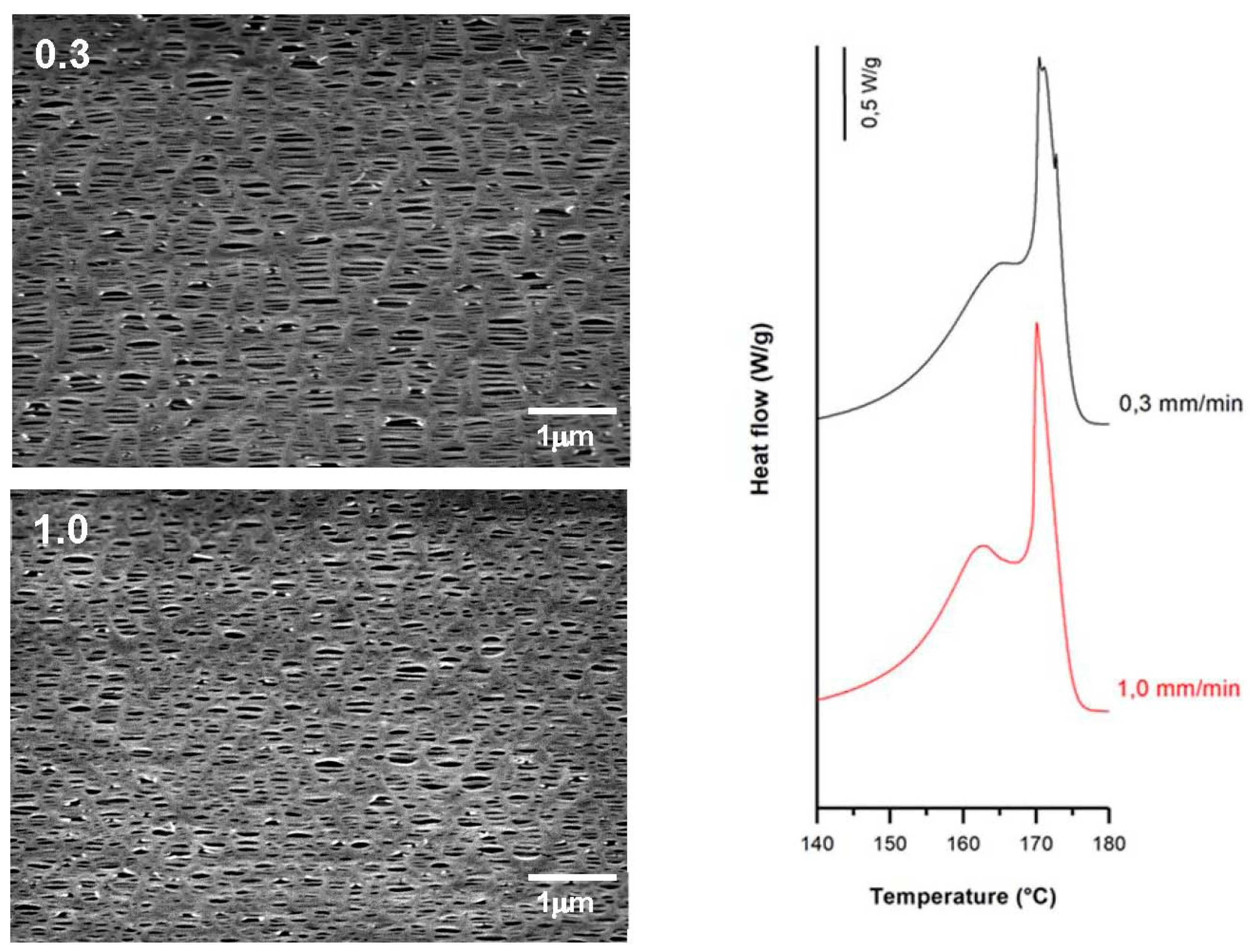
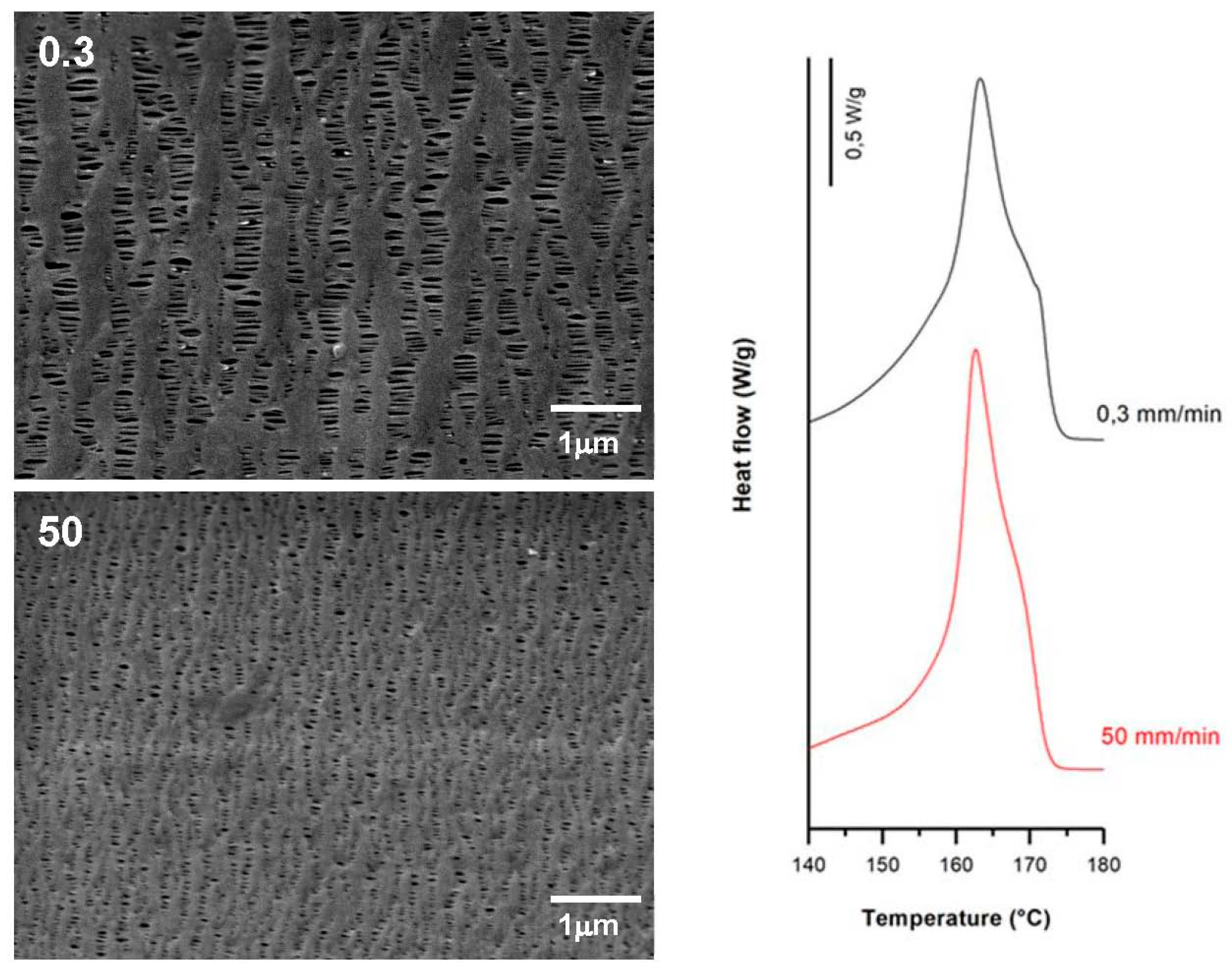
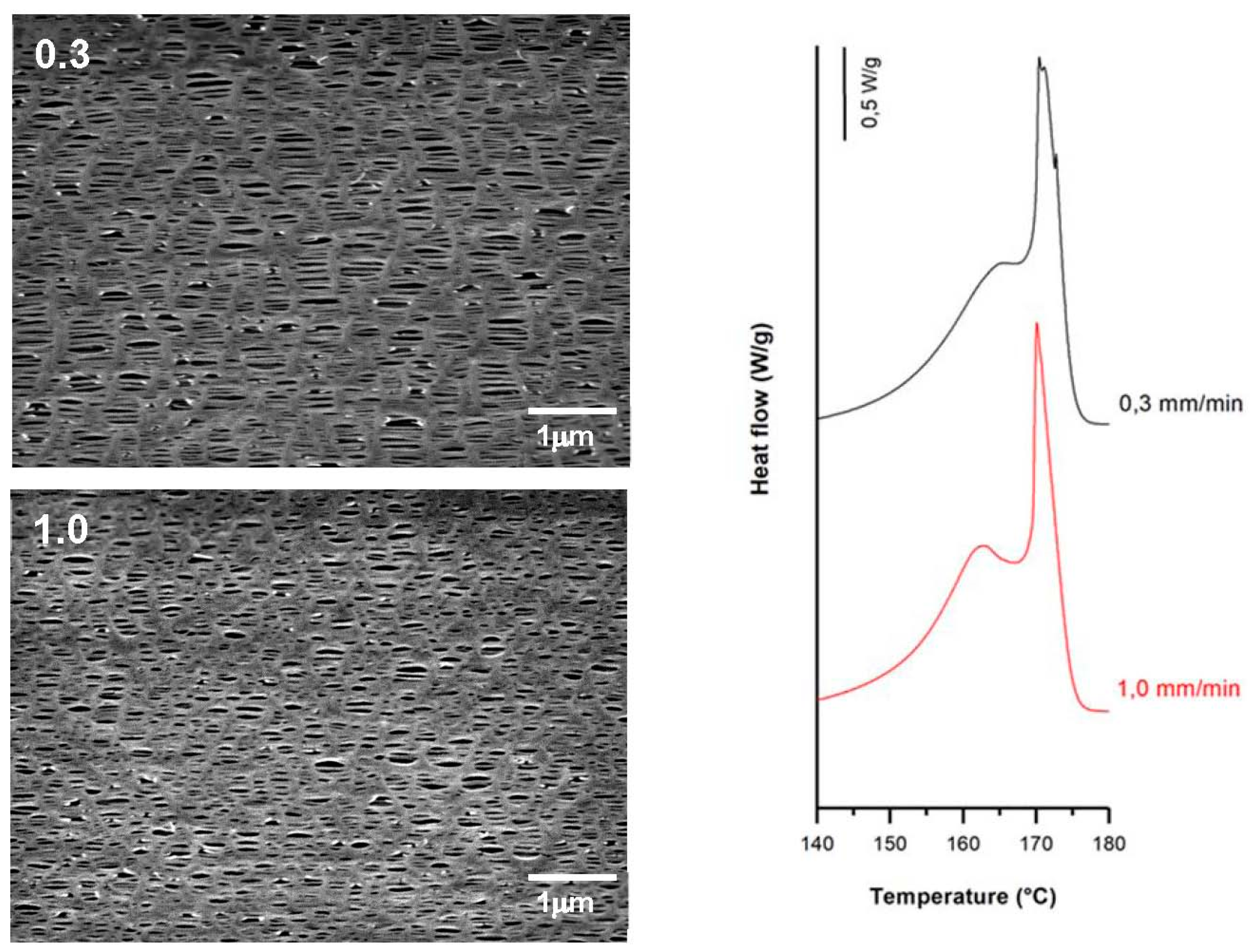
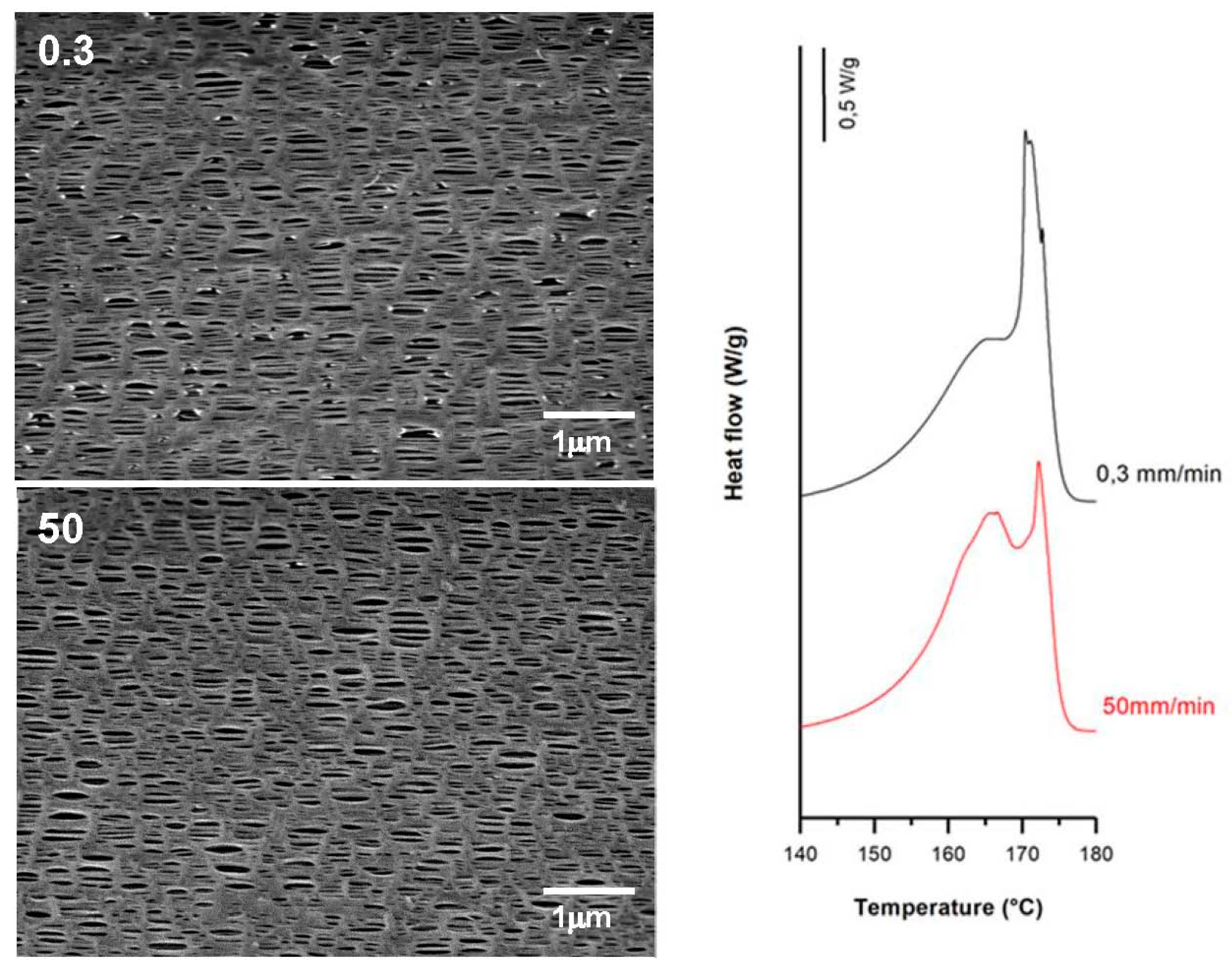
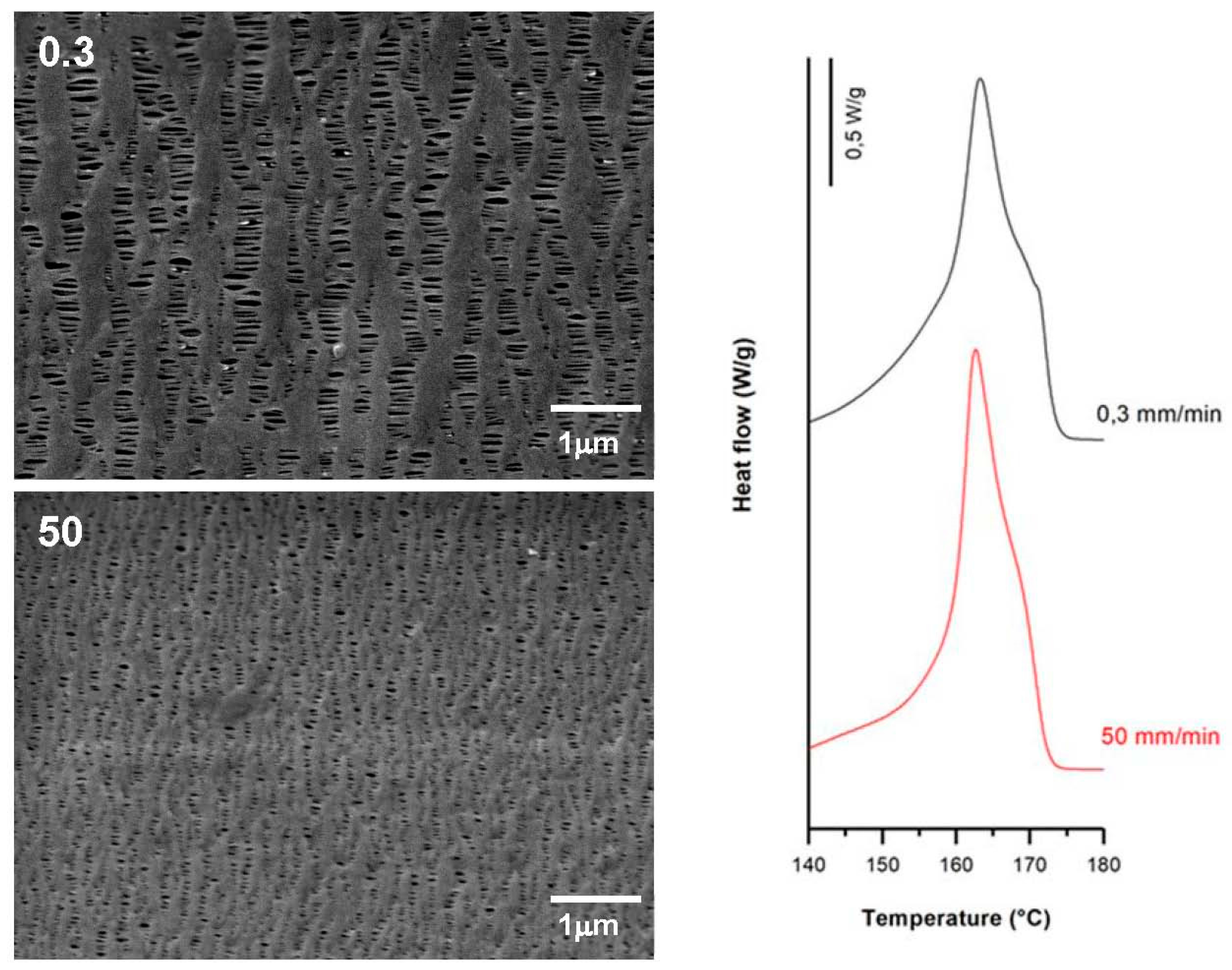
3.3.1. Effect of Strain Rate
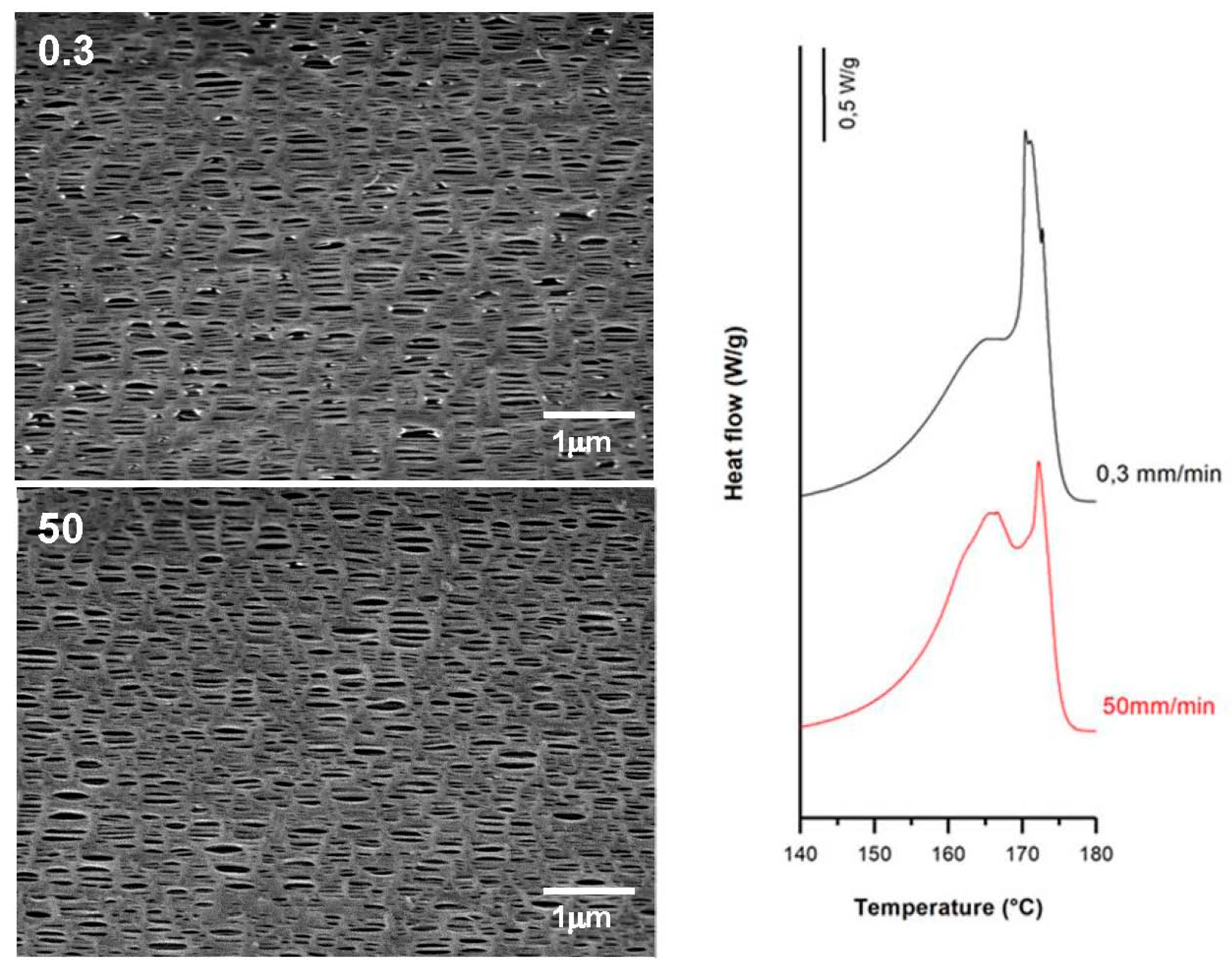
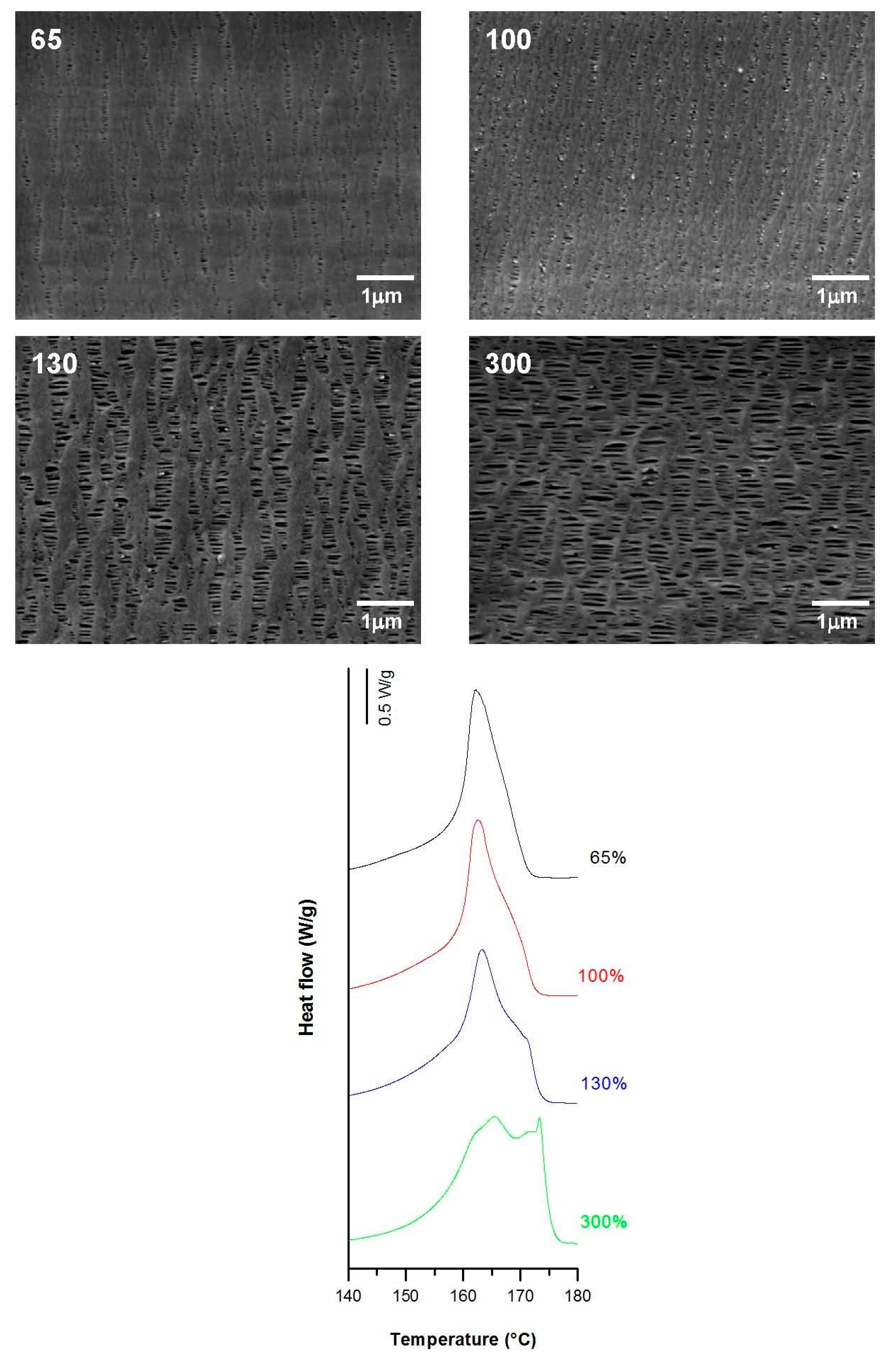
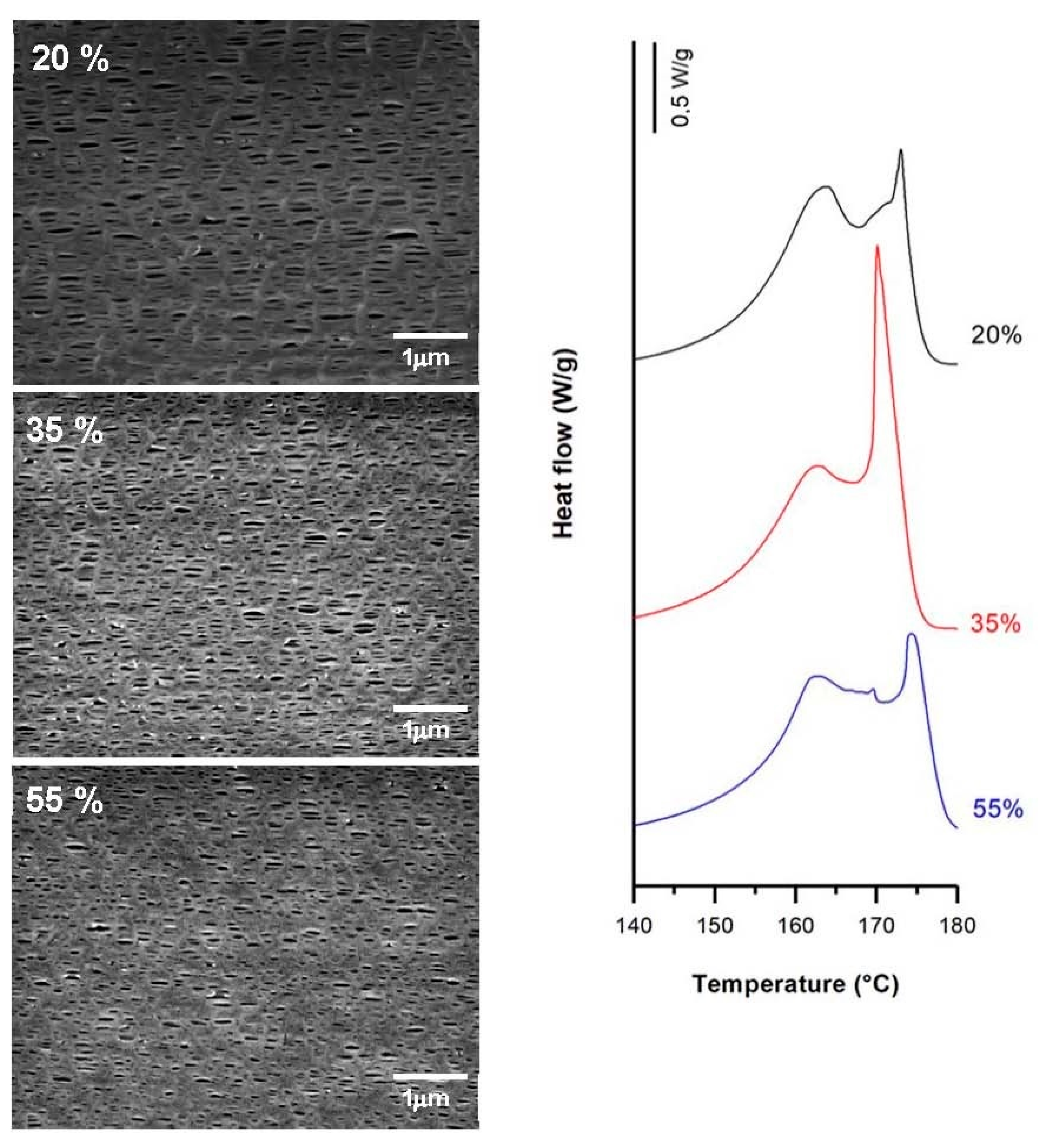
3.3.2. Effect of Strain Extent
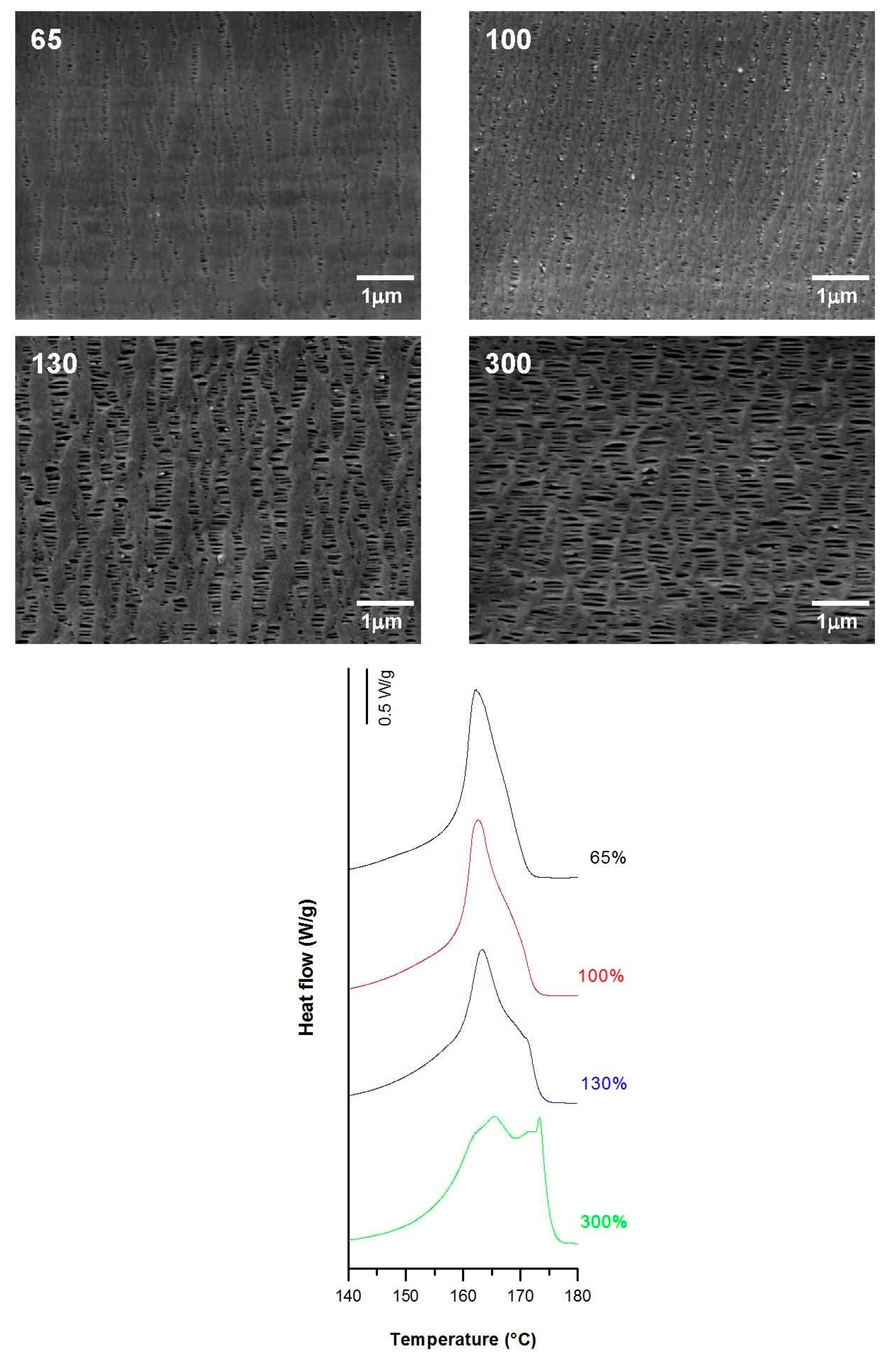
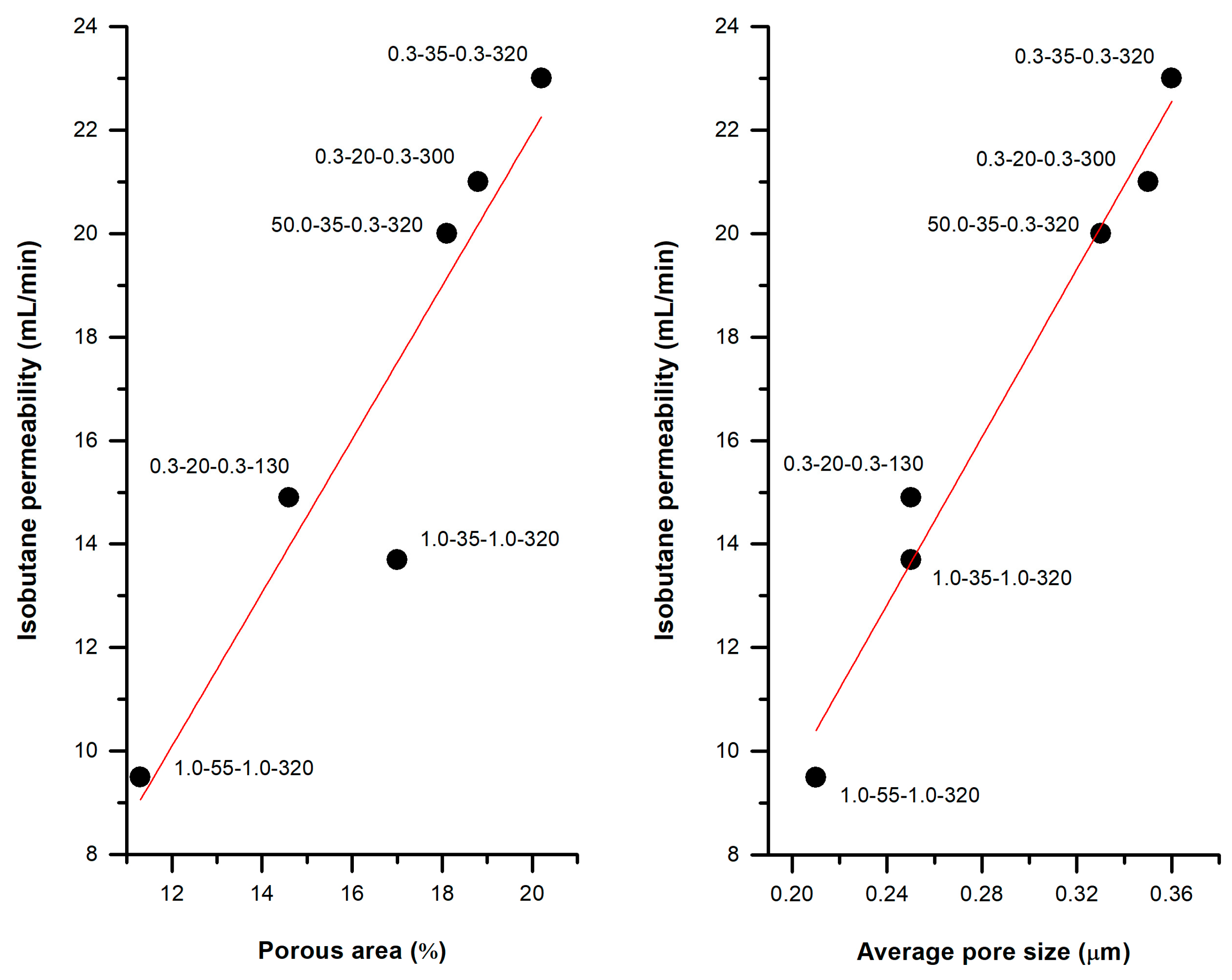
3.3.3. Practical Case: Membranes as Integral Parts of Lighters
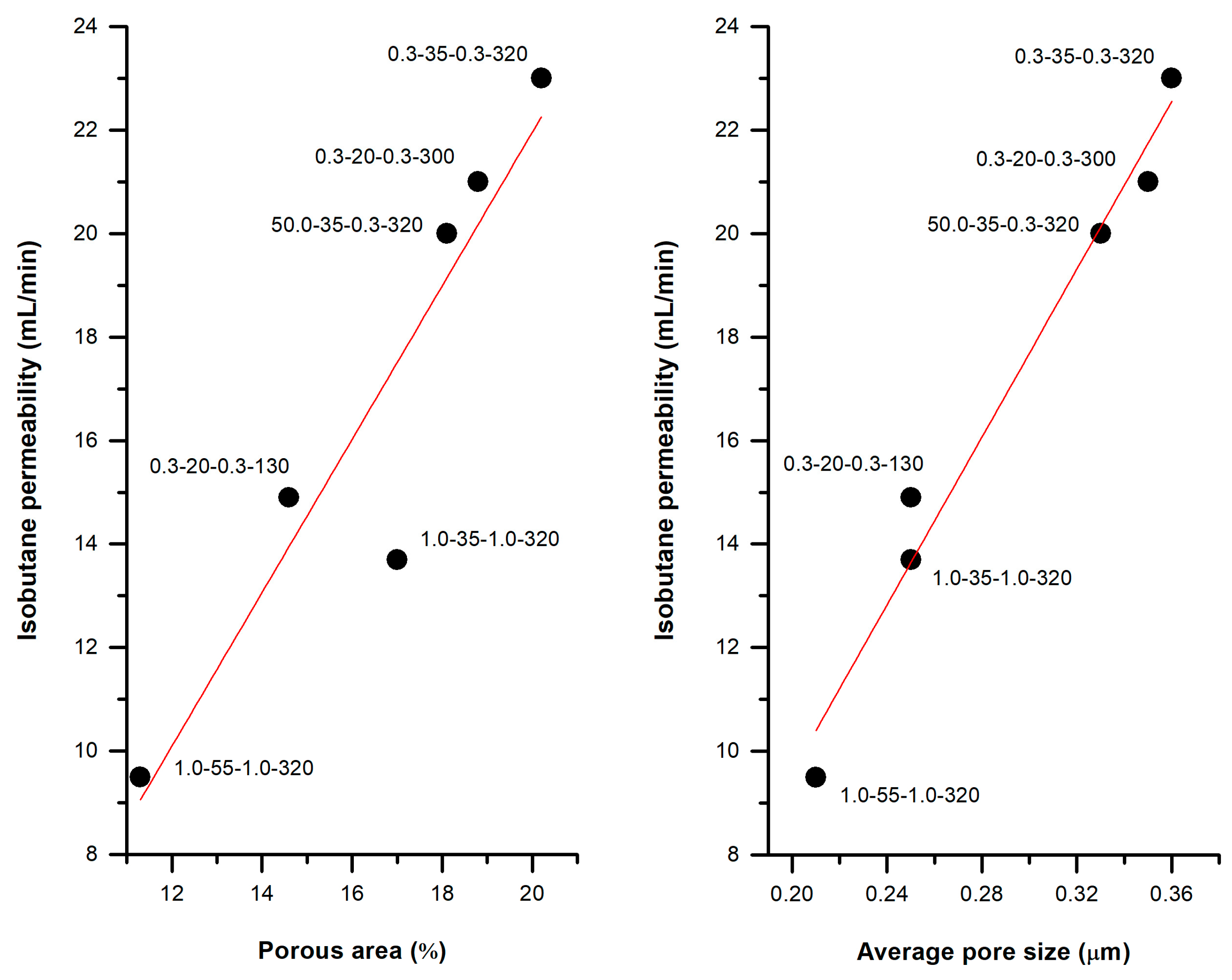
4. Discussion and Main Conclusions
Author Contributions
Conflicts of Interest
References
- Baker, R.W. Membrane Technology and Applications, 2nd ed.; John Wiley & Sons Ltd.: West Sussex, UK, 2004; pp. 3–17. ISBN 0-470-85445-6. [Google Scholar]
- Arora, P.; Zhang, Z. Battery separators. Chem. Rev. 2004, 104, 4419–4462. [Google Scholar] [CrossRef] [PubMed]
- Ulbricht, M. Advanced functional polymer membranes. Polymer 2006, 47, 2217–2262. [Google Scholar] [CrossRef]
- Tabatabaei, S.H.; Carreau, P.J.; Ajji, A. Microporous membranes obtained from polypropylene blend film by stretching. J. Membr. Sci. 2008, 325, 772–782. [Google Scholar] [CrossRef]
- Johnson, M.B.; Wilkes, G.L. Microporous membranes of polyoxymethylene from a melt-extrusion process: (II) Effects of thermal annealing and stretching on porosity. J. Appl. Polym. Sci. 2002, 84, 1762–1780. [Google Scholar] [CrossRef]
- Caihong, L.; Shuqiu, W.; Qi, C.; Ruijie, X.; Bing, H.; Wenqiang, S. Influence of heat-setting temperature on the properties of a stretched polypropylene microporous membrane. Polym. Int. 2014, 63, 584–588. [Google Scholar] [CrossRef]
- Sadeghi, F.; Ajji, A.; Carreau, P.J. Analysis of microporous membranes obtained from polypropylene films by stretching. J. Membr. Sci. 2007, 292, 62–71. [Google Scholar] [CrossRef]
- Sadeghi, F.; Ajji, A.; Carreau, P.J. Microporous membranes obtained from polypropylene blends with superior permeability properties. J. Membr. Sci. Part B Polym. Phys. 2008, 46, 148–157. [Google Scholar] [CrossRef]
- Tabatabaei, S.H.; Carreau, P.J.; Ajji, A. Effect of processing on the crystalline orientation, morphology, and mechanical properties of polypropylene cast films and microporous membrane formation. Polymer 2009, 50, 4228–4240. [Google Scholar] [CrossRef]
- Sadeghi, F.; Ajji, A.; Carreau, P.J. Analysis of row nucleated lamellar morphology of polypropylene obtained from the cast film process: Effect of melt rheology and process conditions. Polym. Eng. Sci. 2007, 47, 1170–1178. [Google Scholar] [CrossRef]
- Saffar, A.; Carreau, P.J.; Ajji, A.; Kamal, M.R. Influence of stretching on the performance of polypropylene-based microporous membranes. Ind. Eng. Chem. Res. 2014, 53, 14014–14021. [Google Scholar] [CrossRef]
- Nakamura, S.; Kaneko, S.; Mizutani, Y. Microporous polypropylene sheets containing CaCO3 filler. J. Appl. Polym. Sci. 1993, 49, 143–150. [Google Scholar] [CrossRef]
- Nagō, S.; Mizutani, Y. Microporous polypropylene sheets containing CaCO3 filler: Effects of stretching ratio and removing CaCO3 filler. J. Appl. Polym. Sci. 1998, 68, 1543–1553. [Google Scholar] [CrossRef]
- Ferrer-Balas, D.; Maspoch, M.L.; Martinez, A.B.; Santana, O.O. Influence of annealing on the microstructural, tensile and fracture properties of polypropylene films. Polymer 2001, 42, 1697–1705. [Google Scholar] [CrossRef]
- Tabatabaei, S.H.; Carreau, P.J.; Ajji, A. Structure and properties of MDO stretched polypropylene. Polymer 2009, 50, 3981–3989. [Google Scholar] [CrossRef]
- Ding, Z.; Bao, R.; Zhao, B.; Yan, J.; Liu, Z.; Yang, M. Effects of annealing on structure and deformation mechanism of isotactic polypropylene film with row-nucleated lamellar structure. J. Appl. Polym. Sci. 2013, 130, 1659–1666. [Google Scholar] [CrossRef]
- Xiande, C.; Ruijie, X.; Jiayi, X.; Yuanfei, L.; Caihong, L.; Liangbin, L. The study of room-temperature stretching of annealed polypropylene cast film with row-nucleated crystalline structure. Polymer 2016, 94, 31–42. [Google Scholar] [CrossRef]
- Wang, S.; Saffar, A.; Ajji, A.; Wu, H.; Guo, S.Y. Fabrication of microporous membranes from melt extruded polypropylene precursor films via stretching: Effect of annealing. Chin. J. Polym. Sci. 2015, 33, 1028–1037. [Google Scholar] [CrossRef]
- Qi, C.; Ruijie, X.; Shuqiu, W.; Changbin, C.; Haibin, M.; Caihong, L.; Li, Z. Influence of annealing temperature on the lamellar and connecting bridge structure of stretched polypropylene microporous membrane. Polym. Int. 2015, 64, 446–452. [Google Scholar] [CrossRef]
- Caihong, L.; Weiliang, H.; Ruijie, X.; Yunqi, X. The correlation between the lower temperature melting plateau endotherm and the stretching-induced pore formation in annealed polypropylene films. J. Plast. Film Sheet 2012, 28, 151–164. [Google Scholar] [CrossRef]
- Saffar, A.; Carreau, P.J.; Ajji, A.; Kamal, M.R. The impact of new crystalline lamellae formation during annealing on the properties of polypropylene based films and membranes. Polymer 2014, 55, 3156–3167. [Google Scholar] [CrossRef]
- Lin, Y.; Meng, L.; Wu, L.; Li, X.; Chen, X.; Zhang, Q.; Li, L. A semi-quantitative deformation model for pore formation in isotactic polypropylene microporous membrane. Polymer 2015, 80, 214–227. [Google Scholar] [CrossRef]
- Caihong, L.; Shuqiu, W.; Ruijie, X.; Qi, C.; Bing, H.; Xinlong, P.; Wenqiang, S. Formation of stable crystalline connecting bridges during the fabrication of polypropylene microporous membrane. Polym. Bull. 2013, 70, 1353–1366. [Google Scholar] [CrossRef]
- Xie, J.; Xu, R.; Chen, X.; Cai, Q.; Lei, C. Influence of heat-treatment temperature on the structure and properties of polypropylene microporous membrane. Polym. Bull. 2016, 73, 265–277. [Google Scholar] [CrossRef]
- Wu, S.; Lei, C.; Cai, Q.; Xu, R.; Hu, B.; Shi, W.; Peng, X. Study of structure and properties of polypropylene microporous membrane by hot stretching. Polym. Bull. 2014, 71, 2205–2217. [Google Scholar] [CrossRef]
- Caihong, L.; Shuqiu, W.; Ruijie, X.; Xinlong, P.; Wenqiang, S.; Bing, H. Influence of low molecular weight tail of polypropylene resin on the pore structure by room-temperature stretching. Polym. Eng. Sci. 2013, 53, 2594–2602. [Google Scholar] [CrossRef]
- Kim, J.; Kim, S.S.; Park, M.; Jang, M. Effects of precursor properties on the preparation of polyethylene hollow fiber membranes by stretching. J. Membr. Sci. 2008, 318, 201–209. [Google Scholar] [CrossRef]
- Supaphol, P.; Harnsiri, W.; Junkasem, J. Effects of calcium carbonate and Its Purity on crystallization and melting behavior, mechanical properties, and processability of syndiotactic polypropylene. J. Appl. Polym. Sci. 2004, 92, 201–212. [Google Scholar]
- Barczewski, M.; Kloziński, A.; Jakubowska, P.; Sterzyński, T. Nonisothermal crystallization of highly-filled polyolefin/calcium carbonate composites. J. Appl. Polym. Sci. 2014, 131, 41201. [Google Scholar] [CrossRef]
- Martínez, M.C.; Benavente, R.; Gómez-Elvira, J.M. Molecular weight dependence and stereoselective chain cleavage during the early stages of the isotactic polypropylene pyrolysis. Polym. Degrad. Stab. 2017, 143, 26–34. [Google Scholar] [CrossRef]
- Rychlý, J.; Matisová-Rychlá, L.; Csmorová, K.; Janigová, I. Non-isothermal thermogravimetry, differential scanning calorimetry and chemiluminiscence in degradation of polyethylene, polypropylene, polystyrene and poly(methyl methacrylate). Polym. Degrad. Stab. 2011, 96, 1573–1581. [Google Scholar]
- Nasrin, R.; Gafur, M.A.; Bhuiyan, A.H. Characterization of isotactic polypropylene talc composites prepared by extrusion cum compression molding technique. Mater. Sci. Appl. 2015, 6, 925–934. [Google Scholar] [CrossRef]
- Arranz-Andrés, J.; Peña, B.; Benavente, R.; Pérez, E.; Cerrada, M.L. Influence of tacticity and molecular weight on the properties of metallocenic isotactic polypropylene. Eur. Polym. J. 2007, 43, 2357–2370. [Google Scholar] [CrossRef]
- Kamal, M.; Sharma, C.S.; Upadhyaya, P.; Verma, V.; Pandey, K.N.; Kumar, V.; Agrawal, D.D. Calcium carbonate (CaCO3) nanoparticle filled polypropylene: Effect of particle Surface treatment on mechanical, thermal, and morphological performance of composites. J. Appl. Polym. Sci. 2012, 124, 2649–2656. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Commercial Tradename | Code | Polymer Architecture | MFI a (dg/min) |
|---|---|---|---|
| ISPLEN PP020 G3E | H1 | Linear | 0.9 |
| ISPLEN PP034 W3F | H2 | Linear | 2.2 |
| ISPLEN PP099 K2M | VF | Linear | 55.0 |
| DAPLOY WB140HMS | BR | Branched | 2.1 |
| Sample | Fc | Xm (%) | Pore Density (pores/μm2) | Porous Area (%) | Average Pore Size (µm) | Porosity (%) | Gurley Permeability [µm/(Pa·s)] |
|---|---|---|---|---|---|---|---|
| H1 | 0.61 | 57.0 | 8.7 | 6.3 | 0.15 | 1.01 | 0.19 |
| H2 | 0.44 | 63.7 | 2.1 | 1.3 | 0.13 | 0.23 | 0.01 |
| H1-2BR | 0.51 | 60.9 | 10.7 | 6.2 | 0.13 | 0.69 | 0.15 |
| H1-2VF | 0.57 | 60.6 | 6.0 | 4.7 | 0.17 | 0.61 | 0.14 |
| H1-10VF | 0.51 | 60.9 | 3.7 | 2.3 | 0.14 | 0.47 | 0.02 |
| H1-C5 | 0.49 | 57.4 | 13.6 | 15.3 | 0.10 | 1.81 | 0.64 |
| H1-C10 | 0.54 | 58.6 | 15.5 | 14.2 | 0.09 | 1.75 | 0.49 |
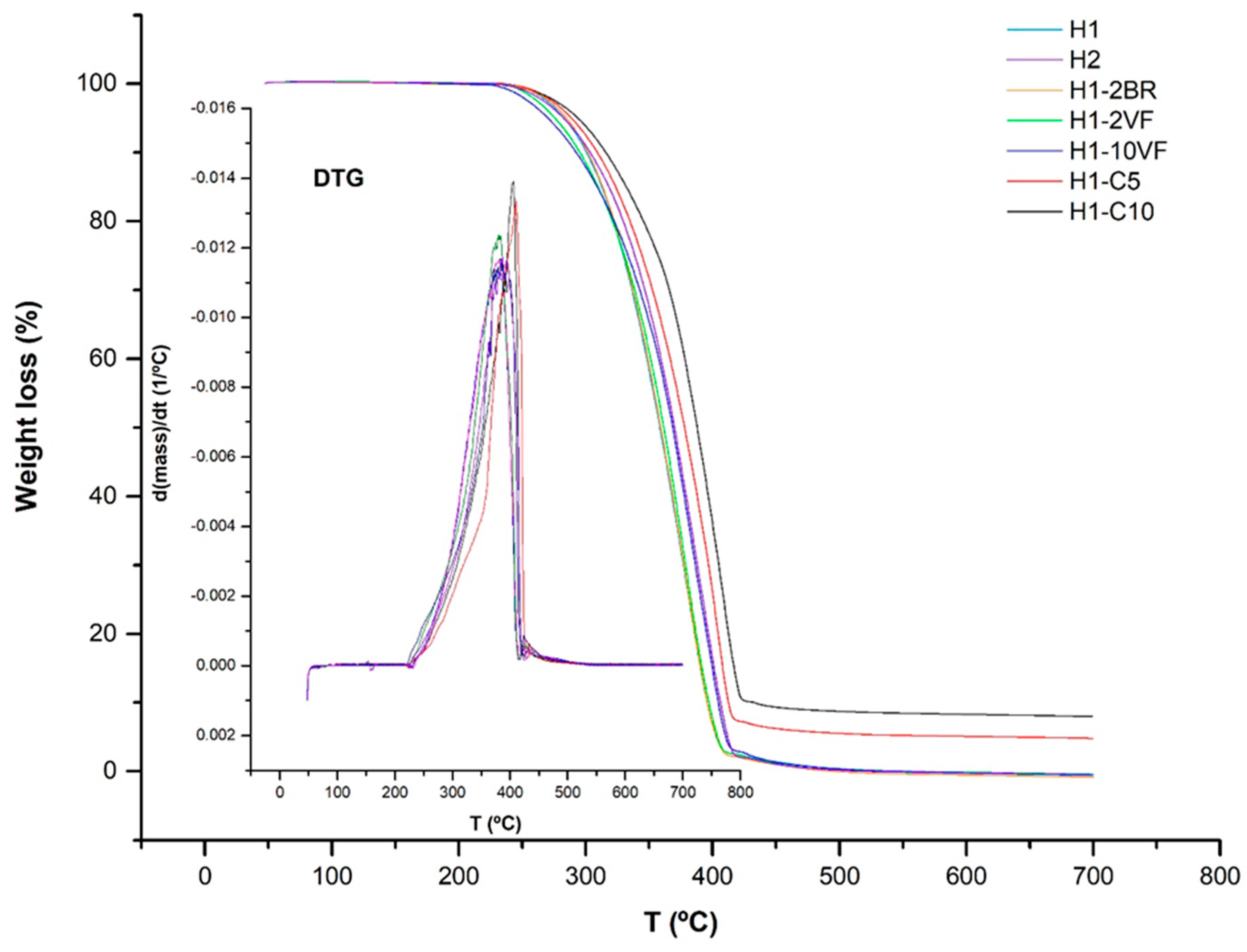
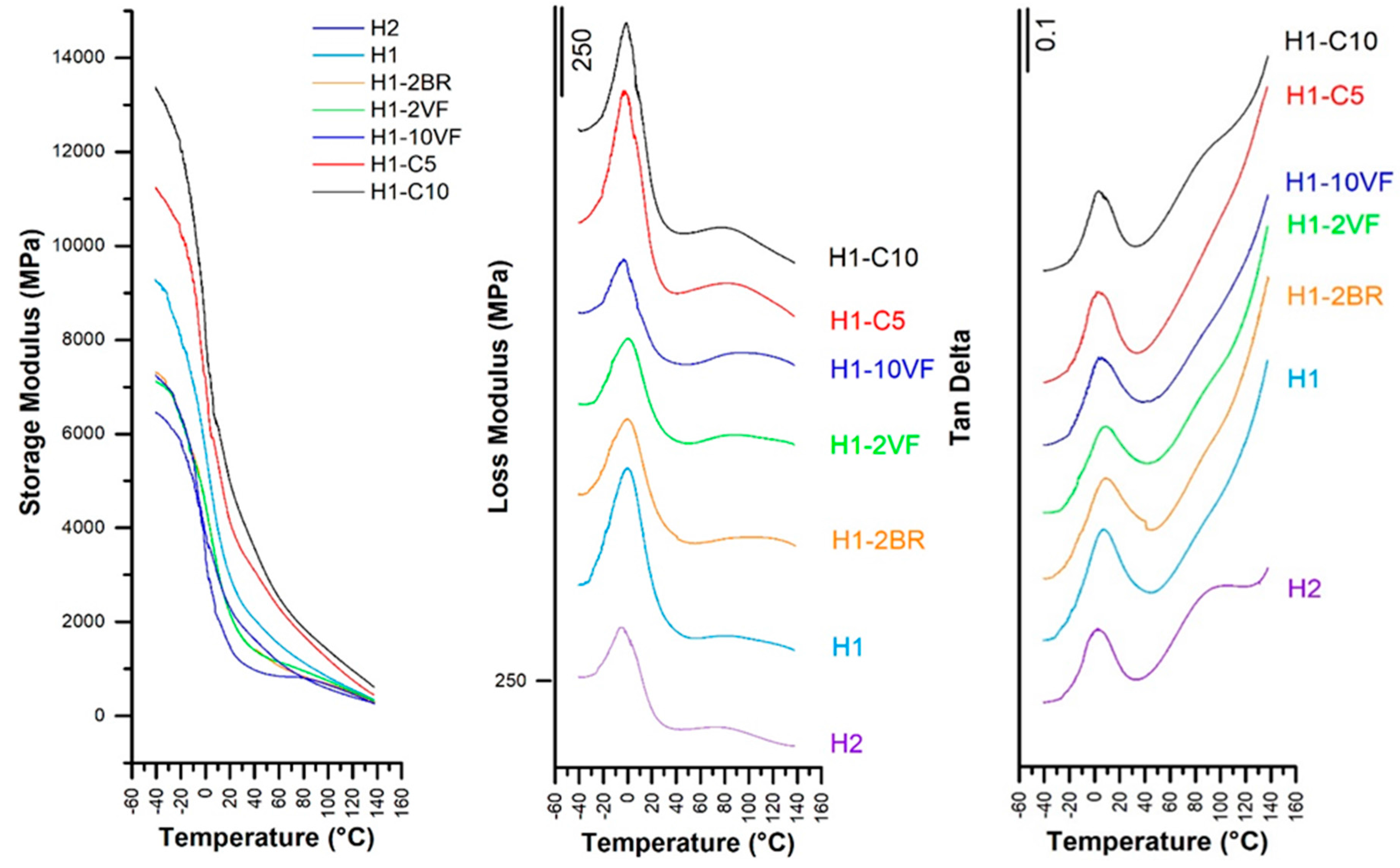
| Membrane | T0.1 (°C) | T0.5 (°C) | Lost Mass 400 °C (%) | Lost Mass 600 °C (%) | Tmax (°C) | E’ −30 °C (MPa) | E’23 °C (MPa) | E’ 100 °C (MPa) | E’’ Peak (°C) | Tan δ (°C) |
|---|---|---|---|---|---|---|---|---|---|---|
| H1 | 300 | 358 | 93.0 | 100 | 381 | 8811 | 2759 | 828 | −0.03 | 0.12 |
| H2 | 303 | 370 | 83.5 | 100 | 387 | 6977 | 2167 | 580 | −5.74 | 0.10 |
| H1-2BR | 301 | 359 | 93.0 | 100 | 381 | 6956 | 1995 | 681 | −0.48 | 0.11 |
| H1-2VF | 295 | 361 | 92.6 | 100 | 380 | 6921 | 1994 | 748 | 0.56 | 0.11 |
| H1-10VF | 292 | 368 | 85.3 | 100 | 383 | 6207 | 1334 | 662 | −3.74 | 0.10 |
| H1-C5 | 310 | 377 | 74.6 | 94.9 | 410 | 10830 | 3898 | 1210 | −3.14 | 0.11 |
| H1-C10 | 317 | 388 | 63.7 | 91.7 | 406 | 12835 | 4705 | 1401 | −0.99 | 0.10 |
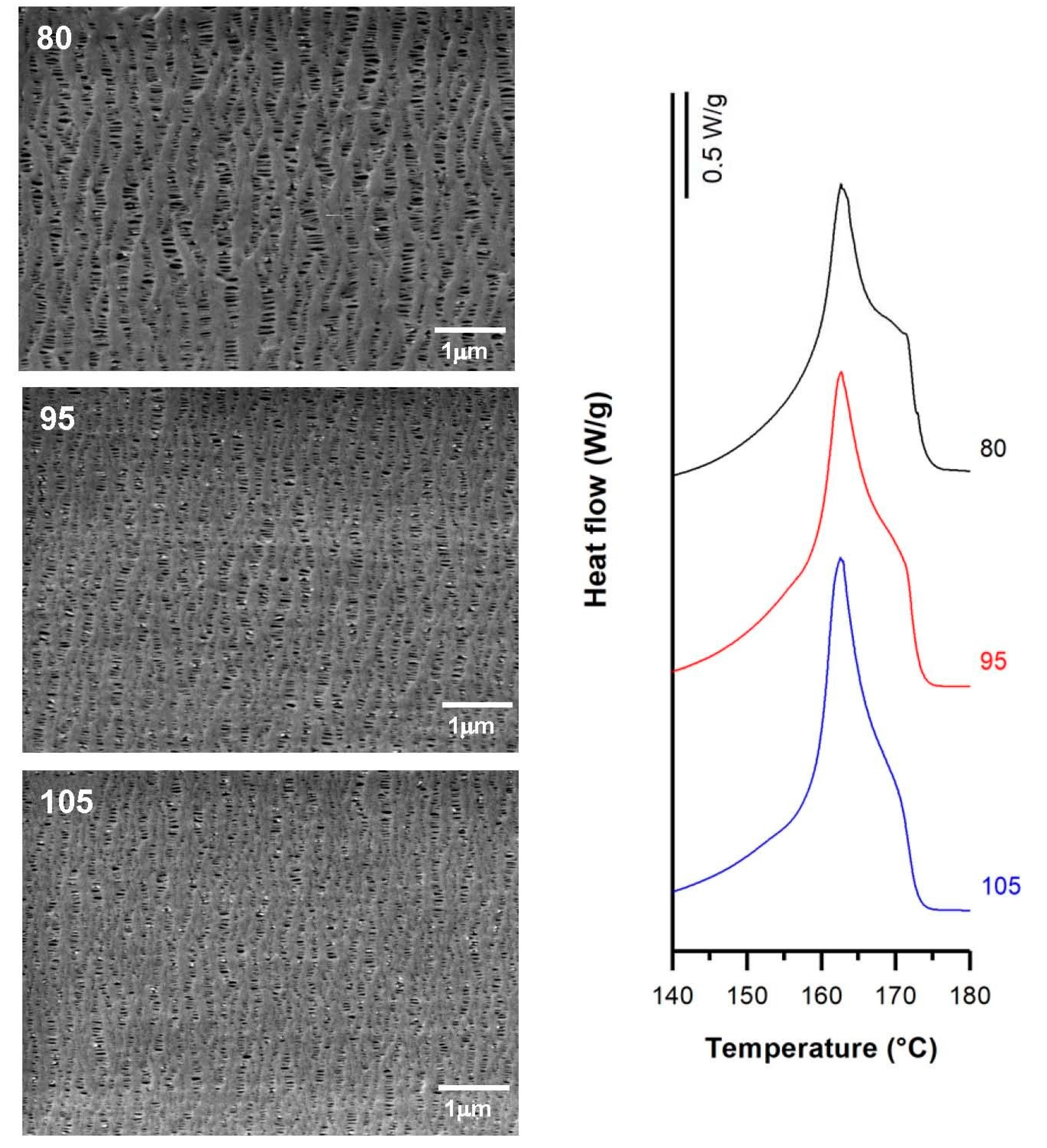
| Sample | Draw Ratio | Fc | Xm(%) | Pore Density (pores/μm2) | Porous Area (%) | Average Pore Size (µm) | Porosity (%) | Gurley Permeability [µm/(Pa·s)] |
|---|---|---|---|---|---|---|---|---|
| H1-2BR | 80 | 0.51 | 49.6 | 12.1 | 9.8 | 0.17 | 0.93 | 0.18 |
| 95 | 0.54 | 60.6 | 12.3 | 5.9 | 0.13 | 0.75 | 0.16 | |
| 105 | 0.55 | 61.9 | 13.4 | 7.7 | 0.14 | 0.71 | 0.16 | |
| H1-C10 | 60 | 0.40 | 45.2 | 11.9 | 12.1 | 0.09 | 0.59 | 0.06 |
| 70 | 0.52 | 55.5 | 10.3 | 11.7 | 0.09 | 0.70 | 0.15 | |
| 80 | 0.54 | 62.9 | 14.2 | 14.8 | 0.10 | 1.42 | 0.49 | |
| 90 | 0.59 | 63.5 | 7.3 | 11.0 | 0.11 | 1.51 | 0.52 | |
| 100 | 0.60 | 63.9 | 7.9 | 10.5 | 0.11 | 1.61 | 0.58 |
| Cold Stage | Hot Stage | Xm(%) | Pore Density (pores/μm2) | Porous Area (%) | Pore Size (µm) | Porosity (%) | Gurley Permeability [µm/(Pa·s)] | ||
|---|---|---|---|---|---|---|---|---|---|
| Strain Rate (mm/min) | Extent (%) | Strain Rate (mm/min) | Extent (%) | ||||||
| 0.3 | 35 | 0.3 | 320 | 62.1 | 9.3 | 20.2 | 0.36 | 1.28 | 0.29 |
| 1.0 | 1.0 | 62.5 | 10.5 | 17.0 | 0.25 | 1.05 | 0.24 | ||
| 0.3 | 35 | 0.3 | 320 | 62.1 | 9.3 | 20.2 | 0.36 | 1.28 | 0.29 |
| 50.0 | 62.2 | 8.9 | 18.1 | 0.33 | 1.22 | 0.25 | |||
| 0.3 | 20 | 0.3 | 130 | 60.7 | 9.4 | 14.6 | 0.25 | 1.21 | 0.22 |
| 50.0 | 59.2 | 12.9 | 4.9 | 0.09 | 0.51 | 0.07 | |||
| 1.0 | 20 | 1.0 | 320 | 57.4 | 6.8 | 13.9 | 0.32 | 0.97 | 0.22 |
| 35 | 62.5 | 10.5 | 17.0 | 0.25 | 1.05 | 0.24 | |||
| 55 | 58.9 | 8.9 | 11.3 | 0.21 | 0.95 | 0.20 | |||
| 0.3 | 20 | 0.3 | 65 | 60.8 | 9.3 | 2.8 | 0.09 | 0.30 | 0.02 |
| 100 | 59.2 | 14.0 | 5.4 | 0.11 | 0.55 | 0.08 | |||
| 130 | 60.7 | 9.4 | 14.6 | 0.25 | 1.21 | 0.22 | |||
| 300 | 65.6 | 7.8 | 18.8 | 0.35 | 1.25 | 0.26 | |||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castejón, P.; Habibi, K.; Saffar, A.; Ajji, A.; Martínez, A.B.; Arencón, D. Polypropylene-Based Porous Membranes: Influence of Polymer Composition, Extrusion Draw Ratio and Uniaxial Strain. Polymers 2018, 10, 33. https://doi.org/10.3390/polym10010033
Castejón P, Habibi K, Saffar A, Ajji A, Martínez AB, Arencón D. Polypropylene-Based Porous Membranes: Influence of Polymer Composition, Extrusion Draw Ratio and Uniaxial Strain. Polymers. 2018; 10(1):33. https://doi.org/10.3390/polym10010033
Chicago/Turabian StyleCastejón, Pilar, Kian Habibi, Amir Saffar, Abdellah Ajji, Antonio B. Martínez, and David Arencón. 2018. "Polypropylene-Based Porous Membranes: Influence of Polymer Composition, Extrusion Draw Ratio and Uniaxial Strain" Polymers 10, no. 1: 33. https://doi.org/10.3390/polym10010033
APA StyleCastejón, P., Habibi, K., Saffar, A., Ajji, A., Martínez, A. B., & Arencón, D. (2018). Polypropylene-Based Porous Membranes: Influence of Polymer Composition, Extrusion Draw Ratio and Uniaxial Strain. Polymers, 10(1), 33. https://doi.org/10.3390/polym10010033